

Abstract
Fifty years ago (in 1964) the psychoactive ingredient of Cannabis sativa, Δ9-tetrahydrocannabinol (THC), was isolated. Nearly 30 years later the endogenous counterparts of THC, collectively termed endocannabinoids (eCBs), were discovered: N-arachidonoylethanolamine (anandamide, AEA) in 1992, and 2-arachidonoylglycerol (2-AG) in 1995. Since then, considerable research has shed light on the impact of eCBs on human health and disease, identifying an ensemble of proteins that bind, synthesize and degrade them, and that altogether form the eCB system. eCBs control basic biological processes, including cell-choice between survival and death, and progenitor/stem cell proliferation and differentiation. Not surprisingly, in the past two decades, eCBs have been recognized as key mediators of several aspects of human pathophysiology, and thus have emerged among the most widespread and versatile signaling molecules ever discovered.
Here, some of the pioneers of this research field review the state-of-the-art of critical eCB functions in peripheral organs. Our community effort is aimed at establishing consensus views on the relevance of the peripheral eCB system for human health and disease pathogenesis, as well as to highlight emerging challenges and therapeutic hopes.
Keywords: bones, cardiovascular system, gastrointestinal tract, immune system, liver, localization, muscles, (female and male) reproductive system, signaling pathways, skin
Historical introduction
Fifty years ago the major psychoactive cannabis (Cannabis sativa) constituent, Δ9-tetrahydrocannabinol (THC; Table 1) was discovered [1]. At first it was believed to exhibit non-specific activity, and few studies investigated its mechanism of action. However, by the mid-1980's, numerous observations suggested that specific receptors were possibly involved. Allyn Howlett found that cannabimimetic drugs inhibit cyclic AMP accumulation in neuronal cells by a receptor-mediated cellular response [2], and THC action was found to be stereospecific, indicating a specific site of action [3]. Indeed, by using a highly potent cannabinoid analogue, a receptor was identified by Howlett's group in 1988 [4]. Later on, Lisa Matsuda and coworkers [5] established the functional expression of the cloned cDNA. This receptor is now known as type 1 cannabinoid receptor (CB1). A second entity, known as CB2, was identified by sequence homology and presumed to be mainly present in the periphery [6]. Today we know that CB2 is also present in the central nervous system (CNS). The next step was the discovery of the endogenous ligands of CB1 and CB2, the so-called endocannabinoids (eCBs). As THC is a lipophilic molecule, it was assumed that any endogenous cannabinoid would probably be a lipid. Indeed, by the use of methods for the separation of lipids it was possible to identify the first eCB, N-arachidonoylethanolamine, named anandamide (AEA; Table 1) [7]. AEA is a derivative of arachidonic acid, a compound which serves as precursor of a large number of endogenous molecules – prostaglandins, prostacyclin, thromboxanes, leukotrienes and others – an example of evolutionary conservation to save energy by the use of the same biochemical pathways for a variety of biologically important molecules. A second eCB – again an arachidonic acid derivative, 2-arachidonoylglycerol (2-AG; Table 1) - was isolated shortly thereafter [8, 9].
Table 1.
Major (endo) cannabinoids, and main metabolic enzymes of the ECS.
| Name (abbreviation) | Chemical structure |
|---|---|
| Δ9-Tetrahydrocannabinol (THC) |

|
| N-Arachidonoylethanolamine (Anandamide, AEA) |

|
| 2-Arachidonoylglycerol (2-AG) |
|
| N-Oleoylethanolamine (OEA) |
|
| N-Palmitoylethanolamine (PEA) |
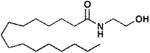
|
| Biosynthetic enzymes of AEA | Intracellular localization |
| Ca2+-dependent N-acyltransferase (NAT) | Integral membrane protein |
| Ca2+-independent N-acyltransferase (iNAT) | Mainly cytosolic |
| N-acyl-phosphatidyl ethanolamines (NAPE)-specific phospholipase D (NAPE-PLD) | Membrane-associated |
| α/β-hydroiase domain 4 (ABHD4) | Membrane-associated |
| Protein tyrosine phosphatase, non-receptor type 22 (PTPN22) | Mainly cytosolic |
| Glycerophosphodiester phosphodiesterase (GDE1) | Integral membrane glycoprotein |
| Degrading enzymes of AEA | Intracellular localization |
| Fatty acid amide hydrolase (FAAH) | Membrane-associated (mainly ER) |
| N-acylethanolamine-hydrolyzing acid amidase (NAAA) | Lysosomal |
| Biosynthetic enzymes of 2-AG | Intracellular localization |
| Diacylglycerol lipase α (DAGLα) | Membrane-associated |
| Diacylglycerol lipase β (DAGLβ) | Membrane-associated |
| Degrading enzymes of 2-AG | Intracellular localization |
| Monoacylglycerol lipase (MAGL) | Membrane-associated and cytosolic |
| α/β-hydrolase domain 6 (ABHD6) | Membrane-associated |
| α/β-hydrolase domain 12 (ABHD12) | Membrane-associated |
| Oxidative enzymes of AEA and 2-AG | Intracellular localization |
| Cyclooxygenase-2 (COX-2) | Membrane-associated |
| Lipoxygenases (LOXs) | Membrane-associated and cytosolic |
| Cytochromes P450 (CYPs) | Membrane-associated |
There are many additional fatty acid amides of ethanol amines and amino acids present in the brain and (in part at least) in the periphery, which do not bind to cannabinoid receptors, but whose actions may be cannabinoid receptor-dependent [10, 11]. They should possibly also be considered to be bona fide components of the eCB family.
Although the discovery of eCBs and the recognition of their ubiquitous activities represent a major advance in biology, we should perhaps wonder why we were for so long unaware of their existence, and why these molecules had not been detected through their activity in so many distinct physiological reactions. 2-AG (and possibly also AEA) is a retrograde synaptic messenger that can prevent the development of excessive neuronal activity, a regulatory action of major importance [12]. eCBs are involved in myriad physiological processes [13-15]. Yet, most of the actions of these endogenous molecules were established only after the dicovery of the proteins that bind and metabolize them, the so-called eCB system (ECS) (for recent reviews see refs 14, 15). Here the main ECS components are presented, and the state-of-the-art of critical eCB functions in peripheral organs is reviewed. Our community effort is aimed at establishing consensus views on the relevance of the peripheral ECS for human health and disease pathogenesis, as well as to highlight emerging challenges and therapeutic hopes.
