

Abstract
The purpose of this communication is to elucidate the key role of NADPH oxidase in NLRP3 inflammasome activation and generation of pulmonary fibrosis by multi-walled carbon nanotubes (MWCNTs). Although it is known that oxidative stress plays a role in pulmonary fibrosis by single-walled CNTs, the role of specific sources of reactive oxygen species (ROS), including NADPH oxidase, in inflammasome activation remains to be clarified. In this study, three long aspect ratio (LAR) materials (MWCNTs, SWCNTs, and silver nanowires) are used to compare with spherical carbon black and silver nanoparticles for their ability to trigger oxygen burst activity and NLRP3 assembly. All LAR materials but not spherical nanoparticles induce robust NADPH oxidase activation and respiratory burst activity in THP-1 cells, which are blunted in p22phox deficient cells. NADPH oxidase is directly involved in lysosome damage by LAR materials, as demonstrated by decreased cathepsin B release and IL-1β production in p22phox deficient cells. Reduced respiratory burst activity and inflammasome activation are also observed in bone marrow-derived macrophages from p47phox deficient mice. Moreover, p47phox deficient mice have reduced IL-1β production and lung collagen deposition in response to MWCNTs. Lung fibrosis is also suppressed by N-acetyl-cysteine (NAC) in wild type animals exposed to MWCNTs.
Keywords: NLRP3 inflammasome, long aspect ratio nanomaterials, oxidative stress, lung fibrosis, NADPH oxidase
1. Introduction
Long aspect ratio (LAR) engineered nanomaterials (ENMs), including single- and multi-walled carbon nanotubes (SWCNTs and MWCNTs), have the capacity to induce chronic inflammation and fibrosis in the lung.[1, 2] While a number of CNT physicochemical properties could contribute to the pathogenesis of pulmonary fibrosis, it has been shown that NLRP3 inflammasome activation and IL-1β production in lung macrophages play an important role in disease pathogenesis in the lung.[1] Moreover, it is known that the production of reactive oxygen species (ROS) contributes to NLRP3 inflammasome activation, which is relevant from the perspective that CNTs engage in oxygen radical generation, including through NADPH oxidase activation by SWCNTs.[2] This raises the question whether the surface membrane assembly of the NADPH oxidase complex that are composed of trans-membrane (gp91phox and p22phox), cytosolic (p40phox, p47phox and p67phox) and GTPase (Rac 1/2) subunits[3] plays a direct role in cellular events leading to NLRP3 inflammasome activation by LAR materials.
We have previously shown that NLRP3 inflammasome assembly in response to LAR materials, including MWCNTs and CeO2 nanowires, involves phagocytic uptake and processing by lysosomes in macrophages.[1, 4] Moreover, lysosomal damage and cathepsin B release contributes directly to the assembly of the NLRP3, ASC and pro-caspase 1 subunits of the NLRP3 inflammasome complex.[1, 4, 5] It is also known that phagocytic uptake by macrophages is accompanied by oxygen burst activity as a result of the inclusion of the activated, multi-subunit NADPH oxidase complex in the phagolysosome.[6] We propose that NADPH oxidase-mediated respiratory burst activity could be directly involved in the lysosomal damage that prestages inflammasome assembly and the generation of pulmonary fibrosis. This information is critical for understanding the role of oxidative stress in the pathogenesis of pulmonary fibrosis as well as possible therapeutic intervention in the pathway by which NADPH oxidase leads to inflammasome activation.
To elucidate the possible contribution of NADPH oxidase to NLRP3 inflammasome activation and pulmonary fibrosis, we compared three LAR materials (MWCNTs, SWCNTs, and silver nanowires) with spherical nanoparticles (carbon black and silver) for their ability to induce IL-1β production. This was accomplished by studying NADPH oxidase activation, respiratory burst activity and NLRP3 inflammasome assembly in wild type and p22phox deficient THP-1 cells as well as wild type and p47phox deficient primary bone marrow-derived macrophages (BMDMs). Based on cellular studies demonstrating a role for NADPH oxidase, we also performed animal studies in which oropharyngeal aspiration of MWCNTs was used to compare lysosomal damage, IL-1β production and induction of pulmonary fibrosis in wild type and p47phox deficient animals. Our data demonstrate that there is direct involvement of NADPH oxidase and oxidative stress with activation of the NLRP3 inflammasome.
