

Abstract
Thoracic aortic diseases that involve progressive enlargement, acute dissection, or rupture are influenced by the hemodynamic loads and mechanical properties of the wall. We have only limited understanding, however, of the mechanobiological processes that lead to these potentially lethal conditions. Homeostasis requires that intramural cells sense their local chemo-mechanical environment and establish, maintain, remodel, or repair the extracellular matrix to provide suitable compliance and yet sufficient strength. Proper sensing, in turn, necessitates both receptors that connect the extracellular matrix to intracellular actomyosin filaments and signaling molecules that transmit the related information to the nucleus. Thoracic aortic aneurysms and dissections are associated with poorly controlled hypertension and mutations in genes for extracellular matrix constituents, membrane receptors, contractile proteins, and associated signaling molecules. This grouping of factors suggests that these thoracic diseases result, in part, from dysfunctional mechanosensing and mechanoregulation of the extracellular matrix by the intramural cells, which leads to a compromised structural integrity of the wall. Thus, improved understanding of the mechanobiology of aortic cells could lead to new therapeutic strategies for thoracic aortic aneurysms and dissections.
Keywords: mechanosensing, actomyosin, focal adhesion, elastic fibers, familial thoracic aortic aneurysms and dissections, Marfan syndrome
Introduction
Thoracic aortic aneurysms and dissections (TAADs) are responsible for significant morbidity and mortality in the young and old alike1,2. Advances over the past 15 years in medical imaging and genetics have increased the number of diagnosed TAADs3,4, which has raised the need to better understand the natural history of these potentially lethal conditions. Progressive dilatation, acute dissection, and rupture of the thoracic aorta are fundamentally biomechanical processes; in particular, dissection and rupture are extreme cases of tissue-level failures that occur when local wall stress exceeds strength. There is a need, therefore, to quantify stresses (i.e., the mechanics) within the aortic wall and to understand how the intramural cells respond to these stresses (i.e., the mechanobiology) by producing, organizing, or degrading the extracellular matrix (ECM) that endows the wall with its structural integrity.
We focus on cell-matrix interactions in TAADs and suggest that the vulnerability of the thoracic aorta to aneurysm, dissection, and ultimately rupture results, in large part, from dysfunctional mechanical homeostasis, that is, a compromised ability of intramural cells to sense their mechanical environment and to regulate the ECM to respond to that environment1,5. We further suggest that actomyosin contractility, independent of tissue-level vasoactivity, is central to both sensing and regulating the mechanical state of the matrix. This interpretation leads naturally to a hypothesis that genetic mutations that affect the mechano-stimulus (e.g., stresses conveyed to cells through the ECM), mechano-sensing (e.g., via membrane receptors or actomyosin functionality), or resultant mechano-signaling (e.g., second messengers) can lead to TAADs. To support this hypothesis, we first review aortic wall composition, vessel-level mechanics and mechanoadaptations, and mechanobiological processes, and then discuss how mutant genes predisposing to TAADs can disrupt mechanical homeostasis.
Aortic Wall Composition
The thoracic aorta is a composite tube consisting of three layers: an intima comprised of endothelial cells on a basement membrane, an elastin- and smooth muscle-rich media, and a collagen- and fibroblast-rich adventitia (Figure 1). The aorta is continually subjected to complex cyclic mechanical loads from pulsatile blood pressure and flow, and, in the case of the ascending aorta, the beating heart itself. Most of the structural integrity of the aortic wall derives from myriad ECM proteins, glycoproteins, and glycosaminoglycans6,7. Of the structural constituents, elastic fibers and fibrillar collagens represent ~60% of the normal wall by dry weight and dominate its elasticity (i.e., ability to distend and recoil without dissipating energy), tensile stiffness (changes in stress associated with changes in strain), and strength (maximum stress sustained without failure). Glycosaminoglycans constitute only ~3 to 5% of the normal wall, but contribute to the compressive stiffness. Most of the remaining ~35% of the wall consists of smooth muscle cells (SMCs). Vessel-level vasomotion induced by SMCs can contribute to mechanoadaptations by elastic arteries such as common carotids, and perhaps the aorta in some circumstances8. Nevertheless, the primary role for aortic SMCs is to govern the mechanical properties by regulating ECM in the media. Fibroblasts similarly regulate matrix in the adventitia.
Figure 1.
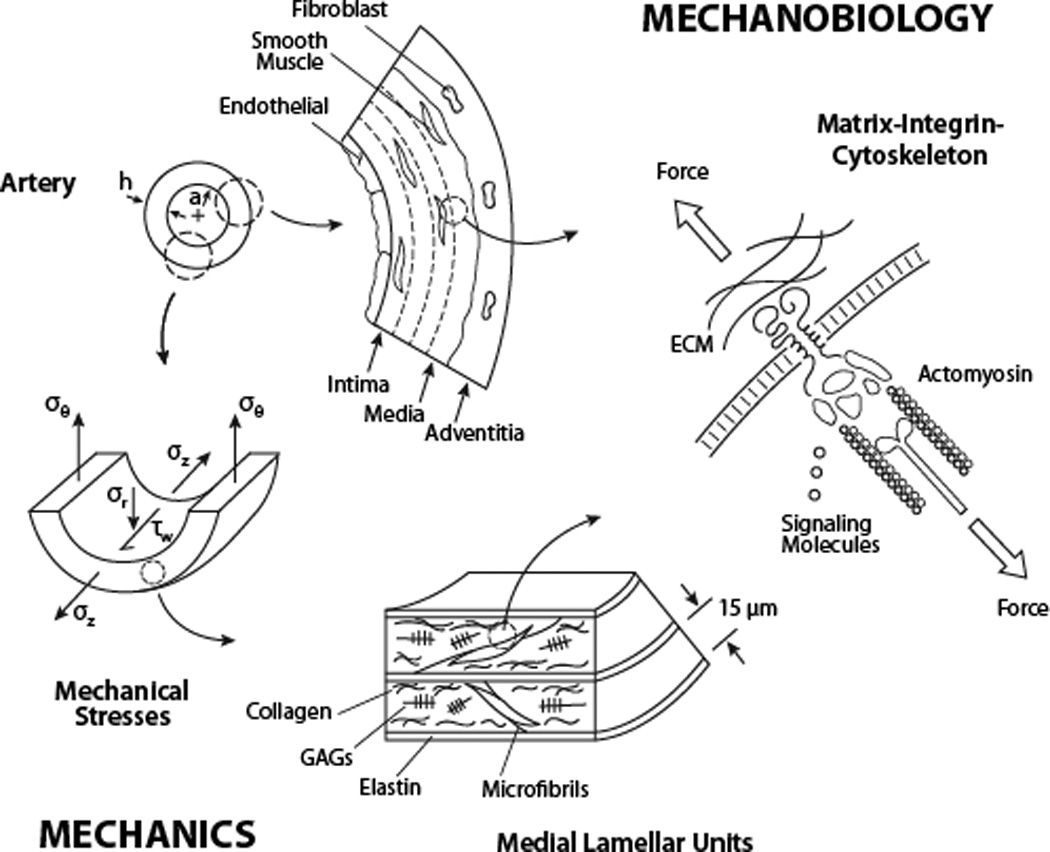
TAADs manifest clinically at the macro (tissue) level but result from mechanisms at the micro (molecular) level. Shown are aortic structure and relevant mechanical stresses (see text) that result from hemodynamic loading. The stressed wall consists of three layers (intima, media, adventitia) populated, respectively, by three cell types (endothelial, smooth muscle, fibroblasts). Medial lamellar units consist of many concentric deliminating elastic laminae (elastin plus microfibrils), with associated smooth muscle cells, collagens, and glycosaminoglycans. A unique contractile-elastic unit consists of co-linear extracellular microfibrils connecting through heterodimeric transmembrane complexes (integrins) with intracellular actomyosin filaments to create load-bearing or sensing functions. Linker proteins within the focal adhesion have structural or signaling roles.
Elastic fibers consist of a core of elastin (90%) surrounded by microfibrils composed primarily of fibrillins, but also fibulins and other glycoproteins. These fibers are organized within the media primarily as concentric laminae, each separated by a layer of SMCs and associated collagens and glycosaminoglycans (Figure 1). The elastin is unique in that, under normal conditions, it is deposited, organized, and cross-linked prior to adulthood and its half-life is ~50 years in humans9. Consequently, elastic fibers become prestressed due to somatic growth10 and typically withstand millions of cycles of loading imposed by the heart (~34 M cycles/year in humans). The elastic fibers are linked structurally to the SMCs though oblique extensions from the elastic laminae. These extensions attach at the cell membrane to dense plaques, or focal adhesions, that connect to intracellular contractile filaments that project across the cell at the same oblique orientation as the elastic extensions (Figure 1). This configuration forms a “contractile-elastic” unit11, which is a tension-bearing or -sensing structure in SMCs. The oblique orientation of this structural unit reverses direction in adjacent elastic lamellae, thus contributing to a uniform distribution of stress across the normal wall12.
It appears that fibrillins contribute to the long term stability of elastic fibers13; this stability may result from connections to SMCs at dense plaques that suppress protease activity14, but the precise mechanisms remain unknown. Long-term stability also appears to result from desmosine and isodesmosine crosslinks, with fibulins promoting effective lysyl oxidase mediated cross-linking during elastogenesis15. Regardless, competent elastic fibers contribute most to aortic compliance (i.e., distensibility) and resilience (i.e., the ability to store energy during systole and to use that energy during diastole to augment blood flow). Damage to (e.g., via mechanical fatigue, that is, a loss of strength due to repeated loading) or degradation of (e.g., loss of mass via proteolysis) elastic fibers causes irreversible changes in wall structure and function.
In contrast, collagen fibers contribute primarily to overall stiffness and strength. The two primary types of collagen within the aortic wall are I (~70%) and III (~30%); the former is found primarily in the adventitia and the latter in the media where it mainly associates with elastic fibers6,7. Both collagens exist as fibers, hence their structural contributions depend upon intra- and inter-molecular cross-links as well as overall fiber diameters, orientations, and interactions with other constituents. Indeed, normal fibrillogenesis requires interactions with fibronectin, other collagens (e.g., type V), and proteoglycans (e.g., biglycan), hence defects in these allied constituents can compromise collagen stiffness or strength and predispose to TAADs16–18. Mutant genes encoding the procollagens for types III and I also lead to TAADs19–21, particularly for type III in vascular Ehlers-Danlos syndrome. It is important that some collagen fibrils run parallel to the aforementioned contractile-elastic units in the media22.
Under normal conditions, the larger collagen fibers tend to undulate slightly and orient in a symmetric-diagonal manner with respect to the long axis of the aorta23. This organization appears to complement the elastic fiber dependent distensibility that is favorable during systole24. Loss of elastic fiber integrity can decrease the undulation of collagen fibers, which manifests at the vessel level as a loss of distensibility/extensibility (i.e., increased stiffness)25. Loss of elastic fibers and increased stiffness are characteristic of aortic aging and thoracic aortic disease26,27. Unlike elastic fibers, collagen fibers in the aorta appear to have a short half-life (perhaps months)28 and are not affected by mechanical fatigue. Rather, appropriate turnover of collagen (deposition and degradation) is critical for aortic homeostasis29, including adaptations to altered hemodynamics or responses to disease or injury30. Beyond its contribution to wall properties during normal cyclic changes in pressure, adventitial collagen plays a special role as a “protective sheath” – it bears an increasing portion of wall stress during acute increases in pressure, thus shielding medial SMCs from excessive stresses31. Not surprisingly, sustained increases in blood pressure can induce significant increases in adventitial collagen, which is mediated in part by increases in local angiotensin-II (Ang-II) signaling32.