The ECS at a glance
The two best characterized eCBs, AEA and 2-AG, bind with different affinities to CB1 and CB2, which are two well-characterized 7-transmembrane G protein-coupled receptors (GPCRs) [15-18]. Accumulated evidence suggests the occurrence of other targets for eCBs, like the purported “CB3” receptor GPR55 [19], and the transient receptor potential vanilloid 1 (TRPV1) ion channel that has an intracellular binding site [20]. Other eCB targets such as the peroxisome proliferator-activated receptors (PPAR) α and γ are localized in the nucleus, where they shuttle from/to the cytosol in a ligand-dependent manner [21]. In addition to distinct receptor targets, the ECS comprises numerous metabolic enzymes. It is widely accepted that eCBs are produced “on demand” from membrane lipid precursors by multiple biosynthetic pathways, i.e. when and where needed upon (patho) physiological stimuli. AEA and 2-AG metabolism occurs through distinct routes, of which several have been described in detail [22, 23]. The canonical view is that AEA is synthesized from membrane phospholipid precursors mainly by the sequential action of N-acyltransferase (NAT) and N-acyl-phosphatidylethanolamines-specific phospholipase D (NAPE-PLD). Instead, two diacylglycerol lipases (DAGLα/β) are responsible for the synthesis of 2-AG. The eCB-mediated effects are terminated by their fast catabolism, mainly through hydrolysis by fatty acid amide hydrolase (FAAH), for AEA, and by monoacylglycerol lipase (MAGL), for 2-AG. Alternatively to hydrolytic routes, AEA and 2-AG can be oxidized by cyclooxygenase-2 (COX-2), distinct lipoxygenases (LOXs), or cytochromes P450 (CYPs). Interestingly, the main oxidative products of AEA and 2-AG are endowed with novel biological activities, that are probably mediated by distinct receptors compared to those that bind eCBs [22, 23].
Overall, it is apparent that these metabolic enzymes regulate the in vivo biological availability of eCBs, which altogether are responsible for keeping the eCB tone [24, 25]. The best characterized of these enzymes are summarized in Table 1, along with their intracellular localization (for a recent review see ref 26).
The complexity of the ECS supports its manifold activities at the periphery, and may offer different targets for the development of selective drugs able to modulate eCB signaling in distinct peripheral cells. In the following sections, current knowledge of the impact of eCB signaling (mainly by activation of the THC-binding CB1 and CB2 receptors) in distinct peripheral organs is presented.
Cardiovascular system
Studies over the past few decades demonstrated that CB1 and CB2, eCBs and their anabolic/catabolic enzymes are present in cardiovascular tissues and may play an important role in the development and/or progression of common cardiovascular disorders [27-29]. Earlier studies focusing on the acute hemodynamic effects in various forms of shock and heart failure have demonstrated that under these pathological conditions eCBs produced by activated monocytes/macrophages contributed to the hypotension and negative inotropy via activation of cardiovascular CB1 [27]. Later studies investigated the signaling mechanisms in murine and human cardiomyocytes, endothelial, vascular smooth muscle cells, and fibroblasts/myofibroblasts, using various clinically relevant cardiomyopathy/heart failure, metabolic syndrome/diabetes, and hypertension models [28-34]. These demonstrated that cardiovascular cells can also generate eCBs under pathological challenges/conditions, which in turn through CB1-dependent and/or -independent pathways may promote the generation of reactive oxygen species (ROS), angiotensin II type 1 receptor signaling, accumulation of advanced glycation end products, β-myosin heavy chain isozyme switch, remodelling/fibrosis, and activation of pro-apoptotic mitogen-activated protein kinases, eventually resulting in decreased function of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase and cell death, leading to cardiac dysfunction/failure [32-35]. In these experimental models, eCBs and/or CB1 receptors were increased/upregulated in the myocardium, and CB1 antagonists ameliorated the contractile dysfunction and all above mentioned characteristic pathological processes. Experimental studies using CB1 antagonists and/or CB1 knockout mice also suggested that eCBs via CB1 in macrophages may promote pro-inflammatory processes and inflammatory cell recruitment, thus contributing to the development/progression of atherosclerosis, as well as to pathological smooth muscle proliferation associated with restenosis [33]. The pro-inflammatory effect of CB1 in the cardiovascular system was also confirmed by using inhibitors and/or knockouts of eCB-metabolizing enzyme FAAH in models of atherosclerosis and cardiomyopathy [33]. In clinical trials of obesity/diabetes the CB1 antagonist/inverse agonist rimonabant decreased multiple cardiovascular risk factors, including inflammatory markers. Increased plasma levels of eCBs were strongly correlated with adverse coronary circulatory events, or impaired coronary endothelial function in human obese subjects [35]. Several human reports using CB1 antagonists clearly established that both marijuana and THC exerted CB1-dependent cardiovascular effects, and clinical trials with peripherally restricted CB1 agonists in pain were discontinued because of development of severe adverse cardiovascular and metabolic consequences of CB1 stimulation [35]. A recent study also raised serious concerns about the cardiovascular safety of marijuana use in humans [36], which is also supported by numerous case reports in young adults consuming marijuana or mixture of synthetic CB1 agonists for recreational use [35].
In contrast to CB1, CB2 signaling appears to be protective in cardiovascular diseases [28]. In endothelium, vascular smooth muscle and fibroblast CB2 activation decreases pathological activation, proliferation or pro-inflammatory/fibrotic response, and it may also exert protective effects in cardiomyocytes [28]. In immune cells, CB2 attenuates chemotaxis, adhesion of inflammatory cells and activation [28]. These effects are responsible for the protective properties of CB2 agonists in preclinical models of atherosclerosis, restenosis, myocardial infarction and stroke [28]. Importantly, in the cardiovascular system eCBs via degradation to arachidonic acid metabolites or through putative novel cannabinoid or other (e.g., TRPV1) receptors, independently of CB1 and CB2, may also exert numerous context-dependent effects, ranging from vasodilation/vasoconstriction to anti-inflammatory/pro-inflammatory effects [35]. The impact of eCB signaling in the cardiovascular system is schematically depicted in Figure 1, whereas Table 2 summarizes therapeutic applications of ECS-related drugs.