2. Results
2.1. Physicochemical Characterizations of ENMs
MWCNTs, SWCNTs, and silver (Ag) wires were selected for comparison to Ag and carbon black spherical nanoparticles. TiO2 nanoparticles were included as a negative control for generation of IL-1β-mediated inflammation based on our previous studies.[7, 8] Physicochemical characterization was performed to assess nanomaterial size, aspect ratio, and zeta potential in different media. The curve-shaped MWCNTs (containing 4.49% Ni and 0.76% Fe) exhibited average diameters of 20–30 nm and lengths of 10–30 μm, with aspect ratios varying from 333 to 1500. The SWCNTs (containing 31.35% Fe) exhibited average diameters of 0.8–1.2 nm and lengths of 1–5 μm, with aspect ratios varying from 1250 to 4167. The Ag wires were long and straight, with an average diameter of 65 nm and a length of 20 μm (aspect ratio=308) (Figure 1 and Table 1). In contrast, Ag, carbon black and TiO2 nanoparticles were spherical, with primary sizes of 7–21, 90 and 25nm, respectively (Figure 1 and Table 1). In addition, we determined the hydrodynamic sizes of these materials in different media (Table 1). Although determination of LAR hydrodynamic size by DLS is only a comparative evaluation that is not accurate for size, we have demonstrated the utility of this approach to compare the agglomeration states of LAR materials.[1, 4, 9] MWCNTs and SWCNTs were better dispersed in complete RPMI-1640 medium than water, which can be explained by the presence of serum proteins (including albumin) that forms a corona that stabilizes the tubes in suspension (Table 1).[10] However, they have a tendency to form large agglomerates in PBS supplemented with BSA and DPPC. Ag wires were better dispersed in water and PBS than RPMI. Carbon black was relatively well dispersed in water. Measurement of the zeta potential demonstrated a negative charge for LAR materials and Ag nanospheres under all media conditions (Table 2). In contrast, carbon black and TiO2 had a positive charge in water, which reverts to negative in RPMI-1640 and PBS (containing BSA and DPPC), while carbon black maintained a positive charge in PBS (Table 2).
Figure 1.

TEM analysis of ENMs. Representative TEM images of ENMs were taken with a JEOL 1200 EX TEM, using an accelerating voltage of 80 kV.
Table 1.
Primary sizes, aspect ratios and hydrodynamic sizes of ENMs in water and exposure media.
| Hydrodynamic Size (nm) |
|||||
|---|---|---|---|---|---|
| ENMs | Primary Size (nm) | Aspect Ratio | Water | RPMI-1640&10% Serum | PBS&0.6 mg/mL BSA&0.01 mg/mL DPPC |
| MWCNTs | 20–30 × 10000–30000 | 333–1500 | 527±45 | 353±35 | 1807±198 |
| SWCNTs | 0.8–1.2 × 1000–5000 | 1250–4167 | 1218±507 | 634±252 | 2447±632 |
| PVP-Ag wire | 65 × 20000 | 308 | 96±6 | 1343±63 | 469±98 |
| PVP-Ag sphere | 7–21 | 1 | 548±84 | 152±52 | 979±57 |
| Carbon black | 90 | 1 | 322±5 | 382±25 | 2437±320 |
| TiO2 (P25) | 25 | 1 | 2077±343 | 312±34 | 750±176 |
Table 2.
Zeta potentials of ENMs in water and exposure media.
| Zeta Potential (mV) |
|||
|---|---|---|---|
| ENMs | Water | RPMI-1640&10% Serum | PBS&0.6 mg/mL BSA&0.01 mg/mL DPPC |
| MWCNTs | −14.7±0.1 | −19.3±2.3 | −9.4±0.6 |
| SWCNTs | −22.3±0.7 | −10.1±2.5 | −16.3±6.2 |
| PVP-Ag wire | −27.3±1.0 | −5.8±2.9 | −7.9±0.8 |
| PVP-Ag sphere | −34.0±1.9 | −4.5±0.8 | −9.7±0.5 |
| Carbon black | 44±3 | −4±7 | 10±5 |
| TiO2 (P25) | 40±7 | −19±1 | −9±1 |
2.2. NLRP3 Inflammasome Activation by LAR ENMs is Dependent on Lysosomal Damage and Oxidative Stress in THP-1 Cells
We used the monocyte-derived THP-1 cell line as an initial tool for assessing NLRP3 inflammasome activation as previously reported.[1, 4, 11] Significant IL-1β production by MWCNTs, SWCNTs, Ag nanowires and monosodium urate (MSU) crystals (positive control for inflammasome activation)[12] was markedly suppressed in NLRP3-deficient (defNLRP3) and ASC-deficient (defASC) THP-1 cells (Figure 2A), confirming the involvement of NLRP3 inflammasome activation. In contrast, spherical Ag, carbon black and TiO2 failed to induce IL-1β production (Figure 2A). Noteworthy, these pro-inflammatory responses was not accompanied by cytotoxicity (Figure S1), or the result of endotoxin contamination (Figure S2). Since we know that cathepsin B release from lysosomes and oxidative stress are involved in NLRP3 inflammasome activation,[1, 4, 5, 11] the cathepsin B inhibitor, CA-074-Me, and N-acetylcysteine (NAC, a thiol antioxidant) were used to show that it is possible to suppress IL-1β production by the LAR materials (Figures 2B and 2C). The data in Figure 2D will be discussed later.
Figure 2.

Long aspect ratio ENMs induce the NLRP3 inflammasome activation and IL-1β production that is dependent on oxidative stress. (A) wt THP-1, NLRP3-deficient (defNLRP3) and ASC-deficient (defASC) THP-1 cells were exposed to ENMs at 50 μg/mL for 6 h. (B–D) wt THP-1 cells were pre-treated with (B) 20 μM of CA-074-Me for 30 min and (C) 25 mM of NAC for 30 min and (D) 20 μM of DPI for 1 h, and then exposed to ENMs (50 μg/mL) for additional 6 h. MSU crystals (100 μg/mL) were used as positive controls. The IL-1β production was quantified by ELISA. *P<0.05 compared to wild type THP-1 cells of same ENM treatment; #P<0.05 compared to control cells without ENM treatment.