Loss of elastic fiber integrity not only affects the cell biology33, it also reduces aortic resilience, which allows the wall to dilate either uniformly, as in aging when the loss is diffuse, or locally, as in aneurysms when the loss is focal34,35. In contrast, loss of collagen fiber integrity decreases strength, which can render the aortic wall vulnerable to rupture16,17,20,36. Glycosaminoglycans accumulate with aging and as part of the pathology in TAADs26,37, with localized pooling potentially contributing to the risk of dissection38. In addition to these and other structural roles, many ECM constituents have instructional roles. Functional elastic fibers contribute to a SMC phenotype that is contractile and quiescent, thus not migratory, proliferative, or highly synthetic33,39. Microfibrils sequester latent transforming growth factor-beta (TGF-β), a cytokine important to many aspects of cellular activity, including SMC differentiation and matrix turnover40,41. Other growth factors and proteases are similarly sequestered by the ECM42. Finally, both mechanically damaged and proteolytically degraded matrix can alter the phenotype of resident cells or lead to an infiltration of inflammatory cells43,44. A compromised ECM thus has mechanical and biological consequences.
In summary, medial SMCs and adventitial fibroblasts are the primary cell types responsible for establishing, maintaining, or restoring the structural integrity, and hence much of the functionality, of the normal thoracic aortic wall (Figure 1). Because the function of the aorta is largely mechanical, it is not surprising that these cells are sensitive to changes in their mechanical environment45,46. Therefore, mechanics is important both for understanding the aorta as a load-bearing structure (e.g., normal distension versus dilatation, dissection, or rupture) and for identifying mechanical stimuli that drive cellular responses (e.g., homeostatic adaptations versus maladaptations or disease progression).
Wall Mechanics and Mechanoadaptations
A useful concept in biomechanics is mechanical stress, a “force intensity” having units of force per area (e.g., N/m2). Two particularly important components of aortic stress are (Figure 1): circumferential stress σθ (due to the distending blood pressure changing the circumference cyclically) and axial stress σz (due to forces in the direction of the long axis, including those arising from somatic growth). Other components include the radial stress σr (due directly to blood pressure and perivascular constraints) and wall shear stress τw (due to frictional interactions between the endothelium and flowing blood). Quantifying stresses as a function of position and time requires solving nonlinear differential equations, but mean values can be estimated easily in straight segments47: σθ = Pa/h, σz = f/πh(2a + h), σr ≅ −P/2, and τw = 4µQ/πa3, where P is the transmural pressure, a the inner radius, h the thickness of the wall, f the axial force, Q the mean volumetric blood flow, and µ the viscosity of the blood. Of these four stresses, σθ and σz are the largest in the aorta: ~150 kPa under normal conditions, where 1 Pa =1 N/m2 and 1 kPa =7.5 mmHg. Abdominal aortic aneurysms may rupture when the larger of these two stresses exceeds 450 kPa48; comparable values of in vivo failure stresses have yet to be suggested for thoracic aneurysms but are likely similar. There are, however, accumulating in vitro data for the human thoracic aorta on biaxial mechanical and failure properties49,50. Finally, the mean radial stress is only about −6 kPa (compressive) and wall shear stress ~1.5 Pa (five orders of magnitude smaller than σθ and σz). Although typically neglected in analyses of wall mechanics, σr may be important in cases of increased glycosaminoglycans38. Regardless of magnitude, all components of stress impact the mechanobiology, with endothelial cells sensitive to changes in τw and SMCs and fibroblasts sensitive to changes in σθ and σz.45,46,51
The seminal paper by Wolinsky and Glagov12 revealed that “the average tension per lamellar unit of [descending thoracic] aortic media was remarkably independent of species and very nearly constant” at a mean value of T ~2 N/m. Hence, the normal number of elastic laminae is fixed for each species, including ~5 in murine to over 60 in human aorta. Because tension T = Pa and stress σθ = T/h, the ~15 µm thickness of a lamellar unit6 yields an average circumferential stress per lamellar unit of ~133 kPa, close to the mean value for the wall. Importantly, this finding suggests that intramural cells deposit and organize ECM during development to establish a preferred mechanical state (via morphogenesis), which they subsequently seek to maintain under normal conditions (via homeostasis). Complementary studies further suggest that these cells seek to restore this state when blood pressure or flow increase from normal52,53. Such mechanoadaptations reveal that the mature aorta retains some of its developmental ability to respond to changes in hemodynamics. Developmental adaptations include differential changes in the ascending aorta and pulmonary trunk following closure of the ductus arteriosus, whereby these two vessels adapt to their different pressures and flows54, and differential changes in the descending thoracic aorta and infrarenal aorta following birth, when increased renal and gastrointestinal flows change the hemodynamics within the infrarenal segment55. The most conspicuous aortic mechanoadaptation in maturity is thickening of the wall to restore intramural stresses in hypertension47,52.
Maintenance of arterial caliber in response to increased blood pressure (to restore wall shear stress toward normal) tends to involve vessel-level changes in vasoactivity30, which are greater in muscular arteries than elastic arteries. Although there is little information on possible basal tone in the human aorta, the murine aorta exhibits vessel-level vasoactivity (i.e., contractility) similar to that of carotids and other elastic arteries56. Aortic contractility is diminished in the thoracic aorta of mice having mutations in genes that predispose to TAADs57–60, hence contributions of shear stress mediation should be considered in these mouse models given that the ascending aorta is relatively thin, having ~8 medial lamellar units. The mechanoadaptive role of shear stress is less clear in human aortas because the thick media consists of 60+ layers of elastic laminae and SMCs.
The human aorta may adapt to changes in pressure and flow like other elastic arteries, yet it exhibits a distinct behavior in extreme exercise61. Marked increases in caliber manifest in the subclavian artery in the dominant arm of professional tennis players and in femoral arteries of professional cyclists while the thoracic and abdominal aorta change modestly62. It may be that the extreme compliance (inverse of stiffness) of the normal aorta accommodates dramatic pressure-induced changes in flow without entrenching changes in caliber. Consistent with this idea, the aorta is responsible for ~65% of total arterial compliance63; values of distensibility (e.g., where d is diameter and PP is pulse pressure) are about 6-fold greater in the proximal aorta than in common carotids in young healthy individuals and 2-fold greater than in the descending thoracic and abdominal aorta64. Yet, the ascending aorta may stiffen more and earlier during normal aging than either the common carotids or descending thoracic aorta64,65 and it exhibits earlier and more dramatic lengthening during aging66. These changes may result from the unique biaxial loading experienced by the ascending aorta – it alone experiences cyclic circumferential stresses due to the distending pressure and cyclic axial stresses from the downward motion of the contracting heart. The ascending aorta also has the highest percentage of elastin, which, of all structural constituents in an artery, is uniquely susceptible to mechanical fatigue because of the inability to repair the elastic fibers9.
The unique structural and mechanical features of the ascending aorta may contribute to its vulnerability to TAADs, but this is complicated by global versus local effects. For example, thickening of the wall could restore mean values of wall stress toward normal in hypertension, but defects within individual medial lamellar units (e.g., accumulated glycosaminoglycans) could still increase stress locally and initiate a delamination38. Loss of functional elastin, whether by mechanical fatigue or proteolytic degradation, would be expected to affect mechanosensing5 as well as contribute to arterial dilatation and lengthening25, with extreme cases of the latter leading to tortuosity24,36. Although decreases in the in vivo axial stretch of an artery tends to off-load the vessel axially and beneficially decrease both the axial and circumferential stress (due to biaxial coupling)67, extreme cases leading to tortuosity are maladaptive. Tortuosity of other arteries, including vertebrals, may also be indicators of thoracic aortic disease risk68. While the most dramatic effects may manifest in the thoracic aorta, other vessels can be affected69.
In summary, aortic cells typically seek to establish, maintain, and restore a homeostatic mechanical state, which can be accomplished via matrix turnover within elastically deformed or vasoaltered states. Such a process requires intramural cells to assess the mechanical state of the ECM, then either to entrench remodeled matrix or to prestress new matrix as it is deposited. Importantly, both of these processes require actomyosin contractility separate from vessel-level vasoactivity, as discussed below. Finally, the ascending aorta appears to have a unique structure and properties consistent with its unique mechanical loading, which may render it particularly vulnerable to fatigue-induced damage of elastic fibers that, in turn, could adversely affect mechanosensing and mechanoregulation of matrix.
Mechanobiology
A seminal finding that cells respond to mechanical stimuli was the observation that SMCs change their production of ECM, including fibrillar collagens and glycosaminoglycans, in response to cyclic mechanical loading70. Later studies showed that this SMC mechanobiological response occurred due to increased Ang-II signaling71, increased sensitivity to Ang-II through its primary receptor, AT1R72, and increased production of a downstream effector molecule, TGF-β73. These findings were supported, respectively, by an attenuated load-induced production of matrix following treatment with an angiotensin converting enzyme inhibitor, an AT1R antagonist, or a TGF-β neutralizing antibody. Another load-induced response by SMCs is altered production of matrix metalloproteinases (MMPs)73, the potency of which can depend on mechanical loading74. Hence, cyclic loading of SMCs affects both matrix production and degradation. Such mechanobiological effects are complicated by related effects. For example, Ang-II increases interleukin-6 and monocyte chemoattractant protein-1, which in vivo facilitates recruitment of monocytes/macrophages that produce cytokines and proteases that contribute to matrix turnover, particularly in adventitial fibrosis75. Nevertheless, cyclic loading also alters SMC expression of platelet derived growth factor76 and cationic channels involved in cell contractility77. Figure 2 summarizes some key responses of SMCs to cyclic loading, where σ(λ(t)) represents the stress σ that depends constitutively on the stretch λ that varies with time t during loading. Such findings clearly link the mechanics and mechanobiology.
Figure 2.
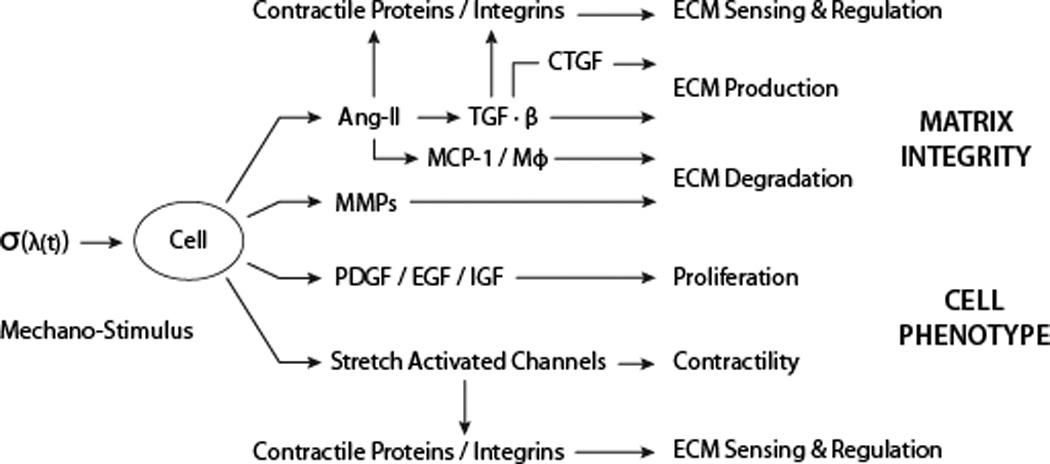
Some of the many smooth muscle cell responses to an applied stress (σ) or stretch (λ). Angiotensin II (Ang-II) is a potent vasoconstrictor that also regulates both the production of intracellular contractile and extracellular matrix (ECM) proteins (partly through transforming growth factor-beta, TGF-β, and connective tissue growth factor, CTGF) and the removal of ECM (partly through monocyte chemoattractant protein-1, MCP-1, and thus monocytes/macrophages, MΦ). Matrix metalloproteinases (MMPs) contribute further to matrix removal whereas platelet derived growth factor, PDGF, epidermal growth factor, EGF, and insulin-like growth factor, IGF, contribute to cell proliferation. Finally, stress/stretch activated ionic channels also play important roles in contraction.