Figure 1.

Role of ECS in cardiovascular injury/disease. Cardiovascular insult inflicted by ischemia, inflammation or hemodynamic overload leads to increased formation of reactive oxygen and/or nitrogen species (ROS/RNS) and inflammation. These processes trigger activation of ECS in cardiovascular system and infiltrating immune cells. eCBs via activation of CB1 in cardiomyocytes, endothelial cells, fibroblasts and certain immune cells promote processes facilitating development of cardiovascular dysfunction, inflammation and pathological remodeling. In contrast, eCBs via activation of CB2 exert opposing protective effects. Moreover, eCBs through their catabolism by FAAH and/or MAGL or oxidation by cyclooxygenases (COXs) or other enzymes may represent a significant source of arachidonic acid (AA) and/or other oxidized eCB metabolites with both pro- and anti-inflammatory effects. Thus, the protective or detrimental effect of eCBs in cardiovascular diseases may largely be context-, time- and pathology-dependent.
Table 2.
Therapeutic applications of ECS-related drugs at the periphery.
| Compound/Category | ECS target | Model (animal/human) | Therapeutic indication(s) | Clinical conditions | References |
|---|---|---|---|---|---|
| (Peripherally-restricted) CB1 antagonists | CB1 | Rodent/human | Cardiomyopathies, heart failure, diabetic cardiovascular complications, atherosclerosis, circulatory shock | Doxorubicin-induced and cirrhotic cardiomyopathies, diabetic cardiomyopathy and vasculopathy, circulatory shock, atherosclerosis, heart failure | [27-29, 31-33, 35, 63, 159, 160] |
|
| |||||
| CB2 agonists | CB2 | Rodent | Myocardial infarction, stroke, myocarditis? cardiomyopathies? | Myocardial infarction, stroke, myocarditis? cardiomyopathies? | [28, 160] |
|
| |||||
| (Peripherally-restricted) CB1 agonists | CB1 | Dog / human | Transient lower esophageal relaxations | Gastroesophageal reflux disease | [161, 162] |
|
| |||||
| (Peripherally-restricted) CB1 agonists | CB1 | Mouse / rat | Diarrhea | Irritable bowel syndrome | [163] |
| Inflammation | Inflammatory bowel disease | [164-166] | |||
| Visceral pain | Gastric ulcer | [167] | |||
|
| |||||
| (Peripherally-restricted) CB1 antagonists | CB1 | Mouse | Metabolic endotoxemia | Obesity | [43] |
| Food intake | [40] | ||||
| Dysmotility | Paralytic ileus | [168, 169] | |||
|
| |||||
| CB2 agonists | CB2 | Mouse / rat | Diarrhea | Irritable bowel syndrome | [44, 170] |
| Inflammation, visceral pain | Inflammatory bowel disease | [44, 170] | |||
|
| |||||
| FAAH inhibitors | CB1 CB2 PPARs | Mouse | Diarrhea | Irritable bowel syndrome | [45, 171] |
| Inflammation | Inflammatory bowel disease | [166, 172] | |||
| Gastric ulcer | [173] | ||||
|
| |||||
| MAGL inhibitors | CB1 CB2 PPARs | Mouse / rat | Diarrhea | Irritable bowel syndrome | [174] |
| Inflammation | Inflammatory bowel disease | [175] | |||
| Gastric ulcer | [47, 176] | ||||
|
| |||||
| Peripherally restricted CB1 antagonists | CB1 | DIO mice, ob/ob mice, db/db mice, ZDF rats | Lipogenesis, inflammation | Obesity/metabolic syndrome, fatty liver disease, type-2 diabetes | [61, 177, 178] |
|
| |||||
| Rimonabant | CB1 | Mouse C2C12 myoblasts | Defective myotube differentiation and muscle regeneration | Muscular dystrophy | [100] |
|
| |||||
| Synthetic CB2 agonists | CB2 | Mouse | Bone mineralization | Osteoporosis | [113] |
|
| |||||
| N-Palmitoylethanolamine | CB1? CB2? TRPV1? PPARs? GPR55? | Human | Vestibulodynia, vulvodynia, proctodynia | Infertility | [179, 180] |
|
| |||||
| THC | CB1 | Mouse | Parturition | Infertility | [125, 128] |
|
| |||||
| N-Palmitoylethanolamine | CB1? CB2? TRPV1? PPARs? GPR55? | Human | Inflammation, pruritus | Atopic dermatitis, prurigo, uremic itch | [140, 141] |
|
| |||||
| (Peripherally-restricted) CB1 antagonists | CB1 | Rodent | Diabetic and other nephropathies and tubulopathies | Diabetic and other nephropathies and tubulopathies | [151, 152, 154] |
|
| |||||
| CB2 agonists | CB2 | Rodent | Diabetic and other nephropathies and tubulopathies | Diabetic and other Nephropathies and tubulopathies | [153, 154] |
DIO (diet-induced obesity), ob/ob (defective in leptin gene), and db/db (defective in leptin receptor gene) mice are models of obesity. Zucker diabetic fatty (ZDF) rats are a model of diabetes.
Collectively, there is strong evidence supporting the pathological function of an overactive ECS (in particular CB1) in cardiometabolic diseases. Indeed, it can be concluded that: i) the ECS is dysregulated in cardiovascular disease; ii) eCBs and synthetic ligands through CB1 and CB2 exert opposing effects on cardiovascular injury and inflammation, with CB1 promoting and CB2 attenuating them; and iii) peripherally restricted CB2 agonists and CB1 anagonists appear as promising targets in cardiovascular disease.
Gastrointestinal tract
The mechanistic basis for the therapeutic effects of cannabis in the gastrointestinal (GI) tract was apparent after the discovery of THC as the major psychoactive constituent of cannabis. Even before a specific cannabinoid receptor was cloned, it was shown that THC inhibited acetylcholine release from enteric nerves [37]. This finding paved the way to an extensive examination of the ECS in the gut and the remarkable discoveries that virtually all gut functions are regulated by eCBs and that the ECS of the gut is critical for CNS control of metabolic and homeostatic functions of the body (Figure 2).
Figure 2.