2.3. Phagocytosis of LAR ENMs Triggers NADPH Oxidase-Dependent Oxygen Burst Activity that Induces NLRP3 Inflammasome Activation
We compared the generation of oxidative burst by ENMs after phagocytosis in THP-1 cells (Figure 3A). We found that after cellular uptake into phagolysosomes (Figure S3), the LAR materials were effective inducers of oxidative burst activity in THP-1 cells, as determined by the rapid increase in the fluorescence intensity of DCF in a time-dependent fashion (Figure 3B). In contrast, no increase in oxidative burst activity was seen in p22phox−/− THP-1 cells (Figures 3C and S4A–B). The involvement of NADPH oxidase was further confirmed in primary bone marrow-derived macrophages (BMDMs) from wild type (wt) and p47phox deficient C57BL/6 mice (Figure S5A), where the oxidative burst was significantly blunted in p47phox−/− BMDMs compared to wt BMDMs (Figures 3D and S5B). In distinction, spherical particles (Ag, carbon black and TiO2) did not induce oxidative burst activity in either THP-1 cells or BMDMs (Figures 3B, 3C, S4A and S5B) in spite of phagolysosomal localization (Figure S3).
Figure 3.


Long aspect ratio ENMs induce NADPH oxidase activation and oxidative burst. (A) A scheme showing the components and activation of NADPH oxidase enzyme complex. NADPH oxidase is composed of two trans-membrane subunits (gp91phox and p22phox), three cytosolic components (p40phox, p47phox and p67phox) and a GTPase (Rac). When activated, the phosphorylated p47phox will bind to p22phox, and the GTP-bound Rac will bind to p67phox, which serves as a switch for NADPH oxidase activation and ROS generation. (B–D) Cells were exposed to ENMs (50 μg/mL) in the presence of 18 μg/mL of Fc OxyBURST. The kinetics of oxidative burst induced by ENMs in (B) wt THP-1 and (C) p22phox−/− THP-1 cells was quantified by monitoring OxyBURST Green fluorescence intensity using microplate reader. *P<0.05 compared to control cells without ENM treatment. (D) The kinetics of oxidative burst in wild type (wt) BMDMs and p47phox−/− BMDMs induced by MWCNTs was quantified by monitoring OxyBURST Green fluorescence intensity using microplate reader. *P<0.05 compared to p47phox−/− BMDMs; #P<0.05 compared to control cells without MWCNT treatment.
We also determined the role of NADPH oxidase in NLRP3 inflammasome activation by ENMs. Compared to wild type cells, IL-1β production by LAR ENMs was significantly attenuated in p22phox−/− THP-1 cells (Figure 4A) as well as p47phox−/− BMDMs (Figure 4B). Furthermore, DPI, a small molecule inhibitor of NADPH oxidase, showed the same effect as knockout of the p22phox and p47phox subunits on reducing IL-1β production (Figure 2D). Collectively, these results demonstrated the important role of NADPH oxidase in LAR ENM-induced NLRP3 inflammasome activation.
Figure 4.
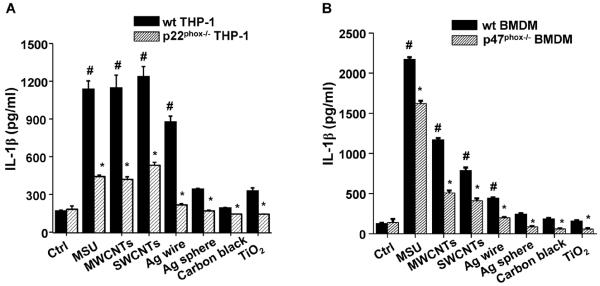
Long aspect ratio ENM-induced NLRP3 inflammasome activation is dependent on NADPH oxidase activation. (A) wt THP-1 and p22phox−/− THP-1 cells were exposed to ENMs (50 μg/mL) for 6 h. (B) Bone marrow-derived macrophages (BMDMs) obtained from wt C57BL/6 and p47phox−/− C57BL/6 mice were exposed to ENMs (50 μg/mL) for 24 h. IL-1β production was quantified by ELISA. *P<0.05 compared to wild type cells of same ENM treatment; #P<0.05 compared to control cells without ENM treatment.
2.4. Oxidative Burst Triggers LAR Material-Induced Lysosomal Damage and Mitochondrial ROS Generation that Lead to NLRP3 Inflammasome Activation
In order to determine if NADPH oxidase plays a role in lysosomal damage, an important pathway involved in ENM-induced NLRP3 inflammasome activation, we studied cathepsin B release in p22phox deficient THP-1 cells. While LAR ENMs induced cathepsin B release in wild type THP-1 cells,[5] as demonstrated by confocal microscopy, p22phox deficiency reduced this damage to the lysosome (Figure 5). In contrast, the enzyme was confined to lysosomes in cells treated with spherical nanoparticles (Figure S6). Since lysosomal damage could lead to mitochondrial ROS generation that plays a role in NLRP3 inflammasome activation,[6] we assessed mitochondrial ROS generation through the use of MitoSOX. This demonstrated that LAR ENMs induced significantly higher levels of mitochondrial superoxide than spherical particles (Figure 6A and S7), while p22phox deficiency resulted in reduced mitochondrial ROS generation by the same materials (Figure 6A–B). MitoTEMPO, a mitochondrial ROS scavenger, reduced superoxide production by LAR materials (Figure S7). MitoTEMPO also reduced IL-1β production by LAR materials (Figure 6C). In addition, NAC interfered in cathepsin B release by LAR materials (Figure 5). In summary, we showed that NADPH oxidase-induced oxidative burst plays an important role in lysosomal damage induced by LAR materials, which lead to subsequent mitochondrial ROS generation that enhances NLRP3 inflammasome activation and IL-1β production.