Similar observations hold for fibroblasts46, which are fundamental to adventitial integrity. Fibroblasts can additionally use mechanical stresses (generated via actomyosin contractility) to activate latent TGF-β sequestered within matrix78. Although long ignored, adventitial fibroblasts are significant contributors to aortic wall homeostasis79, particularly in depositing, organizing, and degrading the collagen fibers that are essential for maintaining overall wall strength and preventing rupture80. Both mechanical stress and TGF-β are required for fibroblasts to differentiate into myofibroblasts78, which are also increasingly recognized as important in aortic mechanobiology, particularly aneurysmal enlargement in the absence of functional SMCs81.
Mechanical stimuli affect multiple signaling pathways (Figure 3), including the mitogen-activated protein kinase (MAPK) pathway downstream of Ang-II82 and the SMAD pathway downstream of TGF-β83. There is also cross-talk between TGF-β and MAPK signaling and between Ang-II and SMAD signaling84. Further complicating such signaling are the following observations: influences of Ang-II on TGF-β can be mediated by epidermal growth factor and its MAPK signaling85; elevated mechanical stress can increase local Ang-II70 and activate latent TGF-β78, both of which affect MMP promoter activity86; Ang-II can also activate latent MMP-2 while MMP-2 can activate latent TGF-β87,88; and SMCs exposed to Ang-II increase their expression of TGF-β89. Therefore, ECM, integrins, and growth factor/cytokine signaling are highly interconnected and responsive to mechanical loading90.
Figure 3.
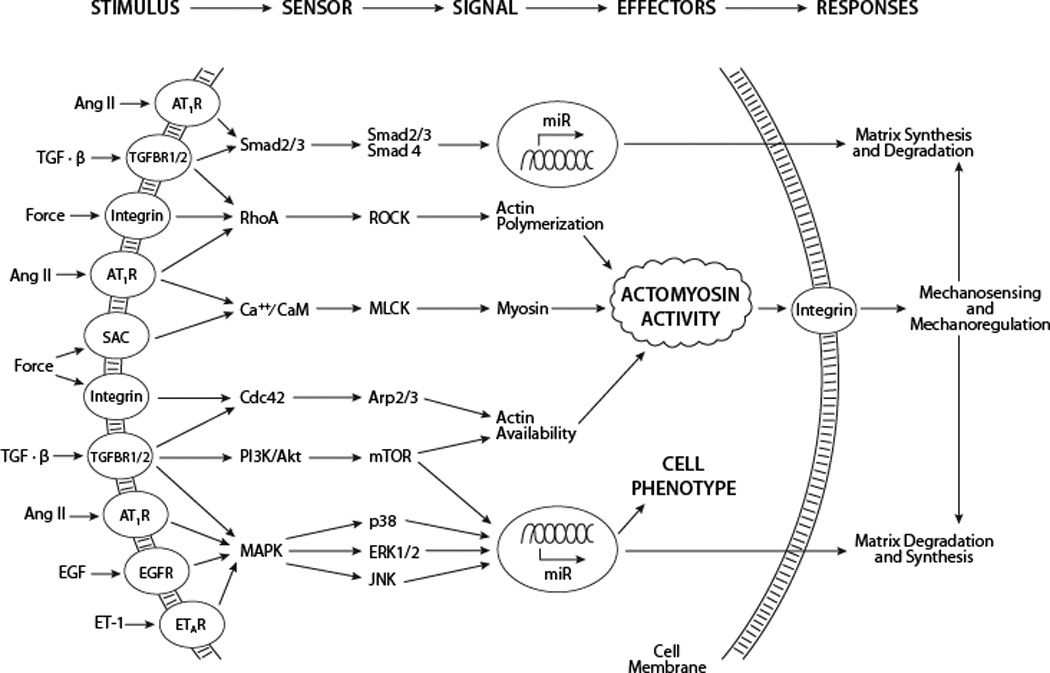
Some of the many membrane receptors and associated signaling pathways that are activated by chemo-mechanical stimuli. In many cases, these diverse pathways alter the actomyosin activity that is fundamental to cellular sensing and regulating of the extracellular matrix, which in turn is fundamental to defining aortic compliance and structural integrity. Other pathways, including NF-κB, are similarly important, particularly because of associated inflammation175.
Notwithstanding the already complex local wall mechanics and cellular mechanosensing and mechanoregulation of ECM in thoracic aortic disease, aging and hypertension are the major risk factors for TAADs3,4,27. Interestingly, Ang-II and TGF-β are also important in aging26 and hypertension91 wherein a switching of SMC phenotype toward synthetic92 can increase the production of matrix that thickens and stiffens the wall. While, thickening of the wall can favorably decrease mean wall stress locally30,52, a global increase in structural stiffness can lead to increased pulse wave velocities and central pulse pressures91,93 that adversely increase proximal aortic loading. Because different vascular disorders manifest in different regions (e.g., Marfan syndrome in the aortic root and vascular Ehlers-Danlos more diffusely), altered hemodynamics due to aortic stiffening need not increase stress on vulnerable regions uniformly and attempts to reduce central pressures could have differential effects. Finally, changes in stress-mediated matrix production that thickens the wall can also potentially diminish structural integrity locally (e.g., via accumulated glycosaminoglycans)37,38. In summary, given the effects of stress on Ang-II and TGF-β production71–73, the complex interactions amongst many related signaling pathways82–89, and the differential effects of these biomolecules on different cell types75,78, it should not be surprising that the roles of Ang-II and TGF-β in TAADs remain controversial41,94 – their roles in the basic mechanobiology and mechanics are similarly complex.
Mechanical Homeostasis Across Scales
Experiments focusing on responses of tissue equivalents, isolated cells, and sub-cellular structures to mechanical stimuli suggest that homeostatic targets also exist at these levels of organization95,96. For example, fibroblasts seeded within initially stress-free but mechanically constrained collagen gels generate an endogenous stress of ~3.2 kPa that reaches steady-state within hours97. The term “tensional homeostasis” was coined to describe this process98. Indeed, if this stress is manually increased or decreased relative to the endogenous level, the cells quickly begin to return the stress toward the level they established originally. Not surprisingly, these cell-mediated processes depend on actomyosin contractility and appropriate integrins attaching to the ECM78,90 and are regulated through RhoA/RhoKinase and MAPK signaling pathways, amongst others99. Recalling that smooth muscle contractility can contribute to vessel-level adaptations to altered hemodynamics30, we see similar signaling (RhoA/RhoKinase, calcium/calmodulin, and MAPK) involved in both low (~3–5 kPa)97,98 and high (~100 kPa)100,101 contractile stress mediated ECM remodeling.
Similar to findings at the tissue level, fibroblasts decrease or increase their stiffness (which reflects changing cytoskeletal stress because of the nonlinear stress-strain behavior) in response to increases or decreases, respectively, in cell stretch102. Such findings suggest that fibroblasts also seek a tensional homeostasis, a concept that was confirmed for equibiaxially stretched SMCs103, suggesting that a rapid disassembly/reassembly of cytoskeletal proteins (i.e., dynamic equilibrium)104 allows cells to return intracellular stiffness, and hence stress, toward a preferred value following perturbations from endogenous levels. These findings are consistent with reports that individual cells generate stresses up to 5 kPa within initially unloaded 3-D hydrogels, with stress localized at focal adhesions105. Such observations suggest that cells not only modify the matrix to establish a preferred micro-environment in which to reside, but also maintain their own stress at a target level (recall Newton’s third law of motion, for every action there is an equal and opposite reaction).
Nearly linear relationships have been observed between cell-induced forces at focal adhesions and focal adhesion area in fibroblasts and SMCs106–108, which suggests a target value of stress around 3–6 nN/µm2 (i.e., 3–6 kPa). Slightly higher values (~10 to 12 kPa) were found for myofibroblasts that exhibit supermature focal adhesions and increased actomyosin contractility relative to synthetic SMCs and fibroblasts108. Nevertheless, these findings collectively suggest the existence of target values of stress. Of course, stresses at focal adhesions relate directly to stresses in the cytoskeleton, including prestresses. Indeed, it was appropriately noted that RhoA-dependent “pre-existing cytoskeletal tension affects the actomyosin apparatus, which in turn coordinates the ability of the cell to adapt to the externally applied stress”.109
In summary, whether at organ (vessel mechanoadaptations), tissue (collagen gels and hydrogels), cellular (isolated non-confluent and embedded within matrix), or sub-cellular (focal adhesions and cytoskeleton) scales, a mechanical homeostasis appears to exist whereby intramural cells try to establish, maintain, or restore a preferred mechanical state95,96. Whether this state is defined locally by stress, strain, stiffness, or another metric is not clear, but stress correlates well with many observations47 (stress and strain are related via constitutive relations, thus, one can be written in terms of the other; stress and stiffness are related linearly for materials such as the aorta that exhibit an exponential stress-strain relation). Interestingly, studies at tissue97, cell105, and sub-cellular106 levels all reveal a preferred level of stress ~5 kPa.
These findings, coupled with the existence of target stresses within the aortic wall of ~150 kPa, suggest that two distinct classes of SMCs (or their behaviors) contribute to arterial mechanics. First, fully contractile SMCs that generate stresses of ~100 kPa can contribute directly to overall load bearing47, including control of caliber (recall that, at least in the mouse, tissue-level aortic contractility is similar to that in other elastic arteries56,58). Second, synthetic SMCs, fibroblasts, and myofibroblasts that appear to seek a target stress of ~5 to 10 kPa cannot contribute directly to overall load bearing, but instead contribute indirectly by regulating the load-bearing ECM. That is, synthetic cells contribute to overall structural integrity by first sensing (literally probing and assessing) their mechanical environment and then producing, actively organizing, or removing ECM to support hemodynamic loads. A corollary, therefore, is that synthetic cells appear to be “stress shielded” by the ECM78 while yet being able to sense and thereby respond to these loads. Stress shielding is consistent with the dynamic nature of mechanosensing, namely, turnover of focal adhesion and cytoskeletal proteins within minutes104,110. That is, although true load-bearing must be sustained, mechanosensing can be intermittent, particularly given that adjustments via matrix turnover occur over periods of hours, days, or weeks.
