

Abstract
Qiliqiangxin (QL), a traditional Chinese medicine, had long been used to treat chronic heart failure. Recent studies revealed that differentiation of cardiac fibroblasts (CFs) into myofibroblasts played an important role in cardiac remodelling and development of heart failure, however, little was known about the underlying mechanism and whether QL treatment being involved. This study aimed to investigate the effects of QL on angiotensin II (AngII)-induced CFs transdifferentiation. Study was performed on in vitro cultured CFs from Sprague–Dawley rats. CFs differentiation was induced by AngII, which was attenuated by QL through reducing transforming growth factor-β1 (TGF-β1) and α-smooth muscle actin (α-SMA). Our data showed that AngII-induced IL-6 mRNA as well as typeI and typeIII collagens were reduced by QL. IL-6 deficiency could suppress TGF-β1 and α-SMA, and both IL-6 siRNA and QL-mediated such effect was reversed by foresed expression of recombined IL-6. Increase in actin stress fibres reflected the process of CFs differentiation, we found stress fibres were enhanced after AngII stimulation, which was attenuated by pre-treating CFs with QL or IL-6 siRNA, and re-enhanced after rIL-6 treatment. Importantly, we showed that calcineurin-dependent NFAT3 nuclear translocation was essential to AngII-mediated IL-6 transcription, QL mimicked the effect of FK506, the calcineurin inhibitor, on suppression of IL-6 expression and stress fibres formation. Collectively, our data demonstrated the negative regulation of CFs differentiation by QL through an IL-6 transcriptional mechanism that depends on inhibition of calcineurin/NFAT3 signalling.
Keywords: qiliqiangxin, cardiac fibroblast, interleukin-6, transdifferentiation, NFAT3
Introduction
Myocardial fibrosis is a multi-factorial process which includes the progressively increased transdifferentiation of cardiac fibroblasts (CFs) into myofibroblasts. The phenotype of ‘persistent myofibroblast’ may lead to increased production of extracellular matrix (ECM), including collagen deposition, enhanced expresson and translocation of α-smooth muscle actin (α-SMA) and transforming growth factor-β1 (TGF-β1) signalling activation. These events are essential for the development of cardiac remodelling and chronic heart failure 1–3.
Qiliqiangxin (QL) is a traditional Chinese herb medicine, including mainly Radix Astragali, Radix Ginseng, Salvia Miltiorrhiza, etc. Recent studies show that QL could improve both systolic and diastolic cardiac function in spontaneously hypertensive rats through inhibiting cardiac chymase-mediated angiotensin II production 4. Pressure overload-induced myocardial hypertrophy and inflammation response is alleviated by QL through inhibiting the ErbB receptors signalling and CBP/p300 nuclear transaction 5. QL also has effect on balancing the pro-inflammatory and anti-inflammatory cytokines during myocardial infarction 6. Recently, QL is proved to be effective on inhibition of calcium signalling by blocking the L-type Ca2+ channel (ICa-L), which weakens myocardial contractility and improves cardiac remodelling 7. However, the protective role of QL in heart, particularly which kind of cardiac cell is involved, is not well understood. CFs are the primary cardiac cells present in myocardium and participate in the formation of the myocardial ECM. Transdifferentiation of CFs into myofibroblast plays a critical role in fibrillar collagens deposition and cardiac fibrosis 8,9. Therefore, we test the hypothesis that QL might improve cardiac function through regulating CFs transdifferentiation.
Recent studies demonstrated that Angiotensin II (AngII) plays a important role in fibroblasts differentiation 10,11, and AngII is also critically involved in chronic inflammation and progessive fibrosis during pressure overload 12–14. AngII infusion significantly increased Interleukin-6(IL-6) expression in rats’ heart, meanwhile elevated IL-6 serum levels promoted cardiac fibrosis through TGF-β1/Smad (TGF-β1/Smad) signalling pathway 15, and promoting transdifferentiation of CFs into myofibroblasts 16,17. Myofibroblasts formation promotes the profibrotic response and cardiac remodelling 18 in pressure overload hearts 5 and cardiomyopathy 19. Meanwhile, evidence show that IL-6 significantly increases collagen deposition and ventricular stiffness 17. Anti-inflammatory therapy might be a promising strategy against CFs transdifferentiation and cardiac fibrosis 20. Regarding to the anti-inflammatory role of QL in pressure overloaded heart, in this study we investigate the effect of QL on AngII-induced CFs, the possible mechanism for QL on CFs transdifferentiation and inflammation targets.
Materials and methods
Materials
Low-glucose DMEM, foetal bovine serum (FBS), Trypsin and HBSS were from Gibco Pasadena, CA (USA). Qiliqiangxin extracts were provided by Shijiazhuang Yiling Pharmaceutical Co., Shijiazhuang Ltd (China); Olmesartan (OLM) was purchased from Shanghai Sankyo Pharmaceutical Co., Shanghai Ltd. (China); Ang II was purchased from Sigma-Aldrich Co., St. Louis, Missouri LLC. (USA); Rabbit anti-rat α-SMA polyclonal antibody was from Bioworld Technology, Nanjing Inc. (China); Rabbit anti-rat TGF-β1 polyclonal antibody was from Santa Cruz Dallas, Texas (USA). Rat IL-6 Quantikine ELISA Kit was from R&D Systems, Minneapolis, MN Inc (USA), IL-6 RNA lentivirus vector was from Shanghai GeneChem Co., Shanghai Ltd (China).