The ECS of the gastrointestinal (GI) tract and liver. AEA, 2-AG, and OEA are synthesized in the gut and liver, where they act locally and in the brain. eCBs regulate gut motility at the level of the enteric neural plexuses, they reduce intestinal inflammation through actions on the immune system and they influence intestinal barrier function at the level of epithelium. eCBs and OEA regulate food intake by actions on enteroendocrine cells in the wall of the gut, the vagus nerve and in the brain. In the liver, CB1 and CB2 have opposing effects, with CB1 promoting steatosis, fibrogenesis, apoptosis and proliferation and CB2 inhibiting these effects.
CB1 and CB2 are highly expressed on enteric nerves and throughout the intestinal mucosa on enteroendocrine cells (CB1), immune cells (CB1 and CB2) and enterocytes (CB1 and CB2), as depicted in Figure 2. Under physiological conditions, the actions of eCBs in the GI tract are largely mediated by CB1 [37]. Activation of the latter stimulates GI motility, suppresses acid and fluid secretion and induces mesenteric vasodilatation [37]. The role of CB1 in the regulation of enteric neurotransmission has recently been revealed. eCBs (and notably 2-AG) are retrograde transmitters in the brain, but in the enteric nervous system they also seem to be involved in a previously unknown form of synaptic control that utilizes two retrograde messengers (eCBs themselves and a purine nucleotide), working in opposite directions to control synaptic strength. Such a phenomenon is known as metaplasticity [38].
The role of the ECS in the regulation of energy balance is multifaceted, and involves the GI tract in at least three ways. First, CB1 is localized at enteroendocrine cells, and it may regulate the release of enteroendocrine peptides such as cholecystokinin, which signals hunger [39]. Second, CB1expression on enterocytes is regulated by enteric microbiota [40]. Activation of this receptor enhances epithelial permeability by reducing the expression of tight junctional proteins, allowing bacterial translocation and metabolic endotoxemia, leading to obesity. Blocking CB1 expression reduces obesity, in part through the enhancement of intestinal barrier function. Third, gut-derived eCBs AEA and 2-AG, and the related compound N-oleoylethanolamine (OEA; Table 1), are key signaling molecules for the control of food intake, as well as for lipid sensing [41]. OEA in particular seems to be positioned to monitor dietary fat intake and does so by activating both homeostatic vagal afferent pathways, and the brain's reward circuitry [42, 43]. In pathophysiological states, both CB1 and CB2 reduce accelerated GI motility, with CB2 activation normalizing motility and serving as a braking mechanism to limit abnormal motility [44]. Interestingly, inhibition of FAAH, thereby elevating endogenous levels of eCBs, also normalizes accelerated GI motility in pathophysiological states, but not in normal animals [45]. The ECS is also an important anti-inflammatory system, and when activated reduces gastric damage and intestinal inflammation [46, 47]. Recent findings have surprisingly revealed that both peripheral and central cannabinoid receptors are required to block the development of colitis [46].
Components of the ECS are altered in many GI disorders, e.g. celiac disease and inflammatory bowel disease [48, 49]. Yet, the therapeutic potential to target this system in their treatment remains to be determined, whereas quite a number of other GI diseases are likely to benefit from ECS-related drugs (Table 2). Whilst cannabis has been used for decades, the clinical utility of the many experimental compounds that have been developed, which target distinct elements of the ECS, remains uncertain and the central effects of many of these compounds will likely limit their usefulness. Peripherally-restricted compounds could overcome these limitations, and appear to have potential in some circumstances [50]. But a note of caution is warranted. In inflammatory bowel disease, cannabis use is common and subjectively improves pain and diarrhea. In a recent short-term (8 week) clinical trial, inflammation was reduced with smoked cannabis in a cohort of patients with Crohn's disease [51]. However, cannabis use is associated with higher risk of surgery in patients with Crohn's disease [52]. This illustrates the problems of pharmacology in a system that is ubiquitous and involved in many diverse functions in the gut (Figure 2).
Fifty years ago, one could not have imagined the remarkable nature of the ECS in the GI tract. To date, we can conclude that: i) eCB signaling contributes to the regulation of motility, barrier function, immune function and the control of food intake and energy balance; ii) CB1 and CB2 located on enteric nerves, enteroendocrine cells, immune cells and the intestinal epithelium mediate the actions of eCBs, by reducing neurotransmitter release, inhibiting release of enteroendocrine hormones, suppressing immune activation and regulating tight junctions, respectively; and iii) the discoveries outlined above provide an exciting array of therapeutic targets and with further developments in this field the opportunities to develop new medicines for GI disorders are immense.
Liver
The ECS of the liver is normally quiescent owing to the low level of cannabinoid receptor expression. However, under pathophysiological conditions receptor expression is increased, and they have now been recognized as having a critical role in liver disease (Figure 2).
Although the liver was initially thought to be devoid of CB1, it is now evident that these receptors are expressed and mediate a number of biological functions in different types of liver cells, including hepatocytes, stellate cells and vascular endothelial cells, and that blockade of these receptors contributes to the beneficial metabolic effects of CB1 blockade.
High-fat feeding promotes hepatic lipogenesis and mice deficient in CB1 are resistant to high-fat diet-induced obesity and the associated hepatic steatosis [53], which suggests that functional CB1 in hepatocytes promote lipogenesis. This was indeed documented by the ability of a CB1 agonist to induce lipogenic gene expression and de novo hepatic fatty acid synthesis in mouse liver in vivo or in isolated hepatocytes in vitro, and by the absence of these effects in CB1 knockout mice or hepatocytes treated with a CB1 antagonist [53]. CB1-mediated hepatic lipogenesis was subsequently documented in human liver cells [54]. In contrast to increasing lipogenesis, hepatic CB1 inhibit fatty acid oxidation, as documented in cultured liver explants [55], and in the liver of wild-type mice but not in mice with hepatocyte-specific deletion of CB1 [56]. Hepatic CB1 also plays a dominant role in hepatic insulin resistance that accompanies high-fat diet-induced obesity: activation of hepatocyte CB1 inhibits insulin clearance by the insulin degrading enzyme, thus contributing to hyperinsulinemia, and also triggers endoplasmic reticulum (ER) stress-dependent suppression of insulin signaling through insulin receptor substrate-1 (IRS1) and Akt-2, resulting in increased glycogenosis [57]. Inhibition of insulin signaling by hepatic CB1 may also involve activation of the CREBH/LIPIN1/diacylglycerol/PKCε signaling pathway that results in increased gluconeogenesis [58]. The expression of CB1 mRNA and protein is very low in normal liver, but is increased substantially in steatotic hepatocytes [53, 56, 59], and chronic CB1 blockade by rimonabant reverses steatosis induced either by high-fat feeding or ethanol and the upregulation of hepatic CB1 associated with both [59, 60]. This indicates not only that increased eCB/CB1 tone mediates the development of fatty liver of various etiologies, but also that CB1 expression is auto-induced under these conditions. The finding that reversal of diet-induced obesity and its hepatic sequelae could be fully replicated by a non-brain-penetrant CB1 antagonist supports the dominant role of peripheral (including hepatic) rather than central CB1 in these effects [61]. The latter receptor is also expressed in hepatic stellate cells, where it is induced by various fibrogenic stimuli, whereas its genetic or pharmacologic ablation protects the liver from fibrosis [62]. In cirrhosis, which represents the advanced stage of liver fibrosis, CB1 are similarly up-regulated in the vascular endothelium where they mediate hepatic vasodilation, which predisposes cirrhotic individuals to ascites formation and variceal hemorrhage [63].