Figure 5.

ENM-induced oxidative burst promotes lysosomal damage. Lysosomal damage and cathepsin B release was identified by Magic Red cathepsin B substrate staining. wt THP-1 cells, p22phox−/− THP-1 cells, and NAC (25 mM, 30 min) pretreated wt THP-1 cells were seeded into 8-well chamber slides and exposed to ENMs (50 μg/mL) for 5 h. Cells were stained with Magic Red reagent for 1 h and visualized under a confocal microscope. The scale bar is 20 μm.
Figure 6.

Long aspect ratio ENMs induce mitochondrial ROS generation. (A) Mitochondrial ROS generation was identified by MitoSOX. After ENM exposure (50 μg/mL) for 3 h, wt THP-1 and p22phox−/− THP-1 cells were stained with 5 μM of MitoSOX for 20 min. The scale bar is 20 μm. Mitochondrial ROS scavenger, MitoTEMPO at 500 μM, decreased mitochondrial ROS generation. (B) Fluorescence intensity was determined by ImageJ software (NIH). *P<0.05 compared to THP-1 cells of same ENM treatment; #P<0.05 compared to control cells without ENM treatment. (C) MitoTEMPO reduced IL-1β production in wt THP-1 cells. Differentiated THP-1 cells were exposed to ENMs (50 μg/mL) in the presence of 500 μM of MitoTEMPO for 6 h. The IL-1β productions were quantified by ELISA. *P<0.05 compared to THP-1 cells of same ENM treatment; #P<0.05 compared to control cells without ENM treatment.
2.5. MWCNT-Induced NLRP3 Inflammasome Activation and Lung Fibrosis are Attenuated in p47phox Deficient Mice Compared to wt Mice
To confirm the role of NADPH oxidase in NLRP3 inflammasome activation in vivo, a proof-of-principle analysis was performed by comparing wt to p47phox deficient mice in response to MWCNT oropharyngeal aspiration for 40 h. After sacrifice of these animals, we observed significant lower IL-1β production in the BAL fluid of p47phox−/− mice compared to wt animals (Figure 7A–B). Following that, we tested the effects of MWCNTs 21 days after oropharyngeal exposure. This demonstrated significant collagen deposition in the lung of wt animals, while p47phox−/− mice failed to show an increase using the Sircol's collagen assay (Figure 7C). The collagen results were confirmed by staining the lung sections with Masson's Trichrome reagents (Figure 7D). In contrast to MWCNTs, TiO2 did not induce either cytokine production or lung fibrosis (Figure S8A–D) in wt mice. These results suggest that NADPH oxidase plays an important role in the MWCNT-induced NLRP3 inflammasome activation and lung fibrosis in vivo.
Figure 7.

Comparison of MWCNT-induced IL-1β production and lung fibrosis in wild type and p47phox−/− mice. wt C57BL/6 (n=6) and p47phox−/− C57BL/6 mice (n=6) were exposed to 2 mg/kg of MWCNTs by oropharyngeal aspiration. (A) IL-1β level in BAL fluid at 40 h. *P<0.05. (B) Fold increases in IL-1β production in MWCNT-treated wt C57BL/6 and p47phox−/− C57BL/6 mice. *P<0.05. To examine the lung fibrosis induced by MWCNTs, wt C57BL/6 (n=6) and p47phox−/− C57BL/6 (n=6) mice were exposed to 2 mg/kg of MWCNTs by oropharyngeal aspiration for 21 days. (C) Total collagen content in murine lung as determined by Sircol's collagen assay. *P<0.05. (D) Masson's Trichrome staining was used to determine the collagen deposition in the lungs of animals treated with MWCNTs. The collagen was stained blue in the micrographs.
2.6. NAC Attenuates MWCNT-Induced Inflammasome Activation and Pulmonary Fibrosis
In order to demonstrate the overall importance of oxidative stress in MWCNT-induced NLRP3 inflammasome activation, wt mice received intraperitoneal (i.p.) NAC administration, which led to a significant reduction in IL-1β production in the BAL fluid compared to animals receiving MWCNT inhalation in the absence of NAC treatment (Figure 8A). This compares well with interference in cathepsin B release in THP-1 cells (Figure 5), as was also confirmed by using Magic Red staining of pulmonary alveolar macrophages obtained from BAL fluid (Figure 8B). We also examined the effect of NAC on the animal lung 21 days after MWCNT exposure. NAC significantly suppressed TGF-β1 production (Figure 9A), which plays a major role in the pathogenesis of pulmonary fibrosis, as well as neutrophil infiltration in the lung (Figure S9A–B). NAC also significantly suppressed collagen deposition, as determined by Sircol's assay (Figure 9B) or Masson's Trichrome staining (Figure 9C). Taken together, these data demonstrate that oxidative stress plays a major role in MWCNT-induced NLRP3 inflammasome activation and lung fibrosis in vivo, and that NAC can reverse the effects of MWCNT-induced inflammasome activation and IL-1β production.