Cardiac fibroblasts isolation and treatment
1–3-day-old neonatal Sprague-Dawley rats were purchased from Shanghai Slac Laboratory Animal Co., Ltd. Cardiac fibroblasts were isolated, as described previously 21, briefly, trypsin digestion and differential attachment, and adherent cells were cultured in low-glucose DMEM supplemented with 10% FBS and 1% penicillin-streptomycin in a humidified incubator with 5% CO2 and 95% air. The CFs of passage 2 were 40–50% confluent, and a portion of the cells transfected with IL-6 small-interfering RNA (IL-6-siRNA with green fluorescence protein labelled) at a multiplicity of infection of 10 and 48 hrs after transfection, CFs were harvested for further analysis. After the cells were 80–90% confluent, the medium was replaced by serum-free DMEM the day before pre-treated with QL (effective intervention dose, 0.5 mg/ml) or OLM (10−7 mol/l) for 30 min. and exposed to AngII (100 nM) for 24 hrs.
Real time polymerase chain reaction
Total RNA was isolated from the CFs using TRIzol reagent (Invitrogen Carlsbad, CA, USA). Primer for IL-6 forward 5′-GCC AGA GTC ATT CAG AGC AAT A-3′ and reverse: 5′-TTA GGA GAG CAT TGG AAG TTG G-3′, for Collagen-I forward 5′-CCA ATG GTG CTC CTG GTA TT-3′ and reverse: 5′-GTT CAC CAC TGT TGC CTT TG-3′, for Collagen III forward 5′-AAT GGC TCT CCA GGA CAA AG-3′ and reverse: 5′-CCA CCA GGA CTG CCA TTA TT-3′, reverse: 55′-GTT CAC CAC TGT TGC CTT TG-3′ and for GAPDH forward 5′-ACT CCC ATT CTT CCA CCT TTG-3′ and reverse: 5′-CCC TGT TGC TGT AGC CAT ATT-3′, the mRNA levels of IL-6, Collagen-I and -III were normalized to GADPH. PCR cycling conditions: 95°C 60 sec., 95°C 15 sec., PCR cycles (×40 cycles): 95°C 15 sec., 65°C 25 sec. All data analyses were used 2−ΔCt method.
Western blot analysis
The proteins in CFs were lysed using RIPA (radioimmunoprecipitation) lysis buffer supplemented with 1 mmol/l PMSF and phosphates inhibitor (Pierce, Rockford, IL, USA). For immunoblotting:equal amounts of proteins were resolved on 12% SDS-polyacrylamide gel and transferred onto poly-vinylidene fluoride membrane (Thermo Scientific), Andingmen East Street, Beijing, China After blocked with 5% bovine serum albumin (BSA) at room temperature for 1 hr, Membranes were exposed to primary antibody anti-GAPDH (1:2000 diluted in TBST), anti-a-SMA (1:1000 dilution), anti-TGF-β1 (1:1000 dilution), over night at 4°C and incubated with HRP-conjugated secondary antibody for 2 hrs at room temperature. The signal was detected with the ECL Plus (Thermo Scientific). Bands were analysed by densitometry using Quantity One software (Hercules, CA, USA).
Immunofluorescence quantification and analyses
The expression levels of α-SMA were also detected by immunofluorescence staining. CFs were treatment with QL (0.5 mg/ml) or OLM (10 nM), and/or stimulated by Ang II (100 nmol/l) for 24 hrs, the cells were washed by PBS, fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. After the cells were incubated with BSA for 30 min., the primary antibody specific for α-SMA was added overnight at 4°C. And the cells were incubated with Fluorescein isothiocyanat (FITC) secondary antibody for 1 hr at room temperature in the dark room, and then DAPI was added for 30 min. The cells were visualized under fluorescence microscope.
Fluorescence intensity of cells was examined and quantified by the specialized measurement incorporated within imaging software. Fluorescence intensities could be compared between regions of the same set of stress fibres and between cells of different treatments. The data were analysed by the mean fluorescence intensities from 10 random selected regions, each with a standard 50-μm2 circular region.
Statistics
All data were expressed as means ± SEM. The statistical significance was assessed by using one-way anova for multiple-group comparisons. Differences between groups were using paired-sample Student's t-test, P < 0.05 was considered statistically significant.