The role of CB2 in the liver was first described by Boris Julien and colleagues who demonstrated an anti-fibrogenic action of its agonists in hepatic myofibroblasts and stellate cells, and that CB2 knockout mice developed enhanced fibrosis in response to the hepatoxin carbon tetrachloride [64]. Since then, CB2 has been implicated in liver fibrosis, fatty liver disease, and acute ischemic liver injury [65]. CB2 is expressed on immune cells, Kupffer cells and myofibroblasts, but not on hepatocytes. Extending the findings of Julien and colleagues, Javier Muñoz-Luque's group [66] also used the carbon tetrachloride model to induce fibrosis of the liver and treated rats with the CB2 agonist JWH-133. The latter substance markedly improved the extent of liver fibrosis leading to reduced portal pressures. This effect is mediated by CB2 on myofibroblasts and is due to reduced interleukin 17 (IL-17) production by Th17 lymphocytes [67].
Inflammation in the liver is an important feature of alcoholic fatty liver disease. Activation of CB2 receptors in this context switches Kupffer cells from a pro-inflammatory (M1) phenotype to an anti-inflammatory (M2) phenotype by the induction of hemeoxygenase-1. Fat accumulation in hepatocytes is also reduced, but this is an indirect effect through reduction in steatogenic cytokines such as IL-1β [68].
After liver transplantation, ischemia-reperfusion injury leads to marked liver damage. In mouse models of ischemia-reperfusion injury, Sandor Bátkai and colleagues revealed a remarkable role of CB2 receptors in reducing inflammatory infiltrates, inflammatory cytokine and chemokine levels and the expression of adhesion molecules on liver sinusoidal endothelial cells [69]. eCBs made in the liver are also capable of regulating the degree of ischemia-reperfusion injury. Wild-type mice given the MAGL inhibitor JZL184 were protected from hepatic injury, as were mice lacking this enzyme [70].
It is apparent that CB1 and CB2 receptors are acting in opposite directions in the liver. CB1 promotes fibrogenesis, steatosis and the cardiovascular complications of liver disease, whereas CB2 is protective, reducing these indices of liver dysfunction. Clinical studies of cannabis use show that it is detrimental, presumably because of its actions at CB1 [71]. Thus, CB1 blockade (with drugs that do not cross the blood-brain barrier) and CB2 agonists have therapeutic potential in liver cirrhosis by targeting both the fibrotic process and its potentially lethal hemodynamic complications (Table 2).
In summary: i) activation of hepatic CB1 promotes vasodilation that can lead to ascites formation, promotes fat accumulation, insulin resistance, and fibrosis; ii) activation of CB2 reduces proinflammatory cytokines, attenuates re-perfusion injury, reduces fat accumulation and is antifibrotic; and iii) CB2 agonists and peripherally restricted CB1 antagonists have therapeutic potential in (non-) alcoholic fatty liver diseases and their long-term consequences.
Immune system
The mammalian immune system can be subdivided into the innate and adaptive immune systems. The innate immune system functions non-specifically, responds to invasive pathogens in a generic fashion, and does not confer long-lasting immunity. Elements of innate immunity include the complement system that elicits a biochemical cascade targeting the surfaces of foreign cells, natural killer (NK) cells that destroy tumor and virus-infected cells, and soluble factors such as eicosanoids and cytokines that are released by injured and infected cells. Typical cytokines that are elicited during this phase of the immune response include interferon-γ, interleukins, and tumor necrosis factor-α. These soluble factors bind to cognate receptors and other cellular targets and set in motion signaling cascades that culminate in the activation of select genes. In contrast to the innate immune system, the adaptive immune system (Figure 3) is antigen-specific, recognizes “non-self antigens during a process referred to as antigen presentation, and confers immunological memory. Principal players in adaptive immunity are B lymphocytes, that are critical players in the humoral immune response through their capacity to produce antibodies, and T lymphocytes that are critical to the cell-mediated immune response. In adaptive immunity, helper T lymphocytes through the facilitation of their CD4 co-recepor (CD4+), recognize antigens (i.e., substances that induce a specific immune response and that react with the products of that response) that are presented by antigen-presenting cells (APCs) in the context of their class II major histocompatability (MHC) molecules. The consequent activation of helper T lymphocytes results in their release of cytokines and other stimulatory signals that activate macrophages, cytotoxic T lymphocytes, and B lymphocytes.
Figure 3.