Figure 8.
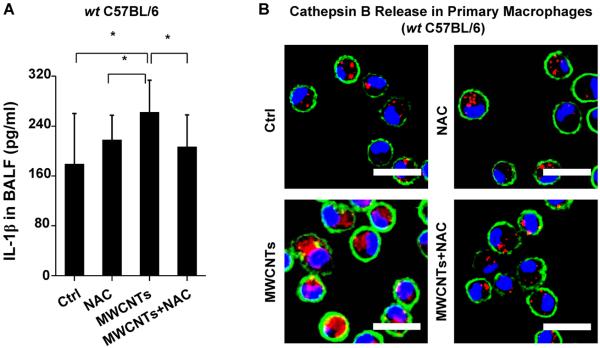
NAC attenuates MWCNT-induced IL-1β production and cathepsin B release in primary macrophages. wt C57BL/6 mice (n=6) were exposed to 2 mg/kg of MWCNTs by oropharyngeal aspiration and NAC administration (i.p.) at 320 mg/kg. (A) IL-1β level in BAL fluid at 40 h. *P<0.05. (B) Cathepsin B release in alveolar macrophages. Alveolar macrophages were collected from the BAL fluid 16 h after MWCNT exposure. The macrophages were stained with Magic Red reagent for 1h. The scale bar is 20 μm.
Figure 9.

NAC attenuates MWCNT-induced lung fibrosis in mice. wt C57BL/6 mice (n=6) were exposed to 2 mg/kg of MWCNTs by oropharyngeal aspiration. NAC administration (i.p.) was performed at 320 mg/kg on every other day for a total of 11 injections in 3 weeks. (A) TGF-β1 in BAL fluid at 21 days. *P<0.05. (B) Total collagen content in murine lung was determined by Sircol's collagen assay. *P<0.05. (C) Masson's Trichrome staining was used to determine the collagen deposition in the lungs of animals treated with MWCNTs and the effect of NAC administration (i.p.). The collagen was stained blue in the micrographs.
3. Discussion
In this paper, we demonstrate that NADPH oxidase-induced oxidative stress is directly involved in NLRP3 inflammasome activation and generation of lung fibrosis by MWCNTs. LAR ENMs exert their effects through NADPH oxidase activation and oxidative burst, which lead to lysosomal damage, mitochondrial ROS generation, oxidative stress and subsequent NLRP3 inflammasome activation. These responses were blunted in NADPH oxidase subunit p22phox deficient THP-1 cells and p47phox deficient BMDMs. Furthermore, we confirmed the important role of NADPH oxidase in NLRP3 inflammasome activation and lung fibrosis in vivo using p47phox deficient mice, which did not develop MWCNT-induced lung fibrosis. Additionally, the role of oxidative stress in MWCNT-induced lung fibrosis was demonstrated through the use of the thiol antioxidant NAC, which interfered in lysosomal damage, NLRP3 inflammasome activation and generation of pulmonary fibrosis in wild type animals.
The key finding in this study is that NADPH oxidase plays a major role in MWCNT-induced NLRP3 inflammasome activation and lung fibrosis. Previously, this biological effect has only been demonstrated for one other LAR, namely asbestos.[13] That effect was linked to the ability of asbestos to induce frustrated phagocytosis in macrophages.[13] We previously observed similar effects for silver nanowires, which were capable of piercing through the cell membrane, with the ability to generate ROS and oxidative stress.[14] However, frustrated phagocytosis was not seen with the CNTs used in this study; these materials did induce lysosomal damage[1] (Figure 5) instead and could be seen in the phagolysosomal compartment by TEM (Figure S3). Lysosomal damage is likely due to the surface catalytic activity of the tubes, which damages the lysosomal membrane. Coating of the tube surface with Pluronic F108, which forms a brush-like layer, prevents lysosomal damage, NLRP3 inflammasome activation, and lung fibrosis.[1]
What is the role of NADPH oxidase in NLRP3 inflammasome activation? Our data suggest that the ability of this enzyme to generate oxidative burst activity plays a role in the lysosomal damage.[15, 16] Not only is it known that ROS can damage the lysosomal membrane,[17, 18] but we also demonstrate that NADPH oxidase deficient cells showed decreased lysosomal damage, cathepsin B release, and IL-1β production by LAR ENMs (Figures 4, 5 and S6). In addition to the release of cathepsin B, lysosomal damage could also lead to mitochondrial perturbation, including permeabilization of the mitochondrial outer membrane and ROS generation.[19–23] Mitochondrial ROS generation is also NADPH oxidase dependent as demonstrated by decreased mitochondrial ROS production in p22phox deficient cells (Figure 6A–B). It is worth noting that mitochondrial ROS production can enhance lysosomal damage through a possible feedback loop.[24] Collectively, lysosomal damage and cathepsin B release (Figures 5 and 8) and mitochondrial ROS (Figure 6), could all contribute to the NLRP3 inflammasome activation.[6, 25]
While it is known that oxidative stress plays a role in NLRP3 inflammasome activation in vitro,[5, 6, 13, 26] in vivo evidence linking oxidative stress to inflammasome activation and lung fibrosis is sparse. One study showed that antioxidant (Vitamin E) deficiency in diet enhances pulmonary inflammatory responses and fibrosis in mouse exposed to SWCNTs.[27] While NAC has been shown to inhibit bleomycin-induced lung fibrosis,[28–30] no previous studies have been undertaken to shown an impact on ENM-induced lung fibrosis. Thus, our data showing the effect of NAC administration (i.p.) on the induction of lung fibrosis by MWCNTs (Figure 9) is novel. Moreover, the NAC data also suggest that it may be possible to consider antioxidant therapy to decrease or overcome MWCNT-induced pulmonary fibrosis.