Results
QL effectively reversed AngII-induced CFs transdifferentiation
To investigate the role of QL in regulating CFs transdifferentiation, we first induced the in vitro cultured CFs by AngII (100 nmol/l), which played an important role in CFs transdifferentiation and the process of epithelial mesenchymal transition. After treatment for 24 hrs, the stage-specific transdifferentiation markers, TGF-β1 and α-SMA, were detected by Western blot. As shown in Figure1A, AngII-induced TGF-β1 and α-SMA in CFs were significantly reduced by pre-treatment with QL (0.5 mg/ml) or OLM (10 nM). The optimized concentration of QL was determined by the effects of concentration gradient on CFs viability and their inhibition of α-SMA (Figs S2 and S3). However, the increases of TGF-β1 and α-SMA were not affected by QL or OLM without the induction of AngII.
Figure 1.
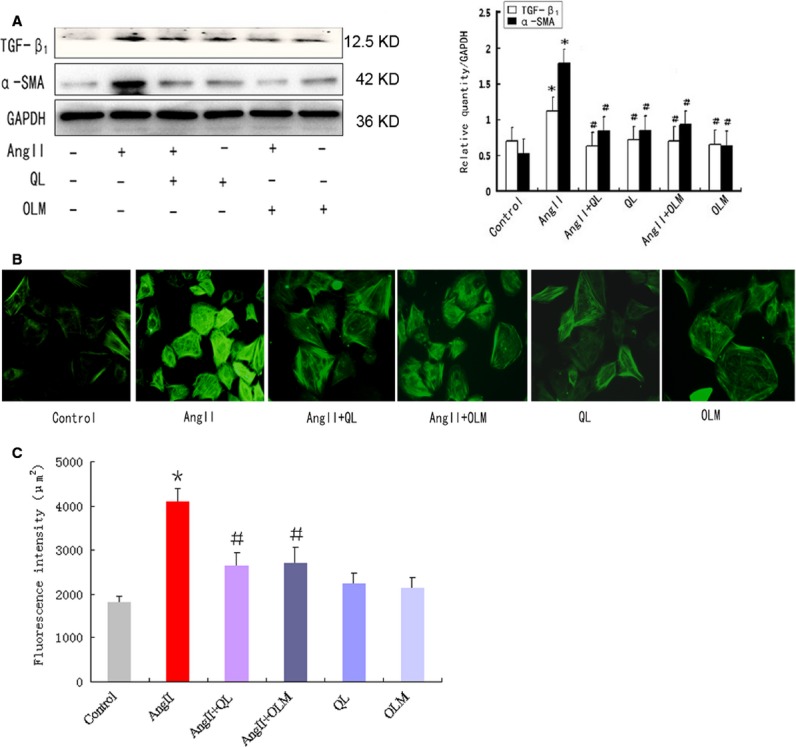
QL effectively reversed AngII-mediated CFs transdiferentiation. CFs were stimulated by AngII (100 nmol/l) for 24 hrs with or without the pre-treatment of QL (0.5 mg/ml) or Olmesartan (10−7 mol/l). (A) The protein levels of TGF-β1 and α-SMA were detected by Western blot. (B) Actin stress fibres expressed in CFs was detected by fluorescent labelling with FITC-conjugated antibody against α-SMA. (C) Quantification of mean fluorescence intensity of stress fibres in CFs with α-SMA staining. *indicates P < 0.05 versus Control group; #indicates P < 0.05 versus AngII-induced group.
Besides the increase in total level of α-SMA, the subcellular localization of α-SMA attached much more importance of functional myofibroblasts that transdifferentiated from CFs. α-SMA was incorporated into actin stress fibres to enhance cell contractility 22. Thus, we next quantified the stress fibres in CFs by immunofluorescence staining with anti-α-SMA (Fig.1B). AngII stimulation for 48 hrs led to enhancement of actin stress fibres, which was diminished by pre-treatment with QL or OLM (Fig.1C), indicating that QL might prevent α-SMA recruitment to stress fibres through inhibition of AngII signalling.
QL reversed AngII-induced CFs transdifferentiation through inhibiting IL-6
Previous studies revealed that IL-6 played an important role in fibroblasts transdifferentiation 12, therefore, we tested the possibility whether IL-6 was regulated by QL. First, our data showed that the transcriptional level of IL-6 was enhanced in CFs by AngII stimulation, and significantly reduced by pre-treating cells with QL or OLM, respectively (Fig.2A and B), suggesting that IL-6 was activated by AngII signalling and could be down-regulated by QL. Importantly, the enhancement of type I and type III collagens in CFs by AngII stimulation for 24 hrs was significantly reversed by QL (Fig.2C), indicating a critical role of QL in reversing CFs transdifferentiation.
Figure 2.