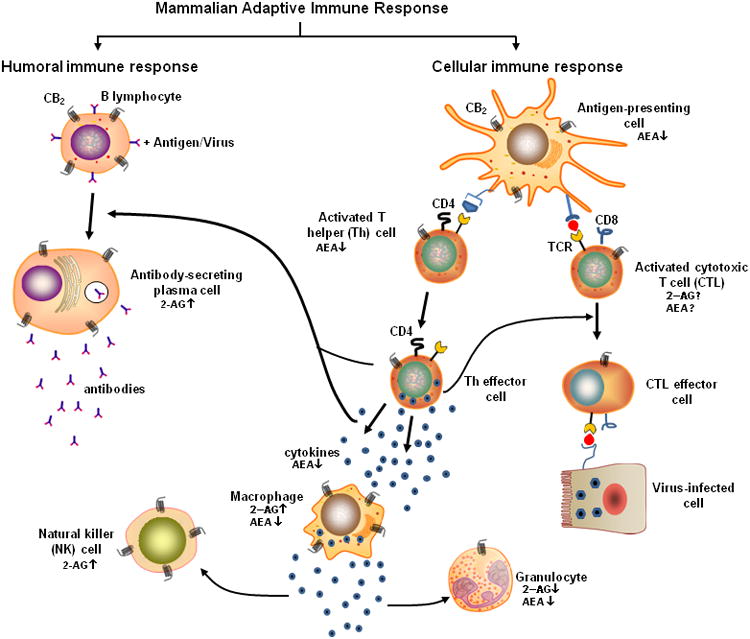
Cell types and cascade of immunological events that come into play in an adaptive mammalian immune reponse, and respective functional activities that have been implicated as modulated by eCBs. The adaptive immune response in mammals is highly specific to a given pathogen, and can provide long-lasting protection by destroying the invading pathogen and the toxic molecules it produces. Cells that are involved in this response are white blood cells known as B lymphocytes and T lymphocytes. These cells, respectively, play a critical role in humoral and cellular immune responses once activated by antigens (i.e., molecules or linear fragments that are recognized by receptors on the lymphocyte surface). In the humoral immune response, activated B lymphocytes secrete antibodies, or immunoglobulins. These antibodies travel through the bloodstream and bind to the cognate antigen resulting in its inactivation. Such an inactivation may include prevention of its attachment to host cells. In the cellular immune response, T lymphocytes are mobilized when they encounter an antigen-presenting cell such as a dendritic cell or B lymphocyte that has digested an antigen and is displaying antigen fragments bound to its major histocompatability (MHC) molecules. During this response, cytokines (small signaling proteins) facilitate T lymphocyte maturation and the growth of more T lymphocytes. The engendered MHC-antigen complex activates T cell receptors resulting in T lymphocyte secretion of additional cytokines. Some T lymphocytes develop into helper (Th) cells and secrete cytokines that attract other immune cells such as macrophages, granulocytes, and natural killer (NK) cells. Some T lymphocytes become cytotoxic cells (CTLs) and lyse tumor cells or host cells infected with viruses. CD4: cluster of differentiation 4, a glycoprotein found on the cell surface and used as marker for Th cells; CD8: cluster of differentiation 8, a glycoprotein found on the cell surface and used as marker for CTLs; TCR: T cell receptor, a molecule found on the surface of T lymphocytes that recgnizes antigens bound to MHC molecules.
Bioactive lipid molecules participate in this cross-talk between different types of immune cells. Included among these are eCBs, that can bind to cannabinoid receptors and cooperate with other signaling molecules to modulate the functional activities of immune cells, particularly at the level of the adaptive immune response [72, 73]. Their ligation to, and activation of, CB1 and CB2 sets in motion a series of signal transduction pathways that converge at the transcriptional level to regulate cell migration and the production of cytokines, chemokines, and other inflammatory factors. Immune cells preponderantly express CB2. Consistent with this observation, there is a large body of data that supports a functional relevance for 2-AG as acting through CB2 to inhibit migratory activities for a diverse array of immune cell types [74, 75]. Indeed, it has been suggested that 2-AG is the cognate functionally relevant eCB for CB2. For example, Takayuki Sugiura and colleagues [76] examined the effect of 2-AG on intracellular free Ca2+ concentrations in human HL-60 macrophage-like cells, and found that it induced a rapid transient increase in levels of intracellular free Ca2+. The induced Ca2+ transient was blocked by a CB2 antagonist, consistent with the involvement of CB2 in this response.
AEA also has been reported to inhibit immune functional activities, particularly the production of pro-inflammatory cytokines. Evgeny Berdyshev and colleagues [77] reported that AEA diminished levels of the cytokines IL-6 and IL-8 from human monocytes. In keeping with these findings, Maria Teresa Cencioni and colleagues [78] showed that AEA suppresses release of cytokines like IL-2, tumor necrosis-α and interferon-γ from activated human peripheral T-lymphocytes, mainly through a CB2-dependent mechanism. Jean-Marie Derocq and colleagues [79], using IL-3-dependent and IL-6-dependent mouse cell lines, proposed that AEA exerted a growth-promotion effect. Luigi Facci and colleagues [80] reported that mast cells, that are bone marrow-derived cells found in mucosal and connective tissues (as well as in the nervous system) and that play a role in tissue inflammation and neuroimmune interactions, expressed a peripheral cannabinoid receptor that was differentially sensitive to AEA. These cells reportedly expressed CB2 that exerted negative regulatory effects on mast cell activation. However, AEA did not down-modulate mast cell activation in vitro. Thus, unequivocal data that supports a functional linkage of AEA to a cannabinoid receptor in mediating immune cell effects remains to be obtained. Indeed, it has been proposed that the immune modulatory effects of AEA could be cannabinoid receptor-independent [77, 79, 81]. eCBs, as typical bioactive lipids, are degraded rapidly, have a short half-life intracellularly and extracellularly, and modulate immune functions in an autocrine and paracrine fashion. Thus, their immediate effective action on immune function may be at localized sites. It is speculated that, in this context, eCBs play an important role in maintaining the overall “fine tuning” of the immune homeostatic balance within the host. A variety of mammalian cell systems have been used as experimental models to validate the in vitro effects of eCBs on immune function. Early experiments involved the exogenous introduction of eCBs to cultures of transformed immune cells or to immune cell sub-populations obtained from mice and humans. Such studies were complemented with those using mixed cell populations that potentially replicated more closely an in vivo condition that integrated crosstalks between different immune cell types. Experimental animal models, such as guinea pigs and mice, also have been used to document effects of eCBs on immune functions [81]. Nevertheless, the conduct of in vivo studies using eCBs has been challenging, partially because these substances are readily degraded, necessitating that they be applied experimentally at relatively high doses. Furthermore, their intracellular fate, compartmentalization, and processing within the host may be distinctive from that of exogenously introduced phytocannabinoids and synthetic cannabinoids. Given these caveats, in vivo studies have converged on an immune functional relevance for eCBs that parallels the one that has been obtained using various in vitro paradigms [82].