4. Conclusion
We conclude that NADPH oxidase is critically important for inflammasome activation and pulmonary fibrosis as shown both in vitro and in vivo using NADPH oxidase deficient models. Mitochondrial ROS are also likely to play an important role perhaps through an interplay between the oxidative burst and mitochondrial ROS production. This finding is further bolstered by demonstration of the failure of lysosomal damage, mitochondrial ROS generation and NLRP3 inflammasome activation in p22phox deficient THP-1 cells and p47phox deficient BMDMs, as well as reduced lung fibrosis in p47phox deficient mice. Further, we show that NAC could attenuate inflammasome activation and lung fibrosis by MWCNTs, and the effect of NAC support a role for ROS.
5. Experimental Section
Reagents and Materials
MSU crystals were purchased from InvivoGen (San Diego, CA); CA-074-Me and MitoTEMPO were obtained from Sigma (St. Louis, MO); N-acetyl-L-cysteine (NAC) was from American Regent, INC. (Shirley, NY). The ELISA kit for human IL-1β was from R&D Systems (Minneapolis, MN). The ELISA kit for mouse IL-1β was from BD Biosciences (San Diego, CA). The ELISA kit for mouse TGF-β1 was from Promega (Madison, WI). Ag wires and Ag nanospheres were from nanoComposix (San Diego, CA); single-walled carbon nanotubes (SWCNTs) were from NanoIntegris (Skokie, IL) (containing 31.35% Fe); and multi-walled carbon nanotubes (MWCNTs) were from Cheap Tubes Inc. (Brattleboro, VT) (containing 4.49% Ni and 0.76% Fe).
Physicochemical Characterizations of ENMs
The ENMs were used as received. The primary average lengths and diameters of the ENMs were assessed by TEM (JEOL 1200 EX transmission electron microscope). The hydrodynamic diameters of ENMs were determined using high-throughput dynamic light scattering (HT-DLS, Dynapro Plate Reader) (Wyatt Technology, Santa Barbara, CA). The zeta (ζ) potentials of ENMs were determined using a ZetaSizer Nano-ZS (Malvern Instruments, Worcestershire WR, UK).
Cell Culture
Human THP-1 cells were grown in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 100 U/mL-100 μg/mL Penicillin-Streptomycin and 50 μM of beta-mercaptoethanol. NLRP3-deficient (defNLRP3) and ASC-deficient (defASC) THP-1 cells were from InvivoGen (San Diego, CA) and grown in RPMI-1640 media supplemented with 10% FBS and antibiotics. p22phox−/− THP-1 cells were grown in RPMI-1640 media supplemented with 10% FBS, 100 U/mL-100 μg/mL Penicillin-Streptomycin, 500 ng/mL of Puromycin and 50 μM of beta-mercaptoethanol.
Determination of NLRP3 Inflammasome Activation and IL-1β Production
THP-1 cells in 100 μL of tissue culture medium were plated at the density of 3×104 cells per well in a 96-well plate, and cells were treated with Phorbol 12-myristate 13-acetate (PMA) (1 μg/mL) for 16 h. Then the medium was replaced with fresh medium and the differentiated THP-1 cells were treated with ENMs (50 μg/mL) in the presence of LPS (10 ng/mL) for additional 6 h. The supernatant of ENM-exposed cells was collected for IL-1β assessment.[1] To test the effects of inhibitors, THP-1 cells were pre-treated with N-acetyl-L-cystein (NAC) (25 mM) or CA-074-Me (20 μM) for 30 min before the exposure of ENMs. For primary BMDMs, cells in 100 μL of tissue culture media were plated at the density of 5×104 per well in a 96-well plate with the addition of LPS (500 ng/mL) for 5 h. The medium was replaced with fresh media and the primed cells treated with ENMs (50 μg/mL) in the presence of LPS (10 ng/mL) for 24 h. The supernatant of the stimulated cells was collected for IL-1β assessment.