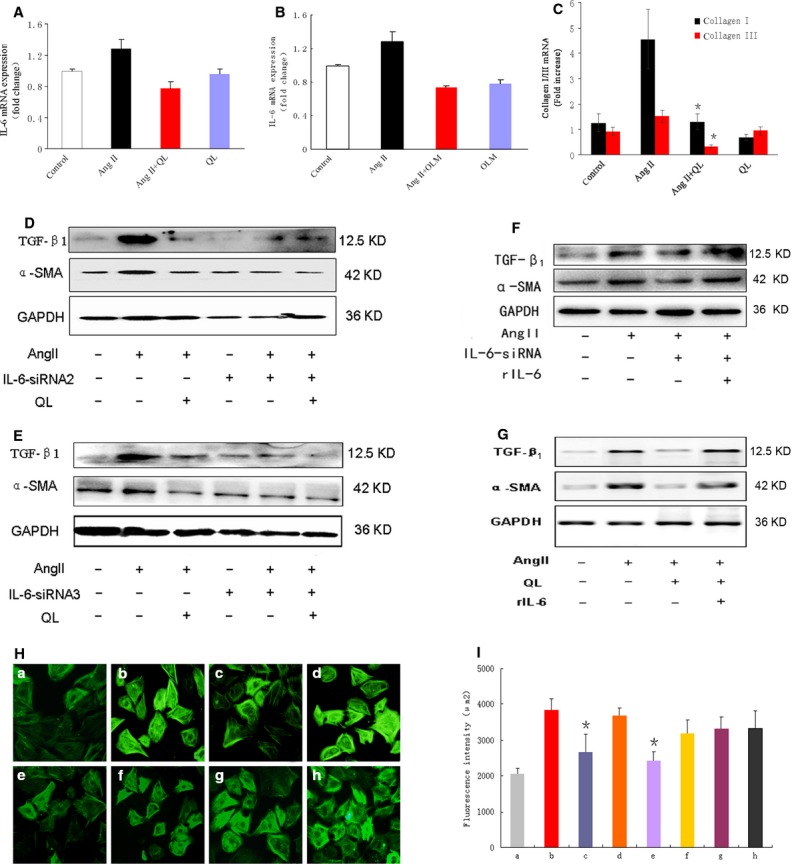
QL reversed AngII-mediated CFs transdiffentation via inhibiting IL-6 transcription. IL-6 in CFs were silenced by small-interfering RNA (siRNA) lentivirus transfection for 48 hrs and stimulated with AngII for another 24 hrs. (A) IL-6 mRNA expression was detected by real time polymerase chain reaction (RT-PCR) in CFs in the presence or absence of QL or (B) OLM. (C) The mRNA levels of Collagen type I and type III in CFs pre-treated with or without QL. (D and E) The effect of QL on AngII-mediated TGF-β1 and α-SMA expressions in the presence or absence of two different kinds of IL-6 siRNA. (F and G) The effect of rIL-6 (20 ng/ml) on QL or IL-6 siRNA-induced suppression of TGF-β1 and α-SMA. (H) Actin stress fibres was detected by α-SMA staining in CFs from different treated groups (a, control; b, AngII; c, AngII+IL-6 siRNA; d, AngII+ control siRNA; e, AngII+QL; f, AngII+QL+rIL-6; g, AngII+IL-6 siRNA+rIL-6; h, rIL-6). (I) Quantification of mean fluorescence intensity of stress fibres in CFs with α-SMA staining. *indicates P < 0.05 versus AngII-induced group.
To confirm the role of IL-6 in CFs transdifferentiation, we knocked down IL-6 by transfection of small-interfering RNA (siRNA) in CFs. We tested the effects of three pairs of siRNA on IL-6 inhibition, the siRNA sequences were shown in Table1. SiRNA1 failed to inhibit IL-6 expression, but both siRNA2 and siRNA3 effectively down-regulated the protein level of IL-6 (Fig. S1). Thus, we chose these two siRNAs against IL-6 in the following experiments.
Table 1.
The list of the designed siRNAs against IL-6
| IL-6 | siRNA sequence | |||
|---|---|---|---|---|
| siRNA1 | S 5′: | GCUAUGCCUAAGCAUAUCA | UU | |
| mRNA: | GT | GCTATGCCTAAGCATATCA | GT | |
| AS 3′: | UU | CGAUACGGAUUCGUAUAGU | ||
| siRNA2 | S 5′: | CAGACCAGUAUAUACCACU | UU | |
| mRNA: | AA | CAGACCAGTATATACCACT | TC | |
| AS 3′: | UU | GUCUGGUCAUAUAUGGUGA | ||
| siRNA3 | S 5′: | CUAAAGGUCACUAUGAGGU | UU | |
| mRNA: | TT | CTAAAGGTCACTATGAGGT | CT | |
| AS 3′: | UU | GAUUUCCAGUGAUACUCCA | ||
Next, we further investigate the key role of IL-6 in inducing CFs transdifferentiation, we determined the expression of TGF-β1 and α-SMA induced by AngII in IL-6 knockingdown CFs. As shown in Figure2, both siRNA2 and siRNA3 against IL-6 could mimic the effect of QL in inhibiting TGF-β1 and α-SMA, importantly OL-induced reduction in TGF-β1 and α-SMA was not further enhanced by adding IL-6 siRNA (Fig.2D and E), indicating that QL and IL-6 might regulate AngII-mediated CFs transdifferentiation via the same pathway which was dependent on IL-6 activation.