Recognition that immune functions can be mediated through the interaction of eCB ligands with specified receptors, such as CB2, should provide novel insights regarding potential target points in signal tansductional pathways that are amenable to therapeutic manipulation. In this fashion, the manipulation and ablation of untoward immunological events, including those possibly associated with neuroinflammatory events linked to infection by the human immunodeficiency virus (HIV) and other pathogens, may be achieved.
In summary: i) eCBs can bind to cannabinoid receptors and cooperate with other signaling molecules to modulate the functional activities of immune cells; ii) the latter cells preponderantly express CB2, and there is a large body of data that supports a functional relevance for 2-AG as acting through this receptor to inhibit migratory activities for a diverse array of immune cell types; and iii) recognition that immune functions can be mediated through the interaction of eCB ligands with cannabinoid receptors should provide novel insights regarding target points for therapeutic manipulation.
Muscles
It is now well-accepted that muscle cells produce eCBs and express CB1, CB2 and eCB metabolic enzymes, all of which seem to be somehow sensitive to the development of overweight and obesity [83-88]. Whilst in primary skeletal muscle myotubes of lean and obese subjects, expression of cnr1 (the gene encoding for CB1) remained unchanged [87], in rats fed a high-fat diet, abdominal wall skeletal muscle expression of cnr2 (encoding for CB2) and magl (encoding for MAGL) genes was decreased and increased, respectively [85]. In genetically obese Zucker rats, CB1 mRNA levels in soleus were decreased [89], whereas AEA levels were increased [90]. Yet, expression of cnr1 in the soleus from mice on a high-fat diet was increased [91] and 2-AG, but not AEA, levels were significantly elevated in parallel to the late development of obesity and hyperglycaemia [92]. These reports indicate that the ECS response to the metabolic state of the organism may have muscle subtype-, species- and diet-dependent differences, and should be interpreted based on the results of pharmacological studies. The latter have shown that systemic pharmacological CB1 antagonism in obese humans or rodents results in increased energy expenditure/oxygen consumption, which in turn is due to elevated fatty acid oxidation [93-96], possibly subsequent to increased muscle mitochondrial biogenesis [97]. In addition, chronic treatment of ob/ob mice and lean or obese Zucker rats with rimonabant results in increased glucose uptake in the soleus muscle [96, 98], whereas administration of a single dose of the CB1/CB2 agonist HU210 caused CB1-dependent reduction of whole body glucose clearance and glucose transport into muscle but not adipose tissue, and decreased insulin-dependent Akt phosphorylation in hind leg muscle [99]. It seems also noteworthy that in soleus muscle explant cultures from lean and insulin-resistant Zucker rats, AEA and rimonabant significantly decrease and increase, respectively, both basal and insulin-dependent glucose import [89]. Finally, AEA has been shown to inhibit insulin-dependent glucose uptake and Akt activation in skeletal muscle cells in a CB1-dependent manner [86]. Overall, these data imply that skeletal muscle eCBs, via activation of CB1, reduce not only mitochondrial activity but also insulin signaling and glucose uptake, possibly through inhibition of IRS1 phosphorylation and insulin-dependent ERK1/2 activation [86, 88]. Thus, the over-activity of the ECS potentially occurring in skeletal muscle during obesity might contribute to defective muscle insulin sensitivity and glucose metabolism, as well as to fatty acid accumulation.
More recently, the ECS was found to play a fundamental role also in skeletal muscle formation (Figure 4a). It was found that, possibly due to changes in the expression of genes involved in its metabolism, levels of 2-AG were decreased both during myotube formation in vitro from murine C2C12 myoblasts and during mouse muscle growth in vivo. 2-AG and a CB1-specific agonist were shown to prevent myotube formation in a manner antagonized by CB1 knockdown and pharmacological antagonism, which instead stimulated differentiation per se [100]. In vivo, muscle fascicles from CB1 knockout embryos were found to contain more muscle fibers, and those from postnatal CB1 null mice exhibited increased diameter relative to those of wild-type littermates. The myoblast differentiation inhibitory action of 2-AG was demonstrated to occur through inhibition of the activity of the Kv7.4 channel, which plays a permissive role in myogenesis and depends on phosphatidylinositol 4,5-bisphosphate (PIP2). Indeed, CB1 stimulation reduced both total and Kv7.4-bound PIP2 levels in C2C12 myoblasts and inhibited Kv7.4 currents in transfected CHO cells. It was suggested that 2-AG might be an endogenous repressor of myoblast differentiation via CB1-mediated inhibition of Kv7.4 channels [100].
Figure 4.
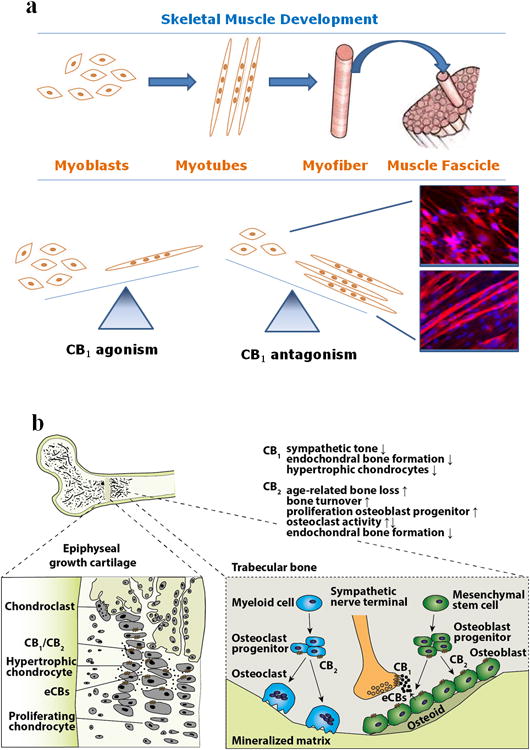
a) Role of ECS in skeletal muscle formation. Simplified representation of the skeletal muscle differentiation process, from myoblasts to myotubes and myofibers and fascicles. Activation of CB1 by eCBs inhibits myoblast to myotube differentiation, whereas CB1 blockade, as in cells treated with a CB1 antagonist or CB1 siRNA, facilitates this process and in vivo, as in CB1 null mice, leads to bigger fibers. b) eCB signaling in bone growth and bone remodeling. CB1 and CB2, as well as eCB synthetic enzymes, are expressed on hypertrophic chondroblasts in the epiphyseal growth cartilage. Mice lacking CB2 develop longer femora and vertebral bodies, resulting in a longer stature, whereas stimulation of CB1 restrains bone growth. Bone remodeling is stimulated in CB2 deficient mice, but with a net loss of bone mass, thus resulting in an age-related osteoporotic phenotype. CB2 signaling stimulates proliferation of osteoblast progenitors, and affects differentiation of osteoclasts. CB1 are most prominently expressed on sympathetic nerve terminals and inhibit release of norepinephrine, thus reducing the sympathetic tone which in turn inhibits bone formation.