Determination of Oxidative Burst
Fc OxyBURST Green assay reagent was used to determine the oxidative burst induced by ENMs in phagosomes. Wild type THP-1 or p22phox−/− THP-1 cells were co-incubated with ENMs for 1 h with the addition of LPS (10 ng/mL) and Fc OxyBURST (18 μg/mL). Kinetics of oxidative burst was monitored using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA). For microscopic analysis, cells were co-incubated with ENMs for 30 minutes with the addition of LPS (10 ng/mL) and Fc OxyBURST (18 μg/mL), then cells were washed twice with PBS and further fixed with 4% of paraformaldehyde (PFA) for 15 min. Following two washes, cells were stained with 10 μM of Hoechst 33342 (Invitrogen, Carlsbad, CA) at room temperature for another 20 min. Finally, cells were washed with PBS twice and examined under fluorescence microscopy. To determine the oxidative burst in primary cells, after wt BMDMs or p47phox−/− BMDMs were harvested and cultured for 7 days in 25% LADMAC conditioned media, cells were plated in a 96-well plate at 5×104 cells/100 μL medium for 4 hours. Then the cells were co-incubated with ENMs for 1 h with the addition of LPS (10 ng/mL) and Fc OxyBURST (18 μg/mL). Kinetics of oxidative burst induced by ENMs in BMDMs or p47phox−/− BMDMs was monitored using a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
Determination of Mitochondrial ROS Generation
Differentiated THP-1 cells were exposed to ENMs at 37 °C and 5% CO2. After 3 h exposure to ENMs, cells were washed twice with PBS and treated with 5 μM of MitoSOX (Invitrogen, Carlsbad, CA) in HBSS at 37 °C and 5% CO2 for 20 min. Then the cells were washed twice with PBS and further fixed with 4% of PFA for 15 min. Following two washes, cells were stained with 10 μM of Hoechst 33342 (Invitrogen, Carlsbad, CA) at room temperature for another 20 min. Finally, cells were washed with PBS for two more times and examined using a Leica confocal SP2 1P-FCS microscope (Advanced Light Microscopy/Spectroscopy Shared Facility, UCLA). High-magnification images were obtained with a 63× objective (Leica, N.A.=1.4). Optical sections were averaged 4 times to reduce background noise. Images were processed using Leica Confocal Software.
Effect of NADPH Oxidase and Oxidative Stress on MWCNT-induced Lung Inflammation and Fibrosis
Eight week old male C57BL/6 mice were purchased from Charles River Laboratories (Hollister, CA). NADPH oxidase subunit (p47phox) deficient C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, Maine). The genotypes of mice used in the experiments were verified by sending ear-clips of mice to Transnetyx Inc. (Cordova, TN). All animals were housed under standard laboratory conditions that have been set up according to UCLA guidelines for care and treatment of laboratory animals as well as NIH guide for the care and use of laboratory animals in research (DHEW78-23). These conditions are approved by the Chancellor's Animal Research Committee at UCLA and include standard operating procedures for animal housing (filter-topped cages; room temperature at 23 ± 2 °C; 60% relative humidity; 12 h light, 12 h dark cycle) and hygiene status (autoclaved food and acidified water). Animal exposure to MWCNTs was carried out by an oropharyngeal aspiration method as described at NIOSH[31]. Briefly, animals were anesthetized by intraperitoneal (i.p.) injection of ketamine (100 mg/kg)/xylazine (10 mg/kg) in a total volume of 100 μL. With the anesthetized animals held in a vertical position, 50 μL of PBS suspension containing 50 μg of MWCNTs was instilled at the back of the tongue to allow pulmonary aspiration. To examine the effect of NAC, NAC was administered i.p. at the dose of 320 mg/kg on every other day. The mice were sacrificed 40 h or 21 days later. Bronchoalveolar lavage (BAL) fluid and lung tissues were collected as previously described.[32] Briefly, the trachea was cannulated, and the lungs were gently lavaged three times with 1 mL of sterile PBS to obtain BAL fluid. The BAL fluid was used for total and differential cell counts and for IL-1β, TGF-β1, Lix and PDGF-AA measurements. Lung tissues were collected and stained with hematoxylin/eosin or with Masson's Trichrome stainings as described before.[33] IL-1β was measured by the mouse IL-1β ELISA kit (BD Biosciences, San Diego, CA). TGF-β1 was measured by the TGF-β1 Emax ImmunoAssay System (Promega Corporation, Madison, WI).
Statistical Analysis
All the experiments were repeated for two to three times. For all the figures and tables, the values shown represent mean ± SD. Statistical significance was determined by two-tailed Student's t-test for two-group analysis.
Supplementary Material
Acknowledgements
This work was primarily supported by the US Public Health Service Grant R01 ES022698, the US Public Health Service Grant U19 ES019528 (UCLA Center for NanoBiology and Predictive Toxicology) and leveraged support from the National Science Foundation and the Environmental Protection Agency under Cooperative Agreement Number DBI 0830117 and 1266377. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation or the Environmental Protection Agency. This work has not been subjected to EPA review and no official endorsement should be inferred. The authors would thank the CNSI Advanced Light Microscopy/Spectroscopy Shared Facility at UCLA for confocal fluorescent microscopy, the use of TEM instruments at the Electron Imaging Center for NanoMachines supported by NIH (1S10RR23057 to Z.H.Z.) and CNSI at UCLA. We also thank Prof. Yang Yang and Tze-Bin Song at UCLA for providing and helping with Confocal Raman Microscope.