Considering that the active form of IL-6 could be secreted into the extracellular, and functioned on the adjacent cells through paracrine modes, we analysed the regain-function of IL-6 by inducing CFs with recommended IL-6 to see if restore IL-6 function that knocking down by siRNA might promote CFs transdifferentiation again. As expected, TGF-β1 and α-SMA were rebounded by rIL-6 treatment in either IL6-siRNA or QL stimulated CFs (Fig.2F and G). Finally, we confirmed that α-SMA staining and stress fibres quantification were significantly reduced by QL or IL-6 siRNA treatment in AngII-induced CFs, these effects were reversed by adding rIL-6 (Fig.2H and I).
QL inhibited IL-6 through regulating nuclear NFAT3 activity
Skeletal muscle cells and CFs are the major sources of IL-6, a growing body of evidence indicated that IL-6 transcription is highly associated with the calcium signalling triggered by endured exercise. Enhanced calcineurin activity is required for the IL-6 secrition both in vitro and in vivo. Here, we found that calcineurin was increased and nuclear NFAT3 was accumulated in AngII-treated CFs, these levels were greatly reduced by QL pre-treatment (Fig.3A and 3B). To confirm whether QL affected IL-6 expression through regulating calcineurin/NFAT3 pathway, we pre-treated CFs with FK506, the calcineurin inhibitor, for 1 hr, then IL-6 mRNA expression was examined after AngII induction for 24 hrs. The results showed that NFAT3 inhibition significantly reduced IL-6 in CFs, and this effect was not further streghtened by adding QL. (Fig.3C), suggesting NFAT3 signalling might be down-regulated by QL, which led to suppression of IL-6 transcription. Furthermore, FK506 pre-treatment attenuated stress fibres in AngII-induced CFs, and there had little difference between groups with or without QL (Fig.3D and E).
Figure 3.
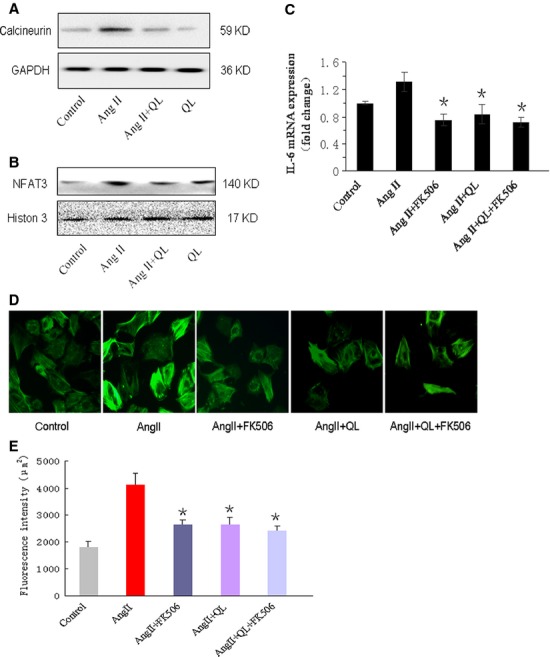
QL suppressed the transcription of IL-6 through regulating calcineurin/NFAT3 signalling pathway. CFs were stimulated by AngII (100 nmol/l) for 24 hrs with or without the pre-treatment of QL (0.5 mg/ml), (A) the cytoplasm level of calcineurin and (B) nuclear level of NFAT3 were detected by Western blot, respectively, in separated protein extracts. (C) CFs were pre-treated with or without FK506 (10−5 mol/l) for 1 hr before AngII stimulation, the effects of QL and FK506 on IL-6 mRNA was detected in different groups. (D) Actin stress fibres were detected by a-SMA staining in CFs from different treated groups (control, AngII, AngII with FK506, AngII with QL, AngII with FK506 and QL). (E) Quantification of mean fluorescence intensity of stress fibres in CFs with a-SMA staining. *indicates P< 0.05 versus AngII-induced group.
Discussion
The major finding of this study was that, (i) QL inhibited AngII-induced CFs transdifferentiation via reducing IL-6 transcription; (ii) NFAT3 nuclear translocation was induced by AngII, which played a critical role in IL-6 secretion and enhancement of TGF-β1 and α-SMA and (iii) QL reversed CFs transdifferentiation possibly through regulating nuclear activity of NFAT3.
AngII, the one of most important renin-angiotensin system components, played a critical role in cardiac remodelling. Recently investigations focused its impact on CFs transdifferentiation 10,23. Because, CFs conversion into myofibroblasts resulted in collagen production and deposition, which might increase the risk of myocardial fibrosis and remodelling 24. In this study, we found that AngII stimulation increased expressions of TGF-β1 and α-SMA, also enhanced actin stress fibres in CFs. Attenuation of these effects by QL suggested its important role in reversion of. In addition, we noted that QL had similar effect as OLM, the AT1R inhibitor, both of which effectively suppressed the process of CFs transdifferentiation induced by AngII, indicating that QL inhibited CFs transdifferentiation through AT1 receptor signalling. Considerable evidence proved that AngII-mediated AT1 receptor activation could transactivated TGF-β1 signalling under pressure overload 23,25. TGF-β1 signalling was essential for cardiac fibrosis, and blocking TGF-β1 effectively reversed CFs transdifferentiation and reduced ECM deposition, which might be the therapeutic target for myocardial fibrosis 26.