Mechanosensing of Matrix
The ability of medial SMCs and adventitial fibroblasts/myofibroblasts to respond to changes in mechanical stimuli requires that they interact directly with the ECM. Their primary mechanosensors are heterodimeric transmembrane complexes called integrins (Figure 1), which are denoted αxβy and cluster to form dense plaques/focal adhesions111. Vascular SMCs, fibroblasts, and myofibroblasts employ a repertoire of integrins78,112, including α1β1, α2β1, α2β1, α5β1, α7β1 and αvβ3 by SMCs and α2β1, α5β1, αvβ3, αvβ5, and αvβ8 by (myo)fibroblasts. These integrins mediate binding to specific constituents of the matrix (e.g., α5β1 for fibronectin and α7β1 for laminin), with some overlap in specificity (e.g., α5β1, αvβ3, and αvβ5 bind fibronectin). Although there is redundancy in genes encoding integrins, deficiency in either σ1 or β1 leads to a vascular phenotype, including reduced mechanoadaptivity113,114. Particular matrix constituents can influence cell phenotype and thus responses to chemo- or mechano-stimulation. For example, fibronectin tends to promote a synthetic smooth muscle phenotype whereas laminin tends to promote a contractile phenotype115. Importantly, microfibrils consisting primarily of fibrillin-1 connect aortic SMCs to the elastic laminae primarily via σ5β1 and σvβ3 integrins116. That these microfibrils tend to align co-axially with actomyosin fibers within the cell11,22 suggests an efficient transferal of loads from cytoskeleton to integrin to matrix and vice versa. Loss of such connections would be expected to alter SMC signaling and phenotype5.
In summary, integrin engagement and actomyosin contractile activity are fundamental to mechanosensing and mechanoregulation of matrix99,110 and thus mechanical homeostasis78,96. Findings at tissue, cellular, and sub-cellular levels of actomyosin-generated stresses of ~5 kPa are consistent with mechanosensing, not gross load-bearing. Although such active stresses are much lower than those involved in vasoactive changes of vessel diameter, the intracellular structures and signaling pathways are similar. Thus, pathways fundamental to SMC mechanobiological changes in general45,117 and mechanosensing in particular99,110 overlap considerably with those in vasoactivity100,101. Similar considerations apply to fibroblasts and myofibroblasts78,81.
Finally, mechanostimulation of cells involves receptor tyrosine kinases (e.g., EGFR or PDGFR), serine-threonine receptors (e.g., TGF-β type I and II receptors), G-protein coupled receptors (e.g., AT1R and ETA), and stretch-activated cationic (e.g., Ca++) channels90,118,119. These pathways may involve ligand-independent, mechanical activation of the receptors120, load-induced secretion of ligand121, opening of channels122, or release of active ligand78. For example, mechanical activation of latent TGF-β can be achieved via actomyosin activity and integrins that bind the latency complex (e.g., σvβ3 and σvβ6), which could impact adventitial remodeling31. Interestingly, cell generated stresses needed to activate TGF-β are ~5 to 9 kPa78, similar to typical stresses at focal adhesions106, in cells placed within otherwise unloaded matrix105, and endogenous levels established in collagen gels by cells97. It may be that mechanosensing and the mechanoregulation of cytokines78, proteases and their action74, or cross-linkers such as transglutaminases123 that are stored within the ECM are highly complementary, again involving the same intracellular structures and signaling pathways. Finally, transmembrane polycystins appear to contribute to mechanosensing in vascular cells; they often co-localize with stretch-activated calcium channels and modulate their activity124. Polycystin-1,2 play important roles in mechanosensing both individually (via filamin-A related interactions with cytoskeletal actin125) and in cooperation with focal adhesions126.
Mechanoregulation of Matrix
Theoretical studies of soft tissue growth and remodeling suggest that, under normal conditions, cells deposit new structural constituents within extant matrix at a homeostatic stress or stretch34,35. This conjecture is appreciated easily in tissue maintenance: if stressed matrix is removed during normal turnover, the only way to maintain the same properties and geometry under with a similar load is to replace this matrix with the same constituents having the same stresses. Such mechanoregulated deposition is supported computationally and experimentally. Computations of arterial mechanoadaptations show that ongoing replacement of stressed matrix with either unstressed or less stressed matrix causes a numerical artery to dilate under a constant pressure, which violates tissue maintenance34. Experiments reveal that SMCs and fibroblasts mediate collagen assembly via integrins (e.g., α2β1) and RhoA activity127,128, which implicate an active process of organization. Indeed, embryonic fibroblasts can produce procollagen independent of actin, but they cannot organize and align collagen fibers without functional actin, thus implying an actomyosin-based mechanoregulation of the fibers via integrins129,130.
Interestingly, TGF-β influences both α2β1 integrin and SMC α-actin levels in cells that remodel collagen gels in vitro131, and focal adhesion kinase is increased in SMCs concomitant with the synthesis of proteins in response to exogenous Ang-II and its associated MAPK signaling132. Fibronectin is also necessary for proper collagen assembly127,128, and fibronectin assembly is mechanoregulated via integrins (α5β1) and actomyosin activity133. Elastic fiber assembly is similarly mediated by cell generated mechanical loads134. Clearly, matrix deposition and organization are mediated by active cell processes that involve force generation95,96.
Cells also actively remodel extant matrix into mechanically preferred states78. For example, fibroblasts build in a “residual matrix tension” (likely ~5 kPa97) in mechanically constrained collagen gels that persists even if subsequent actomyosin activity is blocked135. In other words, residual matrix tension preserves some cell-mediated changes in matrix organization without requiring continued actomyosin activity, which would be energetically favorable. Transglutaminases may play a role in such entrenchment just as in tissue-level arterial remodeling30, noting that matrix bound transglutaminases can be activated by mechanical stress123.
In summary, cells appear to modulate their ECM to match the forces encountered99. Diverse findings suggest that when organizing either newly produced or extant matrix, cells seek to establish, maintain, or restore a preferred (homeostatic) mechanical state that correlates well with a target stress or stiffness. Fundamental to such mechanoregulation of matrix is the ability of the cells first to sense the mechanical state, which requires integrin expression and actomyosin activity. Amongst its many effects, TGF-β can increase the expression and organization of contractile proteins within SMCs and fibroblasts, which correlates with increased integrin clustering78. It seems, therefore, that this cytokine known primarily for increasing the production of matrix simultaneously enables cells to actively organize and sense the deposited matrix through focal adhesions and actomyosin complexes. Establishing and maintaining matrix at a preferred state is fundamental to the overall structural integrity of tissues and the health of the cell. In extreme cases, however, loss of contact with matrix causes cells to undergo a special form of apoptosis called anoikis136. Conversely, simply reducing the load in the arterial wall results in adaptive atrophic remodeling due to degradation of matrix and apoptosis137,138. Clearly then, if dysfunctional mechanosensing leads cells to misinterpret a normal or high stress environment as a low stress environment, they may inadvertently apoptose or elicit an atrophic remodeling response, both of which could compromise overall structural integrity5.
Genes Predisposing to Thoracic Aortic Disease
Since the discovery in 1991 that mutations in FBN1 predispose individuals with Marfan syndrome to TAADs139, mutations in many other genes have similarly been identified1,27,41,69,140–147. In addition, common single base pair variants (SNPs) and genomic copy number variants (CNVs) in some of these same genes increase the risk for TAADs in the general population60,148,149. These genes can be categorized based on their effects on mechanosensing and mechanoregulation of matrix by medial SMCs, particularly through disruption of the contractileelastic unit5.
Numerous mutated genes predisposing to TAADs alter proteins in the microfibril sheath that encapsulates elastin (Figure 1). FBN1 encodes fibrillin-1, the major protein in the microfibrils that connect elastic fibers to dense plaques in SMCs. Fibroblasts explanted from patients with Marfan syndrome consistently exhibit decreased deposition of fibrillin-1 into the ECM150,151, thus suggesting that FBN1 mutations compromise connections between SMCs and elastic fibers. Mutations in microfibril associated glycoprotein 2 (encoded by MFAP5), another component of microfibrils, also predispose to TAAD147. Other mutant genes that lead to thoracic aortic disease affect proteins that normally contribute to the structural integrity of the wall (e.g., fibulin-4 (FNLN4), and collagen III (COL3A1))152,153, which also connect structurally to the SMCs and could alter the mechanical stimuli sensed by these cells as the wall experiences either the same or increased cyclic loads.
Other genes predisposing to TAADs encode proteins that contribute to the structure or function of the SMC contractile unit, as well as cytoskeletal components linking these units to the dense plaques1. The two major proteins in the contractile unit are the SMC-specific isoform of actin, α-actin, and SM-myosin heavy chain, which form thin and thick filaments, respectively. Mutations in the corresponding genes, ACTA2 and MYH11, predispose to thoracic aortic disease60,69,142. Preliminary data suggest that mutations in these genes disrupt the ability of monomers of α-actin and myosin heavy chain to form functional filaments. SMCs also have a dedicated kinase, myosin light chain kinase (MYLK), that phosphorylates the regulatory light chain (RLC) to initiate SMC contraction. MYLK loss of function, predicted to decrease activation of the RLC, also predisposes to TAADs143. Type I cGMP-dependent protein kinase (PRKG1) controls RLC dephosphorylation to relax SMCs. A gain of function mutation in PRKG1 that increases dephosphorylation of the RLC also predisposes to thoracic aortic disease146. Finally, mutations in filamin-A, a cytoplasmic protein that links actin filaments and is strongly implicated in mechanotransduction, predispose to TAADs1,154. All of these mutations in SMC contractile, structural, and signaling genes are predicted to decrease the cells’ ability to generate the force needed to appropriately sense and regulate the mechanical state of the matrix.
Genetic alterations in mouse models have identified additional genes involved in ECM integrity or mechanosensing that predispose to TAADs. These genes encode other proteins and proteoglycans in the matrix (e.g., collagen V, biglycan, and lysyl oxidase)16,17,155, transmembrane structures (e.g., polycystin-1,2)156, intracellular signaling molecules (e.g., integrin linked kinase)157, and calcium binding proteins158. Interestingly, increased S100A12 (a calcium binding protein) resulted in reduced vinculin, SM-myosin heavy chain, and SM-α actin, each of which can contribute to effective sensing and regulation of ECM. Increased S100A12 also increased IL-6 production and activation of TGF-β pathways. Finally, TGF-β signaling in Loeys-Dietz syndrome has been suggested to be Ang-II dependent159, and thoracic aortic disease in the Fbln4SMKO model depends on intramural increases in Ang-II production160.
Genome wide association studies in patients who do not have Marfan syndrome show that common SNPs in FBN1 still associate with thoracic aortic disease149. Whereas altered or diminished fibrillin-1 can contribute to premature mechanical fatigue or elastolysis, consistent with compromised wall properties expected during aneurysmal dilatation14,35,36 and altered mechanosensing5, fibrillin-1 plays additional roles – it sequesters latent TGF-β in the ECM. It has been proposed that mutations in FBN1 can lead to inappropriate release of active TGF-β from the matrix and that excessive TGF-β signaling can cause the increased SMAD and MAPK signaling in aortas of Marfan mouse models161,162. Aortic tissue from patients undergoing surgical repair of familial TAADs also exhibit increased TGF-β signaling based on immunostaining of phosphorylated SMAD2. It is thus surprising that loss of function mutations in genes encoding proteins critical for TGF-β signaling also predispose to TAADs, including those involving genes that encode type 1 and 2 transforming growth factor receptors (TGFBR1 and TGFBR2), one of the three isoforms of TGF-β (TGFB2), and related signaling molecules, SMAD3 (SMAD3) and SMAD4 (SMAD4)140,141,144,145,163. Thus, the paradox of mutations decreasing TGF-β signaling despite evidence of increased TGF-β in end stage aortic tissues raises questions as to the precise role(s) played by TGF-β signaling in thoracic aortic disease.