Footnotes
Supporting Information Supporting Information includes nice figures and Supplemental Experiment Section. Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Wang X, Xia T, Duch M, Ji Z, Zhang H, Li R, Sun B, Lin S, Meng H, Liao Y-P, Wang M, Song T-B, Yang Y, Hersam M, Nel A. Nano Lett. 2012;12:3050. doi: 10.1021/nl300895y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shvedova AA, Kisin ER, Murray AR, Kommineni C, Castranova V, Fadeel B, Kagan VE. Toxicol. Appl. Pharmacol. 2008;231:235. doi: 10.1016/j.taap.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 3.Sumimoto H. FEBS J. 2008;275:3249. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 4.Ji Z, Wang X, Zhang H, Lin S, Meng H, Sun B, George S, Xia T, Nel A, Zink J. ACS Nano. 2012;6:5366. doi: 10.1021/nn3012114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun B, Wang X, Ji Z, Li R, Xia T. Small. 2013;9:1595. doi: 10.1002/smll.201201962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou R, Yazdi AS, Menu P, Tschopp J. Nature. 2011;469:221. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 7.Xia T, Kovochich M, Liong M, Maedler L, Gilbert B, Shi H, Yeh JI, Zink JI, Nel AE. Acs Nano. 2008;2:2121. doi: 10.1021/nn800511k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Ji Z, Chang CH, Zhang H, Wang M, Liao Y-P, Lin S, Meng H, Li R, Sun B, Winkle LV, Pinkerton KE, Zink JI, Xia T, Nel AE. Small. 2014;10:385. doi: 10.1002/smll.201301597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Xia T, Ntim SA, Ji Z, George S, Meng H, Zhang H, Castranova V, Mitra S, Nel AE. ACS Nano. 2010;4:7241. doi: 10.1021/nn102112b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nel AE, Maedler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Nat. Mater. 2009;8:543. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 11.Sun B, Ji Z, Liao Y-P, Wang M, Wang X, Dong J, Chang CH, Li R, Zhang H, Nel AE, Xia T. ACS Nano. 2013;7:10834. doi: 10.1021/nn404211j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Nature. 2006;440:237. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 13.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Science. 2008;320:674. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George S, Lin S, Jo Z, Thomas CR, Li L, Mecklenburg M, Meng H, Wang X, Zhang H, Xia T, Hohman JN, Lin S, Zink JI, Weiss PS, Nel AE. Acs Nano. 2012;6:3745. doi: 10.1021/nn204671v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warren J, Ward P, Johnson K. The Respiratory Burst and Mechanisms of Oxygen Radical-Mediated Tissue Injury. In: Sbarra, Strauss, editor. The Respiratory Burst and Its Physiological Significance. Springer US; 1988. p. 299. [Google Scholar]
- 16.Kalra J, Mantha S, Kumar P, Prasad K. Mol. Cell. Biochem. 1994;136:125. doi: 10.1007/BF00926072. [DOI] [PubMed] [Google Scholar]
- 17.Link G, Pinson A, Hershko C. J. Lab. Clin. Med. 1993;121:127. [PubMed] [Google Scholar]
- 18.Mak IT, Weglicki WB. J. Clin. Invest. 1985;75:58. doi: 10.1172/JCI111697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoka V, Turk B, Schendel SL, Kim TH, Cirman T, Snipas SJ, Ellerby LM, Bredesen D, Freeze H, Abrahamson M, Bromme D, Krajewski S, Reed JC, Yin XM, Turk V, Salvesen GS. J. Biol. Chem. 2001;276:3149. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 20.Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. J. Biol. Chem. 2003;278:31401. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 21.Kushnareva Y, Murphy AN, Andreyev A. Biochem. J. 2002;368:545. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai JY, Jones DP. J. Biol. Chem. 1998;273:11401. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 23.Enoksson M, Robertson JD, Gogvadze V, Bu PL, Kropotov A, Zhivotovsky B, Orrenius S. J. Biol. Chem. 2004;279:49575. doi: 10.1074/jbc.C400374200. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed KA, Sawa T, Ihara H, Kasamatsu S, Yoshitake J, Rahaman MM, Okamoto T, Fujii S, Akaike T. Biochem. J. 2012;441:719. doi: 10.1042/BJ20111130. [DOI] [PubMed] [Google Scholar]
- 25.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Nat. Immunol. 2008;9:847. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Nat. Immunol. 2010;11:136. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 27.Shvedova AA, Kisin ER, Murray AR, Gorelik O, Arepalli S, Castranova V, Young SH, Gao F, Tyurina YY, Oury TD, Kagan VE. Toxicol. Appl. Pharmacol. 2007;221:339. doi: 10.1016/j.taap.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagiwara S, Ishii Y, Kitamura S. Am. J. Respir. Crit. Care Med. 2000;162:225. doi: 10.1164/ajrccm.162.1.9903129. [DOI] [PubMed] [Google Scholar]
- 29.Bonnelye E, Chabadel A, Saltel F, Jurdic P. Bone. 2008;42:129. doi: 10.1016/j.bone.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 30.Booth DK, Floyd SS, Day CS, Glorioso JC, Kovesdi I, Huard J. Tissue Eng. 1997;3:125. [Google Scholar]
- 31.Shvedova AA, Kisin E, Murray AR, Johnson VJ, Gorelik O, Arepalli S, Hubbs AF, Mercer RR, Keohavong P, Sussman N, Jin J, Yin J, Stone S, Chen BT, Deye G, Maynard A, Castranova V, Baron PA, Kagan VE. Am. J. Physiol-Lung C. 2008;295:L552. doi: 10.1152/ajplung.90287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hao M, Comier S, Wang M, Lee JJ, Nel A. J. Allergy Clin. Immunol. 2003;112:905. doi: 10.1016/j.jaci.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Xia T, Addo Ntim S, Ji Z, Lin S, Meng H, Chung C-H, George S, Zhang H, Wang M, Li N, Yang Y, Castranova V, Mitra S, Bonner JC, Nel AE. ACS Nano. 2011;5:9772. doi: 10.1021/nn2033055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.