Association between inflammation response and CFs transdifferentiation was crucial for the development of cardiac fibrosis 27. Inflammatory infiltrates produce various cytokines including prototypical inflammatory cytokines such as TNF-α, IL-6 and IL-1β. Several studies proved that IL-6 played a key role in collagen deposition, fibrosis and CFs transdifferentiation 15,17,28. IL-6-mediated myofibroblasts conversion was causal for the process of cardiac fibrosis and remodelling. TGF-β1 and α-SMA were significantly lower or even absent in hearts of interleukin-6 knockout (IL-6−/−) mice than in wild-type mice after AngII infusion. Gene analysis showed that IL-6 was mainly present in CFs rather than in cardiomyocytes and macrophages after AngII administration 15. In this experiment, we also observed the expression of IL-6 in CFs was significantly increased by AngII induction. Importantly, QL effectively attenuated expression of IL-6 in AngII-stimulated CFs, and QL mimicked the effect of IL-6 siRNA on inhibition of TGF-β1 and α-SMA expressions, reduction in actin stress fibres, and reversion of collagen deposition. But QL did not amplify the inhibitory effect on CFs transdifferentiation in the presence of IL-6 siRNA, suggesting IL-6 activation and downstream signalling pathway was critically involved in AngII-induced TGF-β1 signalling and CFs transdifferentiation, QL inhibited CFs transdifferentiation, at least in part, via down-regulating IL-6/TGF-β1 signalling pathway.
AT1R inhibitor was postulated to have therapeutic effects on inflammatory conditions. Nishio et al. 29 previously demonstrated that OLM could improve the LV hypertrophy, fibrosis and diastolic dysfunction though attenuation of inflammatory response in a mouse model of diastolic heart failure. Candesartan prevented expressions of TNF-α, cyclooxygenase-2 and IL-6. The increase in inflammatory cytokines, such as IL-6, was highly associated with cardiac remodelling 30 and dilated cardiomyopathy 31. In our study, we found that IL-6 was significantly down-regulated by QL, especially knocking down of IL-6 could reverse the process of CFs transdifferentiation.
Another important finding of this study was that QL suppressed the IL-6 expression via negative regulation of calcineurin/NFAT3 signalling. Despite the large populations of immune cells, skeletal muscle cells and CFs might be the major sources of IL-6 production. However, the precise mechanism of CFs-mediated IL-6 activation was quite different from that mediated by macrophage or Th2 lymphocytes 32,33. A growing body of evidence indicated that IL-6 transcription is highly associated with the calcium signalling triggered by endured exercise. Enhanced calcineurin activity is required for the IL-6 secrition both in vitro and in vivo 34–36. We previously proved that AngII activated calcineurin and led to cardiac hypertrophy in a pressure overload-induced heart remodelling model 37. In this study, we found that QL effectively inhibited AngII-mediated calcineurin activation and NFAT3 nuclear translocation, which might contribute to the inhibition of IL-6 expression, because FK506 pre-treatment could reverse the transcription of IL-6. Wei et al. reported that L-type Ca2+ channel (ICa-L) was blocked by QL and myocardial contractility was weakened because of reduced Ca2+ influx 7. Thus, we assumed that calcineurin might be the key point for QL-mediated inhibition of ICa-L, which not only attenuated cardioamyocytes hypertrophy but also suppressed CFs transdifferentiation. QL-mediated reducing of Ca2+ influx affected localization of α-SMA at actin stress fibres, and blunted the process of conversion from CFs to myofibroblasts.
Several nuclear transcriptional factors could be activated by calcineurin, such MEF2, NFAT3 and CREB, these factors were all involved in cardiac remodelling 38–40. But here, we focused on the change in nuclear NFAT3 that suppressed by QL, NFAT3 inhibition by FK506 effectively attenuated IL-6 expression. Although there was no evidence provided that IL-6 was regulated by NFAT3, cotransfection of a constitutively active or wide-type form of NFAT3 did increase the IL-6 promoter activity. IL-6 secretion is highly associated with the calcineurin activity during muscle contraction. In our data, we proved that QL could suppress calcineurin, but till now, it was hard to prove which herb mainly contributed to the effect on ICa-L channel, our further investigation would attach the technique to separate the effective intergradient of QL capsules, and their impacts on calcium signalling, IL-6 expression and CFs transdifferentiation.
In conclusion, our present data supported a role for QL in the reversion of CFs transdifferentiation, which was achieved by suppressing the calcineurin activity, down-regulating IL-6 transcription and downstream TGF-β1 and α-SMA expressions. The experiment indicated a new perspective in the molecular mechanisms of QL on CFs transdifferentiation and cardiac fibrosis.