Studies in mice demonstrate that TGF-β signaling is critical for proper development of the aorta. Loss of TGF-β signaling disrupts the differentiation of neural crest-derived SMCs that populate the ascending aorta and the secondary heart field epithelial-to-mesenchymal transition involved in forming SMCs in the aortic root during development164,165. The importance of TGF-β in the differentiation of aortic SMCs is illustrated by the finding that ascending aortas in Tgfb2−/− mice are small and thin-walled166, and mice with the Tgfbr2 gene conditionally deleted in vascular SMCs display arterial dilation and dissection80. These studies showed that TGF-β gene mutations do not disrupt cardiovascular development; migration or homing of cells to the aorta during development was normal. Rather, decreased TGF-β signaling predisposes to slowly progressing, primarily adult-onset vascular diseases initially involving the aortic root and ascending aorta. TGF-β may be required for full differentiation of aortic SMCs during development, and specifically the development of a full complement of contractile-elastic units to withstand life-long mechanical loading of the aorta. Supporting this hypothesis, SMCs explanted from patients with TGFBR2 mutations show reduced expression of proteins in the SMC contractile unit when compared with control SMCs, and they differentiate to a lesser degree than control SMCs in response to TGF-β1176. Thus, failure to build the proper number of SMC contractile-elastic units during development due to defects in TGF-β pathway genes may lead to altered sensing of stress by the aortic SMCs and secondary compensatory increases in Ang II and TGF-β signaling.
A Unifying Hypothesis
An emerging concept is that altered cell-matrix connections, especially via the contractile-elastic unit, play important roles in TAADs1,14,27,58,161,167–169. Given that such connections are fundamental determinants of cell phenotype112 and cell survival136, this hypothesis is intuitive. Based on our review of the mechanics and mechanobiology, both computational and experimental, we submit further that many of the identified genetic mutations in TAADs implicate two specific, complementary aspects of cell-matrix interactions that directly impact the structural integrity of the aortic wall. Functional connections must exist between SMCs and surrounding ECM to enable these cells (i) to actively assess their mechanical environment and express appropriate mechanosensitive genes, and (ii) to actively remodel extant matrix or prestress newly produced matrix as it is incorporated within the wall. Both of these cell-mediated functions are necessary for establishing, maintaining, or restoring structural integrity in response to altered biomechanical loads. Moreover, cell-level actomyosin contractile activity is fundamental to such mechanosensing and mechanoregulating of the matrix, as suggested by the convergence of TAAD-related signaling pathways with mechanisms of cell sensing and regulation of matrix (Figure 3). Consequently, if any one of these three essential links – matrix stiffness, transmembrane structures, or cytoskeletal structures, including the actomyosin apparatus and proper signaling to express and assemble these links – is defective (Figure 4), mechanosensing and mechanoregulation will be compromised. That is, mechanical homeostasis will be dysfunctional and the aorta will become vulnerable to structural failure.
Figure 4.
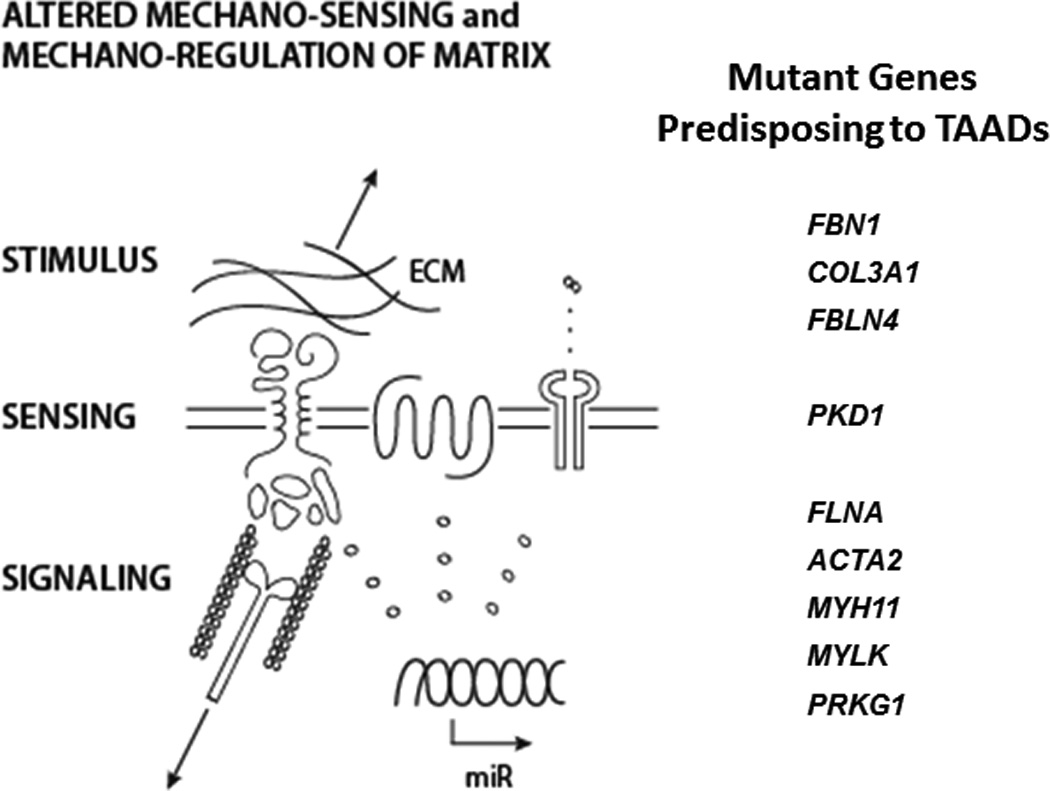
Confluence of some of the gene mutations that predispose to thoracic aortic aneurysms and dissections and affect the: mechano-stimulus (e.g., transferal of stress from the matrix to the cell), the mechano-sensing of this stimulus, or the associated mechano-sensitive signaling pathways. See text for information on the affected genes and related references.
Given the apparent importance of TGF-β and possibly Ang-II in TAADs1,27,41,161,170, their mechanobiological roles must be considered. To this end, in vitro and in vivo studies reveal direct relationships between mechanical stimuli on SMCs and both Ang-II and TGF-β activity71–73,117. These results are consistent with the influence these biomolecules have on matrix production/degradation78,87,89 and the actomyosin-integrin structures82,83 that are needed to organize a matrix capable of supporting hemodynamically induced loads. Indeed, Ang-II and TGF-β play similarly important roles in hypertension and aging wherein the wall can thicken to counteract increased pressure-induced wall stress26,32,91,93, though not all matrix production need be structurally protective37,38. Interestingly, a transcriptional analysis of vascular aging identified altered cell-matrix interactions as particularly important171. Mechanical stress and TGF-β signaling are also complementary partners in the differentiation of fibroblasts to myofibroblasts, which enables increased synthesis and organization of matrix78,81. These processes may play important roles in adventitial remodeling in severe cases of increased mechanical stress32,75,80, which, when the media degenerates in TAADs, is engaged as a protective sheath35.
Closure
The normal thoracic aorta can maintain its geometry, structure, and function over decades and adapt well to modest sustained changes in hemodynamics. Such maintenance and adaptivity are achieved, in large part, by intramural cells properly sensing their chemo-mechanical environment and actively regulating the ECM to ensure appropriate compliance and structural integrity. However, aging results in diffusely diminished smooth muscle functionality, lost elastic fiber integrity, remodeled collagen, increased glycosaminoglycans, and increased intramural Ang-II and TGF-β26,171. These changes can manifest clinically as decreased arterial distensibility, increased pulse wave velocity, and increased central systolic and pulse pressure, all of which feedback to increase the hemodynamic loads93. Many of these same changes are found at tissue, cellular, and molecular levels in some individuals having TAADs142,172. In some cases, therefore, TAADs may represent an accelerated, exaggerated “localized aging” of the aorta. In other cases, mutations (e.g., in TGFBR1/2) likely compromise the developing matrix and differentiation of SMCs, which affects subsequent function and structural integrity80, including arterial tortuousity at young ages2. Thus, ineffective cell-matrix interactions likely render the wall vulnerable to increases in hemodynamic loading, even during development.
The aortic wall will fail as a structure, namely dissect or rupture, if and only if local mechanical stresses exceed mechanical strength. There is a pressing need, therefore, to focus on local, not global, mechanics173. In contrast, mechanobiological responses by aortic cells need not depend on the actual mechanical environment – responses depend on what the cell can perceive (i.e., sense) and achieve (e.g., genes that can be expressed). Based on the diverse findings collected together herein, we submit that TAADs arise, in large part, due to an inappropriate mechanosensing and mechanoregulation of ECM by medial SMCs that renders the aortic wall vulnerable to dilatation and dissection. An associated or subsequent inability of adventitial fibroblasts to maintain or restore a sufficiently strong adventitia can further lead to rupture.
Future research should thus focus on local, not global, mechanics; mechanisms by which cells sense changes in local mechanical stimuli; and associated signaling and gene expression pathways that govern the structural integrity of the aortic wall. Some guidance forward is found in the recent elucidation of abnormal mechanosignaling in the heart in Marfan syndrome174. Elucidation of these pathways may identify new therapeutic targets to decrease destructive (low-stress) or enhance constructive (high-stress) mechano-sensitive remodeling of matrix. On this foundation, translational efforts to prevent TAAD may be successful in preventing aneurysms and dissections of the thoracic aorta.
Acknowledgments
Sources of funding
This work was supported by grants from the National Marfan Foundation (JDH and GT), the John Ritter Foundation (DMM) and the NIH: R01 HL062594 and P50 HL083794 (DMM), R01 HL075092 (MAS), and U01 HL116323 (JDH and GT).
Nonstandard Abbreviations and Acronyms
- Ang-II
angiotensin-II
- AT1R
angiotensin-II type I receptor
- CNV
copy number variant
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- RLC
regulatory light chain
- TAAD
thoracic aortic aneurysms and aortic dissections
- TGF-β
transforming growth factor - β
- SMC
smooth muscle cell
- SNP
single nucleotide polymorphism;
Footnotes
Disclosures:
None.