Acknowledgments
This study was supported by grants from the National Basic Research Program of China (973 Program, 2012CB518605) and National Natural Science Foundation of China (81070105).
Disclosure
The authors confirm that there are no conflicts of interest.
Supporting Information
Figure S1 Western blot analysis of the effects of different siRNAs on IL-6 inhibiton.
Figure S2 Cell viability was determined in culured CFs stimulated by indicated concentrations of QL.
Figure S3 a-SMA expression in culured CFs stimulated by indicated concentrations of QL.
References
- Poobalarahi F, Baicu CF, Bradshaw AD. Cardiac myofibroblasts differentiated in 3D culture exhibit distinct changes in collagen I production, processing, and matrix deposition. Am J Physiol Heart Circ Physiol. 2006;291:H2924–32. doi: 10.1152/ajpheart.00153.2006. [DOI] [PubMed] [Google Scholar]
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–78. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Vellaichamy E, Young D, et al. Influence of cytokines and growth factors in ANG II-mediated collagen upregulation by fibroblasts in rats: role of myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H107–17. doi: 10.1152/ajpheart.00763.2003. [DOI] [PubMed] [Google Scholar]
- Liu W, Chen J, Xu T, et al. Qiliqiangxin improves cardiac function in spontaneously hypertensive rats through the inhibition of cardiac chymase. Am J Hypertens. 2012;25:250–60. doi: 10.1038/ajh.2011.219. [DOI] [PubMed] [Google Scholar]
- Zou Y, Lin L, Ye Y, et al. Qiliqiangxin inhibits the development of cardiac hypertrophy, remodeling, and dysfunction during 4 weeks of pressure overload in mice. J Cardiovasc Pharmacol. 2012;59:268–80. doi: 10.1097/FJC.0b013e31823f888f. [DOI] [PubMed] [Google Scholar]
- Xiao H, Song Y, Li Y, et al. Qiliqiangxin regulates the balance between tumor necrosis factor-alpha and interleukin-10 and improves cardiac function in rats with myocardial infarction. Cell Immunol. 2009;260:51–5. doi: 10.1016/j.cellimm.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Wei Y, Liu X, Hou L, et al. Qiliqiangxin affects L type Ca(2+) current in the normal and hypertrophied rat heart. Evid Based Complement Alternat Med. 2012;2012:131830. doi: 10.1155/2012/131830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago JJ, Dangerfield AL, Rattan SG, et al. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010;239:1573–84. doi: 10.1002/dvdy.22280. [DOI] [PubMed] [Google Scholar]
- Chen LX, Yang K, Sun M, et al. Fluorofenidone inhibits transforming growth factor-beta1-induced cardiac myofibroblast differentiation. Pharmazie. 2012;67:452–6. [PubMed] [Google Scholar]
- Wang XF, Gao GD, Liu J, et al. Identification of differentially expressed genes induced by angiotensin II in rat cardiac fibroblasts. Clin Exp Pharmacol Physiol. 2006;33:41–6. doi: 10.1111/j.1440-1681.2006.04321.x. [DOI] [PubMed] [Google Scholar]
- Oguro H, Tsuchikawa K. The effect of angiotensin II on the proliferation of human gingival fibroblasts. Odontology. 2009;97:97–102. doi: 10.1007/s10266-009-0102-y. [DOI] [PubMed] [Google Scholar]
- Levick SP, McLarty JL, Murray DB, et al. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension. 2009;53:1041–7. doi: 10.1161/HYPERTENSIONAHA.108.123158. [DOI] [PubMed] [Google Scholar]
- Kudo H, Kai H, Kajimoto H, et al. Exaggerated blood pressure variability superimposed on hypertension aggravates cardiac remodeling in rats via angiotensin II system-mediated chronic inflammation. Hypertension. 2009;54:832–8. doi: 10.1161/HYPERTENSIONAHA.109.135905. [DOI] [PubMed] [Google Scholar]
- Levick SP, Murray DB, Janicki JS, et al. Sympathetic nervous system modulation of inflammation and remodeling in the hypertensive heart. Hypertension. 2010;55:270–6. doi: 10.1161/HYPERTENSIONAHA.109.142042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Li Y, Jia L, et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE. 2012;7:e35144. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meléndez GC, McLarty JL, Levick SP, et al. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–31. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainscough JF, Drinkhill MJ, Sedo A, et al. Angiotensin II type-1 receptor activation in the adult heart causes blood pressure-independent hypertrophy and cardiac dysfunction. Cardiovasc Res. 2009;81:592–600. doi: 10.1093/cvr/cvn230. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Matsumoto K, Mizuno S, et al. Hepatocyte growth factor prevents tissue fibrosis, remodeling, and dysfunction in cardiomyopathic hamster hearts. Am J Physiol Heart Circ Physiol. 2005;288:H2131–9. doi: 10.1152/ajpheart.01239.2003. [DOI] [PubMed] [Google Scholar]
- Taqueti VR, Mitchell RN, Lichtman AH. Protecting the pump: controlling myocardial inflammatory responses. Annu Rev Physiol. 2006;68:67–95. doi: 10.1146/annurev.physiol.68.040104.124611. [DOI] [PubMed] [Google Scholar]
- Teunissen BE, Smeets PJ, Willemsen PH, et al. Activation of PPARdelta inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc Res. 2007;75:519–29. doi: 10.1016/j.cardiores.2007.04.026. [DOI] [PubMed] [Google Scholar]
- Meyer-ter-Vehn T, Sieprath S, Katzenberger B, et al. Contractility as a prerequisite for TGF-beta-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci. 2006;47:4895–904. doi: 10.1167/iovs.06-0118. [DOI] [PubMed] [Google Scholar]
- Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–32. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Hein S, Arnon E, Kostin S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–91. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- Schultz Jel J, Witt SA, Glascock BJ, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–96. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Zhu YZ. Role of transforming growth factor-beta in the progression of heart failure. Cell Mol Life Sci. 2006;63:2584–96. doi: 10.1007/s00018-006-6085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi K, Li X, Coker M, et al. Cross-regulation between the renin-angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc Res. 2004;63:433–42. doi: 10.1016/j.cardiores.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- Nishio M, Sakata Y, Mano T, et al. Therapeutic effects of angiotensin II type 1 receptor blocker at an advanced stage of hypertensive diastolic heart failure. J Hypertens. 2007;25:455–61. doi: 10.1097/HJH.0b013e328010d635. [DOI] [PubMed] [Google Scholar]
- Sukumaran V, Watanabe K, Veeraveedu PT, et al. Beneficial effects of olmesartan, an angiotensin II receptor type 1 antagonist, in rats with dilated cardiomyopathy. Exp Biol Med. 2010;235:1338–46. doi: 10.1258/ebm.2010.010016. [DOI] [PubMed] [Google Scholar]
- Sandmann S, Li J, Fritzenkötter C, et al. Differential effects of olmesartan and ramipril on inflammatory response after myocardial infarction in rats. Blood Press. 2006;15:116–28. doi: 10.1080/08037050600586593. [DOI] [PubMed] [Google Scholar]
- Lissoni P, Rovelli F, Tisi E, et al. Relation between macrophage and T helper-2 lymphocyte functions in human neoplasms: neopterin, interleukin-10 and interleukin-6 blood levels in early or advanced solid tumors. J Biol Regul Homeost Agents. 1995;9:146–9. [PubMed] [Google Scholar]
- Chuang CC, Chuang YC, Chang WT, et al. Macrophage migration inhibitory factor regulates interleukin-6 production by facilitating nuclear factor-kappa B activation during Vibrio vulnificus infection. BMC Immunol. 2010;11:50. doi: 10.1186/1471-2172-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banzet S, Koulmann N, Simler N, et al. Fibre-type specificity of interleukin-6 gene transcription during muscle contraction in rat: association with calcineurin activity. J Physiol. 2005;566:839–47. doi: 10.1113/jphysiol.2005.089193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banzet S, Koulmann N, Sanchez H, et al. Contraction-induced interleukin-6 transcription in rat slow-type muscle is partly dependent on calcineurin activation. J Cell Physiol. 2007;210:596–601. doi: 10.1002/jcp.20854. [DOI] [PubMed] [Google Scholar]
- Weigert C, Düfer M, Simon P, et al. Upregulation of IL-6 mRNA by IL-6 in skeletal muscle cells: role of IL-6 mRNA stabilization and Ca2+-dependent mechanisms. Am J Physiol Cell Physiol. 2007;293:C1139–47. doi: 10.1152/ajpcell.00142.2007. [DOI] [PubMed] [Google Scholar]
- Zhou N, Li L, Wu J, et al. Mechanical stress-evoked but angiotensin II-independent activation of angiotensin II type 1 receptor induces cardiac hypertrophy through calcineurin pathway. Biochem Biophys Res Commun. 2010;397:263–9. doi: 10.1016/j.bbrc.2010.05.097. [DOI] [PubMed] [Google Scholar]
- Ambra R, Di Nardo P, Fantini C, et al. Changes in DNA binding activity of transcription factors in UM-X7.1 cardiomyopathic hamsters. Life Sci. 2002;71:2369–81. doi: 10.1016/s0024-3205(02)02020-9. [DOI] [PubMed] [Google Scholar]
- van Oort RJ, van Rooij E, Bourajjaj M, et al. MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation. 2006;114:298–308. doi: 10.1161/CIRCULATIONAHA.105.608968. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Teg Pipes GC, McAnally J, et al. Modulation of adverse cardiac remodeling by STARS, a mediator of MEF2 signaling and SRF activity. J Clin Invest. 2007;117:1324–34. doi: 10.1172/JCI31240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Western blot analysis of the effects of different siRNAs on IL-6 inhibiton.
Figure S2 Cell viability was determined in culured CFs stimulated by indicated concentrations of QL.
Figure S3 a-SMA expression in culured CFs stimulated by indicated concentrations of QL.