References
- 1.Milewicz DM, Gou DC, Fadulu VT, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 2.Jain D, Dietz HC, Oswald GL, Maleszewski JJ, Halushka MK. Causes and histopathology of ascending aortic disease in children and young adults. Cardiovasc Path. 2011;20:15–75. doi: 10.1016/j.carpath.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elefteriades JA, Farkas EA. Thoracic aortic aneurysm: Clinically pertinent controversies and uncertainties. J Am Coll Cardiol. 2010;55:841–857. doi: 10.1016/j.jacc.2009.08.084. [DOI] [PubMed] [Google Scholar]
- 4.Howard DP, Banerjee A, Fairhead JF, Perkins J, Silver LE, Rothwell PM. Population-based study of incidence and outcome of acute aortic dissection and premorbid risk factor control: 10-year results from the Oxford Vascular Study. Circulation. 2013;127:2013–2037. doi: 10.1161/CIRCULATIONAHA.112.000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Dysfunctional mechanosensing in aneurysms. Science. 2014;344:477–479. doi: 10.1126/science.1253026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dingemans RP, Teeling P, Lagendijk JH, Becker AE. Extracellular matrix of the human aortic media: An ultrastructural histochemical and immunohistochemical study of the adult aortic media. Anat Record. 2000;258:1–14. doi: 10.1002/(SICI)1097-0185(20000101)258:1<1::AID-AR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89:957–989. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonker FH, van Keulen JW, Schlossen FJ, Indes JE, Moll FL, Verhagen HJ, Muhs BE. Thoracic aortic pulsatility decreases during hypovolumic shock: Implications for stent-graft sizing. J Endovasc Ther. 2011;18:491–496. doi: 10.1583/10-3374.1. [DOI] [PubMed] [Google Scholar]
- 9.Arribas SM, Hinek A, González MC. Elastic fibres and vascular structure in hypertension. Pharmacol Therapeu. 2006;111:771–791. doi: 10.1016/j.pharmthera.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Davis EC. Elastic lamina growth in the developing mouse aorta. J Histochem Cytochem. 1995;43:1115–1123. doi: 10.1177/43.11.7560894. [DOI] [PubMed] [Google Scholar]
- 11.Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta: Role in aortic medial organization. Lab Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- 12.Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20:99–111. doi: 10.1161/01.res.20.1.99. [DOI] [PubMed] [Google Scholar]
- 13.Pereira L, Lee SY, Gayraud B, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunton TE, Biery J, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 15.Yanagisawa H, Davis EC. Unraveling the mechanism of elastic fiber assembly: the roles of short fibulins. Intl J Biochem Cell Biol. 2010;42:1084–1093. doi: 10.1016/j.biocel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wenstrup RJ, Florer JB, Davidson JM, Phillips CL, Pfeiffer BJ, Menezes DW, Chervoneva I, Birk DE. Murine model of the Ehlers-Danlos syndrome. J Biol Chem. 2006;281:12888–12895. doi: 10.1074/jbc.M511528200. [DOI] [PubMed] [Google Scholar]
- 17.Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, Young MF, Bianco P. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115:2731–2738. doi: 10.1161/CIRCULATIONAHA.106.653980. [DOI] [PubMed] [Google Scholar]
- 18.Lindeman JHN, Ashcroft BA, Beenakker JWM, et al. Distinct defects in collagen microarchitecture underlie vessel-wall failure in advanced abdominal aneurysms and aneurysms in Marfan syndrome. PNAS. 2010;107:862–865. doi: 10.1073/pnas.0910312107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rahkonen O, Su M, Hakovirta H, Koskivirta I, Hormuzdi SG, Vuorio E, Bornstein P, Penttinen R. Mice with a deletion I the first intron of the Collal gene develop age-dependent aortic dissection and rupture. Circ Res. 2004;94:83–90. doi: 10.1161/01.RES.0000108263.74520.15. [DOI] [PubMed] [Google Scholar]
- 20.Byra P, Chillag S, Petit S. Osteogenesis imperfecta and aortic dissection. Am J Med Sci. 2008;336:70–72. doi: 10.1097/MAJ.0b013e318158e981. [DOI] [PubMed] [Google Scholar]
- 21.Smith LB, Hadoke PWF, Dyer E, Denvir MA, Brownstein D, Miller E, Nelson N, Wells S, Cheeseman M, Greenfield A. Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome. Cardiovasc Res. 2011;90:182–190. doi: 10.1093/cvr/cvq356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark J, Glagov S. Structural integration of the arterial wall Relationships and attachments of medial smooth muscle cells in normally distended and hyperdistended aortas. Lab Invest. 1979;40:587–602. [PubMed] [Google Scholar]
- 23.Schriefl AJ, Zeindlinger G, Pierce DM, Regitnig P, Holzapfel GA. Determination of the layer-specific distributed collagen fibre orientations in human thoracic and abdominal aortas and common iliac arteries. J R Soc Interface. 2012;9:1275–1286. doi: 10.1098/rsif.2011.0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cyron C, Humphrey JD. Preferred fiber orientations in arteries and veins understood from netting analysis. Math Mech Solids. 2015 in-press. [Google Scholar]
- 25.Ferruzzi J, Collins MJ, Yeh AT, Humphrey JD. Mechanical assessment of elastin integrity in fibrillin-1 deficient carotid arteries: Implications for Marfan syndrome. Cardiovasc Res. 2011;92:287–295. doi: 10.1093/cvr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin N Am. 2009;93:583–604. doi: 10.1016/j.mcna.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev – Cardiol. 2009;6:771–786. doi: 10.1038/nrcardio.2009.191. [DOI] [PubMed] [Google Scholar]
- 28.Nissen R, Cardinale GJ. Udenfriend Increased turnover of arterial collagen in hypertensive rats. PNAS. 1978;75:451–453. doi: 10.1073/pnas.75.1.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Feo JA, Sluijter JPG, de Kleijn DPV, Pasterkamp G. Modulation of collagen turnover in cardiovascular disease. Curr Pharmacol Design. 2005;11:2501–2514. doi: 10.2174/1381612054367544. [DOI] [PubMed] [Google Scholar]
- 30.Dajnowiec D, Langille BL. Arterial adaptations to chronic changes in haemodynamic function: coupling vasomotor tone to structural remodeling. Clin Sci. 2007;113:15–23. doi: 10.1042/CS20060337. [DOI] [PubMed] [Google Scholar]
- 31.Bellini C, Ferruzzi J, Roccabianca S, DiMartino E, Humphrey JD. A microstructurally-motivated model of arterial wall mechanics with mechanobiological implications. Annl Biomed Engr. 2014;42:488–502. doi: 10.1007/s10439-013-0928-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuang SQ, Geng L, Prakash SK, Cao JM, Guo S, Villamizar C, Kwartler CS, Peters AM, Brasier AR, Milewicz DM. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler Thromb Vasc Biol. 2013;33:2172–2179. doi: 10.1161/ATVBAHA.113.301624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 34.Valentin A, Humphrey JD, Holzapfel GA. A multi-layered computational model of coupled elastin degradation, vasoactive dysfunction, and collagenous stiffening in aortic aging. Annl Biomed Engr. 2011;39:2027–2045. doi: 10.1007/s10439-011-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson JS, Baek S, Humphrey JD. Importance of initial aortic properties on the evolving regional anisotropy, stiffness, and wall thickness of human abdominal aortic aneurysms. J R Soc Interface. 2012;9:2047–2058. doi: 10.1098/rsif.2012.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobrin PB, Schwarcz TH, Baker WH. Mechanisms of arterial and aneurysmal tortuosity. Surgery. 1998;104:568–571. [PubMed] [Google Scholar]
- 37.Humphrey JD. Possible roles of glycosaminoglycans in aortic dissections, with implications to dysfunctional TGF-β. J Vasc Res. 2013;50:1–10. doi: 10.1159/000342436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roccabianca S, Bellini C, Humphrey JD. Computational modeling suggests good, bad, and ugly roles of glycosaminoglycans in arterial mechanics and mechanobiology. J Roy Soc Interface. 2014 doi: 10.1098/rsif.2014.0397. (ePub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. Development and disease A critical role for elastin signaling in vascular morphogenesis and disease. Develop. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 40.ten Dijke P, Arthur HM. Extracellular control of TGF-β signaling in vascular development and disease. Nat Rev Molc Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 41.Doyle JJ, Gerber EE, Dietz HC. Matrix-dependent perturbation of TGFβ signaling and disease. FEBS Letters. 2012;586:2003–2015. doi: 10.1016/j.febslet.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hynes RO. The extracellular matrix: Not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hornebeck W, Emonard H. The cell-elastin-elaastase(s) interacting triade dictates elastolysis. Front Biosci. 2011;16:707–722. doi: 10.2741/3714. [DOI] [PubMed] [Google Scholar]
- 44.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3:a005058. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li C, Xu Q. Mechanical stress-initiated signal transduction in vascular smooth muscle cells in vitro and in vivo. Cell Signal. 2007;19:881–891. doi: 10.1016/j.cellsig.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Chiquet M, Gelman L, Lutz R, Maier S. From mechanotransduction to extracellular matrix gene expression in fibroblasts. Biochim Biophysic Acta. 2009;1793:911–920. doi: 10.1016/j.bbamcr.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey JD. Cardiovascular Solid Mechanics. NY: Springer; 2002. [Google Scholar]
- 48.Fillinger MF. Who should we operate on and how do we decide: predicting rupture and survival in patients with aortic aneurysms. Sem Vasc Surg. 2007;20:121–127. doi: 10.1053/j.semvascsurg.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 49.Roccabianca S, Figueroa CA, Tellides G, Humphrey JD. Quantification of regional differences in aortic stiffness in the aging human aorta. J Biomech Behav Biomed Matl. 2014;29:618–634. doi: 10.1016/j.jmbbm.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Herrera CM, Atienza JM, Rojo FJ, Claes E, Guinea GV, Celentano DJ, Garcia-Montero C, Burgos RL. Mechanical behavior and rupture of normal and pathological human ascending aortic wall. Med Biol Eng Comput. 2012;50:559–566. doi: 10.1007/s11517-012-0876-x. [DOI] [PubMed] [Google Scholar]
- 51.Chiu J-J, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashi K, Naiki Adaptation and remodeling of vascular wall; biomechanical response to hypertension. J Mech Behav Biomed Matl. 2009;2:3–19. doi: 10.1016/j.jmbbm.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 53.Driss AB, Benessian J, Pitevin P, Levy BI, Michel JB. Arterial expansive remodeling induced by high flow rates. Am J Physiol. 1997;272:H851–H858. doi: 10.1152/ajpheart.1997.272.2.H851. [DOI] [PubMed] [Google Scholar]
- 54.Leung DYM, Glagov S, Mathews MB. Elastin and collagen accumulation in rabbit ascending aorta and pulmonary trunk during postnatal growth. Circ Res. 1977;41:316–323. doi: 10.1161/01.res.41.3.316. [DOI] [PubMed] [Google Scholar]
- 55.Langille BL, Brownlee RD, Adamson SL. Perinatal aortic growth in lambs: relation to blood flow changes at birth. Am J Physiol. 1990;259:H1247–H1253. doi: 10.1152/ajpheart.1990.259.4.H1247. [DOI] [PubMed] [Google Scholar]
- 56.Agianniotis A, Rachev A, Stergiopulos N. Active stress in mouse aorta. J Biomech. 20122012:451924–1927. doi: 10.1016/j.jbiomech.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 57.Schildmeyer LA, Braun R, Taffet G, Debiasi M, Burns AE, Bradley A, Schwartz RJ. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J. 2000;14:2213–2220. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- 58.Chung AWY, Yeung KA, Sandor GGS, Judge DP, Dietz HC, van Breeman C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and −9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101:512–522. doi: 10.1161/CIRCRESAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- 59.He W-Q, Qiao Y-N, Zhang C-H, et al. Role of myosin light chain kinase in regulation of basal blood pressure and maintenance of salt-induced hypertension. Am J Physiol. 2011;301:H584–H591. doi: 10.1152/ajpheart.01212.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuang S-Q, Kwartler CS, Byanova KL, Pham J, Gong L, Prakash SK, Huang J, Kamm KE, Stull JT, Sweeney HL, Milewicz DM. Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ Res. 2012;110:1411–1422. doi: 10.1161/CIRCRESAHA.111.261743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pelliccia A, Di Paolo FM, Quattrini FM. Aortic root dilatation in athletic population. Prog Cardiovasc Dis. 2012;54:432–437. doi: 10.1016/j.pcad.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 62.Huonker M, Schmid A, Schmidt-Trucksa B, Grathwohl D, Keul J. Size and blood flow of central and peripheral arteries in highly trained able-bodied and disabled athletes. Am J Physiol. 2003;95:685–691. doi: 10.1152/japplphysiol.00710.2001. [DOI] [PubMed] [Google Scholar]
- 63.Saouti N, Marcus JT, vonk Noordegraaf A, Westerhof N. Aortic function quantified: the heart’s essential cushion. J Appl Physiol. 1985;113:1285–1291. doi: 10.1152/japplphysiol.00432.2012. [DOI] [PubMed] [Google Scholar]
- 64.Redheuil A, Yu WC, Mousseaux E, de Cesare A, Yan R, Kachenoura N, Bluemke D, Lima JA. Reduced ascending aortic strain and distensibility: earliest manifestations of vascular aging in humans. Hypertension. 2010;55:319–326. doi: 10.1161/HYPERTENSIONAHA.109.141275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paini A, Boutouyrie P, Calvet D, Tropeano AI, Laloux B, Laurent S. Carotid and aortic stiffness: determinants of discrepancies. Hypertension. 2006;47:371–376. doi: 10.1161/01.HYP.0000202052.25238.68. [DOI] [PubMed] [Google Scholar]
- 66.Sugawara J, Hayashi K, Yokoi T, Tanaka H. Age-associated elongation of the ascending aorta in adults. JACC Cardiovasc Imag. 2008;1:739–748. doi: 10.1016/j.jcmg.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 67.Humphrey JD, Eberth JF, Dye WW, Gleason RL. Fundamental role of axial stress in compensatory adaptations by arteries. J Biomech. 2009;42:1–8. doi: 10.1016/j.jbiomech.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morris SA, Orbach DB, Geva T, Singh MN, Gauvreau K, Lacro RV. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation. 2011;124:388–396. doi: 10.1161/CIRCULATIONAHA.110.990549. [DOI] [PubMed] [Google Scholar]
- 69.Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leung DYM, Glagov S, Matthews MB. Cyclic stretching stimulates synthesis of matrix components by arterial smooth muscle cells in vitro. Science. 1976;191:475–477. doi: 10.1126/science.128820. [DOI] [PubMed] [Google Scholar]
- 71.Li Q, Muragaki Y, Hatamura I, Ueno H, Ooshima A. Stretch-induced collagen synthesis in cultured smooth muscle cells from rabbit aortic media and a possible involvement of angiotensin-II and transforming growth factor-beta. J Vas Res. 1998;35:93–103. doi: 10.1159/000025570. [DOI] [PubMed] [Google Scholar]
- 72.Stanley AG, Patel H, Knight AL, Williams B. Mechanical strain-induced human vascular matrix synthesis: the role of angiotensin II. JRAAS. 2000;1:32–35. doi: 10.3317/jraas.2000.007. [DOI] [PubMed] [Google Scholar]
- 73.O’Callaghan CJ, Williams B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells. Hypertension. 2000;36:319–324. doi: 10.1161/01.hyp.36.3.319. [DOI] [PubMed] [Google Scholar]
- 74.Ruberti JW, Hallab NJ. Strain-controlled enzymatic cleavage of collagen in loaded matrix. Biochem Biophys Res Comm. 2005;336:483–489. doi: 10.1016/j.bbrc.2005.08.128. [DOI] [PubMed] [Google Scholar]
- 75.Tieu BC, Ju X, Lee C, Sun H, Lejeune W, Recinos A, III, Brasier AR, Tilton RG. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J Vasc Res. 2011;48:261–272. doi: 10.1159/000320358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilson E, Mai Q, Sudhir K, Weiss RH, Ives HE. Mechanical strain induces growth of vascular smooth muscle cells via autocrine action of PDGF. J Cell Biol. 1993;123:741–747. doi: 10.1083/jcb.123.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davis MJ, Donovitz JA, Hood JD. Stretch-activated single-channel and whole cell currents in vascular smooth muscle cells. Am J Physiol. 1992;262:C1083–C1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- 78.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodeling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 79.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. doi: 10.1016/j.cardiores.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–767. doi: 10.1172/JCI69942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Forte A, Corte AD, De Feo M, Cerasuolo F, Cipollaro M. Role of myofibroblasts in vascular remodeling: focus on restenosis and aneurysm. Cardiovasc Res. 2010;88:395–405. doi: 10.1093/cvr/cvq224. [DOI] [PubMed] [Google Scholar]
- 82.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 83.Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-β signaling in vascular fibrosis. Cardiovasc Res. 2007;74:196–206. doi: 10.1016/j.cardiores.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 84.Zhang YE. Non-Smad pathways in TGF-B signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schlumberger W, Thie M, Rauterberg J, Robenek H. Collagen synthesis in cultured aortic smooth muscle cells. Arterioscl Thromb. 1991;11:1660–1666. doi: 10.1161/01.atv.11.6.1660. [DOI] [PubMed] [Google Scholar]
- 86.Ruddy JM, Jones JA, Stroud RE, Mukherjee, Spinale FG, Ikonomidis JS. Differential effects of mechanical and biological stimuli on matrix metalloproteinase promoter activity in the thoracic aorta. Circulation. 2009;120:S262–S268. doi: 10.1161/CIRCULATIONAHA.108.843581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang M, Zhang J, Spinetti G, Jiang L-Q, Monticone R, Zhao D, Cheng L, Krawczyk M, Talan M, Pintus G, Lakatta EG. Angiotensin II activates matrix metalloproteinase type 2 and minics age-associated carotid arterial remodeling in young rats. Am J Path. 2005;167:1429–1442. doi: 10.1016/S0002-9440(10)61229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang M, Zhao D, Spinetti G, Zhang J, Jiang LQ, Pintus G, Monticone R, Lakatta EG. Matrix metalloproteinase 2 activation of transforming growth factor-β1 (TGF-β) and TGF-β1-type II receptor signaling within the aged arterial wall. Arterioscl Thromb Vasc Biol. 2006;26:1503–1509. doi: 10.1161/01.ATV.0000225777.58488.f2. [DOI] [PubMed] [Google Scholar]
- 89.Tharaux P-L, Chatziantoniou C, Fakhouri F, Dussaule J-C. Angiotensin II activates collagen I gene through a mechanism involving the MAPK/ER kinase pathway. Hypertension. 2000;36:330–336. doi: 10.1161/01.hyp.36.3.330. [DOI] [PubMed] [Google Scholar]
- 90.Munger JS, Sheppard D. Crosstalk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011;3:1–6. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lacolley P, Challande P, Osborne-Pellegrin M, Regnault V. Genetics and pathophysiology of arterial stiffness. Cardiovasc Res. 2009;81:637–648. doi: 10.1093/cvr/cvn353. [DOI] [PubMed] [Google Scholar]
- 92.Reusch P, Wagdy H, Reusch R, Wilson E, Ives HE. Mechanical strain increases smooth muscle and decreases nonmuscle myosin expression in rat vascular smooth muscle cells. Circ Res. 1996;79:1046–1053. doi: 10.1161/01.res.79.5.1046. [DOI] [PubMed] [Google Scholar]
- 93.Safar ME. Arterial aging - hemodynamic changes and therapeutic options. Nat Rev. 2010;7:442–449. doi: 10.1038/nrcardio.2010.96. [DOI] [PubMed] [Google Scholar]
- 94.Chen X, Lu H, Rateri DL, Cassis LA, Daugherty A. Conundrum of angiotensin II and TGF-β interactions in aortic aneurysms. Curr Opin Pharmacol. 2013;13:180–185. doi: 10.1016/j.coph.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Humphrey JD. Vascular adaptation and mechanical homeostasis at tissue, cellular, and sub-cellular levels. Cell Biochem Biophys. 2008;50:53–78. doi: 10.1007/s12013-007-9002-3. [DOI] [PubMed] [Google Scholar]
- 96.DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nat Rev-Mol Cell Biol. 2011;12:308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kolodney MS, Wysolmerski RB. Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. J Cell Biol. 1992;117:73–82. doi: 10.1083/jcb.117.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brown RA, Projapati R, McGrouther DA, Yannas IV, Eastwood M. Tensional homeostasis in dermal fibroblasts: mechanical responses to mechanical loading in three-dimensional substrates. J Cell Physiol. 1998;175:323–332. doi: 10.1002/(SICI)1097-4652(199806)175:3<323::AID-JCP10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 99.Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2:a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim HR, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signaling pathways in health and disease. J Cell Mol Med. 2008;12:2165–2180. doi: 10.1111/j.1582-4934.2008.00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiol. 2009;24:342–356. doi: 10.1152/physiol.00023.2009. [DOI] [PubMed] [Google Scholar]
- 102.Mizutani T, Haga H, Kawabata K. Cellular stiffness response to external deformation: tensional homeostasis in a single fibroblast. Cell Motil Cytoskel. 2004;59:242–248. doi: 10.1002/cm.20037. [DOI] [PubMed] [Google Scholar]
- 103.Na S, Trache A, Trzeciakowski J, Sun Z, Meininger GA, Humphrey JD. Time-dependent changes in smooth muscle stiffness and focal adhesion area in response to cyclic equibiaxial stretch. Annl Biomed Engr. 2008;36:369–380. doi: 10.1007/s10439-008-9438-7. [DOI] [PubMed] [Google Scholar]
- 104.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Legant WR, Miller JS, Blakely BL, Cohen DM, Genin GM, Chen CS. Measurement of mechanical tractions exerted by cells in three dimensional matrices. Nat Meth. 2010;7:969–971. doi: 10.1038/nmeth.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3:466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- 107.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. PNAS. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goffin JM, Pittet P, Csucs G, Lussi JW, Meister J-J, Hinz B. Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers. J Cell Biol. 2006;172:259–268. doi: 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lim S-M, Trzeciakowski JP, Sreenivasappa H, Dangott LJ, Trache A. Rho-A induced cytoskeletal tension controls adaptive cellular remodeling to mechanical signaling. Integr Biol. 2012;4:615–627. doi: 10.1039/c2ib20008b. [DOI] [PubMed] [Google Scholar]
- 110.Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev-Mol Cell Biol. 2006;7:265–275. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 111.Ross TD, Coon BG, Yun S, Baeyens N, Tanaka K, Ouyang M, Schwartz MA. Integrins in mechanotransduction. Curr Opin Cell Biol. 2013;25:613–618. doi: 10.1016/j.ceb.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res. 2001;52:372–386. doi: 10.1016/s0008-6363(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 113.Louis H, Kakou A, Regnault V, Labat C, Bressenot A, Lo JG, Gardner H, Thornton SN, Challande P, Li Z, Lacolley P. Role of α1β1 - integrin in arterial stiffness and angiotensin-induced arterial wall hypertrophy in mice. Am J Physiol. 2007;293:H2597–H2604. doi: 10.1152/ajpheart.00299.2007. [DOI] [PubMed] [Google Scholar]
- 114.Turlo KA, Noel ODV, Vora R, LaRussa M, Fassler R, Hall-Glenn F, Iruela-Arispe ML. An essential requirement for β1 integrin in the assembly of extracellular matrix proteins within the vascular wall. Develop Biol. 2012;365:23–35. doi: 10.1016/j.ydbio.2012.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Roy J, Kazi M, Hedin U, Thyberg Phenotypic modulation of arterial smooth muscle cells is associated with prolonged activation of ERK1/2. Differentiation. 2000;67:50–58. doi: 10.1046/j.1432-0436.2001.067001050.x. [DOI] [PubMed] [Google Scholar]
- 116.Bax DV, Bernard SE, Lomas A, Morgan A, Humphries J, Shuttleworth CA, Humphries MJ, Kielty CM. Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by α5β1 and αvβ3 integrins. J Biol Chem. 2003;278:34605–34616. doi: 10.1074/jbc.M303159200. [DOI] [PubMed] [Google Scholar]
- 117.Lacolley P, Regnault V, Nicoletti A, Li Z, Michel J-B. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;15:194–204. doi: 10.1093/cvr/cvs135. [DOI] [PubMed] [Google Scholar]
- 118.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4:83–90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 119.Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signaling spreads. Nat Cell Biol. 2002;4:E65–E68. doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- 120.Storch U, Mederos Y, Schnitzler M, Gudermann T. G protein-mediated stretch reception. Am J Physiol. 2012;302:H1241–H1249. doi: 10.1152/ajpheart.00818.2011. [DOI] [PubMed] [Google Scholar]
- 121.Van Wamel AJ, Ruwhof C, van der Valk-Kokshoom LE, Schrier PI, van der Laarse A. The role of angiotensin II, endothelin-1, and transforming growth factor-beta as autocrine / paracrine mediators of stretch-induced cardiomyocyte hypertrophy. Mol Cell Biochem. 2001;218:113–124. doi: 10.1023/a:1007279700705. [DOI] [PubMed] [Google Scholar]
- 122.Inoue R, Jian Z, Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther. 2009;123:371–385. doi: 10.1016/j.pharmthera.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 123.Huelsz-Prince G, Belkin A, van Bavel E, Bakker ENTP. Activation of extracellular transglutaminase 2 by mechanical force in the arterial wall. J Vasc Res. 2013;50:383–395. doi: 10.1159/000354222. [DOI] [PubMed] [Google Scholar]
- 124.Sharif-Naeini, Folgering JH, Bichet D, Duprat F, Delmas P, Patel A, Honore E. Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol. 2010;48:83–89. doi: 10.1016/j.yjmcc.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 125.Patel A, Honore E. Polycystins and renovascular mechanosensory transduction. Nat Rev: Nephrol. 2010;6:530–538. doi: 10.1038/nrneph.2010.97. [DOI] [PubMed] [Google Scholar]
- 126.Israeli S, Amsler K, Zheleznova N, Wilson PD. Abnormalities in focal adhesion complex formation, regulation, and function in human autosomal recessive polycystic kidney disease epithelial cells. Am J Physiol. 2010;298:C831–C846. doi: 10.1152/ajpcell.00032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins α11β1 and α2β1 . J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- 128.Li S, Van den Diepstraten C, D’Souza SJ, Chan BMC, Pickering JG. Vascular smooth muscle cells orchestrate the assembly of type I collagen via α2β1 integrin, RhoA, and fibronectin polymerization. Am J Path. 2003;163:1045–1056. doi: 10.1016/s0002-9440(10)63464-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Canty EG, Starborg T, Lu Y, Humphries SM, Holmes DF, Meadows RS, Huffman A, O’Toole ET, Kadler KE. Actin filaments are required for fibripositor-mediated collagen fibril alignment in tendon. J Biol Chem. 2006;281:38592–38598. doi: 10.1074/jbc.M607581200. [DOI] [PubMed] [Google Scholar]
- 130.Kalson NS, Starborg T, Lu Y, Mironov A, Humphries SM, Holmes DF, Kadler KE. Nonmuscle myosin II powered transport of newly formed collagen fibrils at the plasma membrane. PNAS. 2013;110:E4743–E4752. doi: 10.1073/pnas.1314348110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arora PD, Narani N, McCulloch DA. The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am J Path. 1999;154:871–882. doi: 10.1016/s0002-9440(10)65334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Govindarajan G, Eble DM, Lucchesi PA, Samarel AM. Focal adhesion kinase is involved in angiotensin-II mediated protein synthesis in cultured vascular smooth muscle cells. Circ Res. 2000;87:710–716. doi: 10.1161/01.res.87.8.710. [DOI] [PubMed] [Google Scholar]
- 133.Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24:389–399. doi: 10.1016/j.matbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 134.Kozel BA, Rongish BJ, Czirok A, Zach J, Little CD, Davis EC, Knutsen RH, Wagenseil JE, Levy MA, Mecham RP. Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol. 2006;207:87–96. doi: 10.1002/jcp.20546. [DOI] [PubMed] [Google Scholar]
- 135.Marenzana M, Wilson-Jones N, Mudera V, Brown RA. The origins and regulation of tissue tension: identification of collagen tension-fixation process in vitro. Experiment Cell Res. 2006;312:423–433. doi: 10.1016/j.yexcr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 136.Michel J-B. Anoikis in the cardiovascular system Known and unknown extracellular mediators. Arterioscler Thromb Vasc Biol. 2003;23:2146–2154. doi: 10.1161/01.ATV.0000099882.52647.E4. [DOI] [PubMed] [Google Scholar]
- 137.Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–1375. doi: 10.1056/NEJM198705283162204. [DOI] [PubMed] [Google Scholar]
- 138.Bayer IM, Adamson SL, Langille BL. Atrophic remodeling of the artery-cuffed artery. Arterioscl Thromb Vasc Biol. 1999;19:1499–1505. doi: 10.1161/01.atv.19.6.1499. [DOI] [PubMed] [Google Scholar]
- 139.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 140.Mizuguchi T, Collod-Beroud G, Akiyama T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 142.Zhu L, Vranckx R, Khau, van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 143.Wang L, Guo D-C, Cao J, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Regalado ES, Guo DC, Villamizar C, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneuyrsms and dissections associated with mild systemic features of Marfan syndrome. Nat Gen. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Guo DC, Regalado E, Casteel DE, et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93:398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Barbier M, Gross M-S, Aubart M, et al. MFAP5 loss of function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. 2014;95:736–743. doi: 10.1016/j.ajhg.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Prakash SK, LeMaire SA, Guo DC, et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Human Gen. 2010;87:743–756. doi: 10.1016/j.ajhg.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lemaire SA, McDonald ML, Guo DC, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Milewicz DM, et al. Marfan syndrome: defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J Clin Invest. 1992;89:79–86. doi: 10.1172/JCI115589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hollister DW, et al. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med. 1990;323:152–159. doi: 10.1056/NEJM199007193230303. [DOI] [PubMed] [Google Scholar]
- 152.Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting the structure, stability, and processing of type III collagen. J Biol Chem. 1988;263:6226–6232. [PubMed] [Google Scholar]
- 153.Dasouki M, et al. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007;143A:2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- 154.Sheen VL, Walsh CA. Periventricular heterotopioa: new insights into Ehlers-Danlos syndrome. Clin Med Res. 2005;3:229–233. doi: 10.3121/cmr.3.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Maki JM, Rasanen J, Tikkanen H, Sormunen R, Makikallio K, Kivirikko KI, Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 156.Hassane S, Claij N, Lantinga-van Leeuwen IS, Van Munsteren JC, Van Lent N, Hanemaaijer R, Breuning MH, Peters DJM, DeRuiter MC. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler Thromb Vasc Biol. 2007;27:2177–2183. doi: 10.1161/ATVBAHA.107.149252. [DOI] [PubMed] [Google Scholar]
- 157.Shen D, Li J, Lepore JJ, et al. Aortic aneurysm generation in mice with targeted deletion of integrin-linked kinase in vascular smooth muscle cells. Circ Res. 2011;109:616–628. doi: 10.1161/CIRCRESAHA.110.239343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hofmann Bowman M, Wilk J, Heydemann A, Kim G, Rehman J, Lodato JA, Raman J, McNally EM. S100A12 mediates aortic wall remodeling and aortic aneurysm. Circ Res. 2010;106:145–154. doi: 10.1161/CIRCRESAHA.109.209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gallo Em, Loch DC, Habashi JP, et al. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathologies. J Clin Invest. 2014;124:448–460. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huang J, Yamashiro Y, Papke CL, Ikeda Y, Lin Y, Patel M, Inagami T, Le VP, Wagenseil JE, Yanagisawa H. Angiotensin-coverting enzyme-induced activation of local angiotensin signaling is required for ascending aortic aneurysms in Fibulin-4-deficient mice. Science Translational Med. 2013;5:1–11. doi: 10.1126/scitranslmed.3005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lindsay ME, Dietz HC. The genetic basis of aortic aneurysm. Cold Spring Harb Perspect Med. 2014;4:a015909. doi: 10.1101/cshperspect.a015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Teekakirikul P, et al. Thoracic aortic disease in two patients with juvenile polyposis syndrome and SMAD4 mutations. Am J Med Genet. 2013;A161:185–191. doi: 10.1002/ajmg.a.35659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen S, Lechleider RJ. Transforming growth factor-beta induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94:1195–1202. doi: 10.1161/01.RES.0000126897.41658.81. [DOI] [PubMed] [Google Scholar]
- 165.Choudhary B, Zhou J, Li P, Thomas S, Kaartinen V, Sucov HM. Absence of TGFβ signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47:115–121. doi: 10.1002/dvg.20466. [DOI] [PubMed] [Google Scholar]
- 166.Sanford LP, et al. TGF-beta2 knockout mice have multiple developmental defects that are non-overlapping with other TGF-beta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Moltzer E, te Riet L, Swagemakers SMA, et al. Impaired vascular contractility and aortic wall degeneration in Fibulin-4 deficient mice: effect of angiotensin II type 1 (AT1) receptor blockade. PLoS ONE. 2011;6:1–12. doi: 10.1371/journal.pone.0023411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jeremy RW, Robertson E, Lu Y, Hambly BD. Perturbations in mechanotransduction and aneurysm formation in heritable aortopathies. Int J Cardiol. 2013;169:7–16. doi: 10.1016/j.ijcard.2013.08.056. [DOI] [PubMed] [Google Scholar]
- 169.Cook JR, Carta L, Galatioto J, Ramirez F. Cardiovascular manifestations in Marfan syndrome and related diseases; multiple genes causing similar phenotypes. Clin Genet. 2015;87:11–20. doi: 10.1111/cge.12436. [DOI] [PubMed] [Google Scholar]
- 170.Chen X, Lu H, Rateri DL, Cassis LA, Daugherty A. Conundrum of angiotensin II and TGF-b interactions in aortic aneurysms. Curr Opin Pharmacol. 2013;13:180–185. doi: 10.1016/j.coph.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rammos C, Hendgen-Cotta UB, Deenen R, Pohl J, Stock P, Hinzmann C, Kelm M, Rassaf T. Age-related vascular gene expression profiling in mice. Mech Ageing Dev. 2014;135:15–23. doi: 10.1016/j.mad.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 172.Jondeau G, Boutouyrie P, Lacolley P, Laloux B, Dubourg O, Bourdarias J-P, Laurent S. Central pulse pressure is a major determinant of ascending aorta dilatation in Marfan syndrome. Circulation. 1999;99:2677–2681. doi: 10.1161/01.cir.99.20.2677. [DOI] [PubMed] [Google Scholar]
- 173.Bellini C, Wang S, Milewicz DM, Humphrey JD. Myh11(R247C/R247C) mutations in thoracic aortic vulnerability to intramural damage despite a general biomechanical adaptivity. J Biomech. 2015;48:113–121. doi: 10.1016/j.jbiomech.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Cook JR, Carta L, Benard L, et al. Abnormal muscle mechanosensing triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 2014;124:1329–1339. doi: 10.1172/JCI71059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ju X, Ijaz T, Sun H, et al. IL-6 regulates extracellular matrix remodeling associated with aortic dilatation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J Am Heart Assoc. 2014;3:e000476. doi: 10.1161/JAHA.113.000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Inamoto S, Kwartler CS, Lafont AL, et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. 2010;88(3):520–529. doi: 10.1093/cvr/cvq230. [DOI] [PMC free article] [PubMed] [Google Scholar]