Finally, it is important to remember that AEA, and to a lesser extent 2-AG, also activate TRPV1 channels, which were shown to stimulate both skeletal muscle mitochondrial biogenesis [101] and hypertrophy [102]. However, no evidence exists on the possibility that eCBs modulate these two aspects of skeletal muscle function via TRPV1 activation. Against this background, it is not surprising that the therapeutic exploitation of ECS-related drugs in mucle diseases is still in its infancy, showing as yet only one potential application (Table 2).
Taken together, it can be concluded that: i) eCBs are deeply involved in both the control of energy metabolism by the skeletal muscle and the formation of new muscular fibers; ii) both these fundamental actions are mediated preferentially via CB1 and involve AEA and 2-AG to varying degrees; and iii) the ECS ultimately controls the utilization of energy in the skeletal muscle, by either reducing glucose oxidation by myofibers or the extent of muscle formation.
Bones
The skeleton provides a mechanical support to soft tissues and drives body growth. It is constantly adapted to changing mechanical needs by a remodeling process, in which bone is removed by osteoclasts and newly formed by osteoblasts. The lack of appropriate remodeling leads to excessive mineral deposition, which reduces the flexibility of the bone and results in an accumulation of fatigue damage and stress fractures. The remodeling process is tightly coordinated by an intricate system that involves eCB signaling [103-114], as depicted in Figure 4b. Thus, AEA and 2-AG are locally produced by bone cells and reach concentrations similar to brain tissues [115]. Stimulation of CB2 on osteoblast precursor cells has a mitogenic effect and results in an expansion of the preosteoblastic cell pool, while mature osteoblasts respond with increased alkaline phosphatase activity and matrix mineralization [112, 113]. The role of CB2 signaling in osteoclastogenic cultures and RAW 264.7 cells is more complex, as both inhibitory and stimulatory effects of CB2 agonists on osteoclast formation have been reported under different experimental conditions [103, 109, 110, 113]. Interestingly, expression of CB2 on osteoclasts is modulated by TRPV1 channel, which is another target for AEA and has profound effects on bone remodeling [116]. Mice lacking CB2 displayed a strikingly enhanced age-related loss of trabecular bone volume density with a concomitantly increased bone turnover, a phenotype reminiscent to human postmenopausal osteoporosis [113]. Human genetic studies have also identified polymorphisms in the CB2 coding region that are associated with low bone mass and osteoporosis [117, 118]. Together, these findings implicate CB2 signaling in pro-ostogenic processes along the osteoblast lineage and indicate a therapeutic potential of CB2 agonists in the anabolic therapy of osteoporosis (Table 2). Indeed, studies in mice have shown that CB2 agonists rescue ovariectomy-induced bone loss [113].
eCB signaling is also important in the central nervous system regulation of bone remodeling via antagonistic sympathetic and parasympathetic projections. Norepinephrine released from sympathetic fibers inhibits bone formation and stimulates bone resorption [119, 120], whereas parasympathetic acetylcholine decreases bone resorption [121]. CB1 is mostly expressed on sympathetic nerve endings [115], and its activation by 2-AG produced from closely apposed osteoblasts inhibited norepinephrine release, thus alleviating tonic inhibition of bone formation by the sympathetic nervous system. Deletion of CB1 in congenic C57BL/6J mice resulted in a low bone mass phenotype [115]. A detailed phenotype analysis indicated that CB1 signaling increased trabecular bone mass and radial diaphyseal growth by up-regulating bone formation and down-regulating bone resorption [115]. The phenotype was masked, however, in a different CB1 deficient mouse line maintained on an outbred CD1 genetic background, which showed no changes in bone remodeling parameters [110, 114].
Recently, a functional ECS was also discovered in the epiphyseal growth cartilage (EGC) [122], which drives bone and consequently body growth by endochondral bone formation. In this process, chondrocyte progenitors differentiate into proliferative and then hypertrophic chondrocytes. The extracellular matrix separating the hypertrophic chondrocytes is then calcified, resorbed by osteoclasts/chondroclasts and replaced by bone. CB1 and CB2, as well as DAGLα and DAGLβ are expressed in EGC hypertrophic chondrocytes, and 2-AG was detected at significant levels in the EGC. Femora of mice lacking CB1 or CB2 were considerably longer at the end of the rapid growth phase compared to wild-type animals. Importantly, THC slowed skeletal elongation of wild-type and CB2 knockouts, but not that of CB1 deficient mice. This was probably due to a direct effect of THC on hypertrophic chondrocytes, because THC inhibited EGC chondrocyte hypertrophy in ex vivo cultures, and reduced the hypertrophic cell zone thickness of treated animals [122]. These experimental findings are in line with human studies, reporting that babies born to marijuana using mothers have a reduced fetal growth rate, resulting in a reduced birth weight, a shorter stature and a reduced head size at birth [123, 124].
Considering the profound effects of THC on murine bone growth and the highly prevalent consumption of marihuana by adolescents during their growth phase, it will be important to examine the possible postnatal growth retarding effects of THC in humans. Overall, it can be stated that eCB signaling regulates bone elongation and bone remodeling by modulating: i) proliferation and differentiation of bone cells; ii) communication between bone cells; and iii) neuronal control of bone remodelling.
Reproductive system
Anecdotal and epidemiological evidence suggested adverse effects of marijuana on male and female reproduction, well before direct molecular evidence that phytocannabinoids affect female reproduction was provided. CB1 and CB2 were shown to be expressed in the preimplantation mouse embryos, where CB1 was primarily localized in the blastocyst trophectoderm (reviewed in ref. 125). It was also shown that THC, as well as uterine AEA, can activate CB1 and interfere with embryo development. Notably CB1, but not CB2, was primarily expressed in the mouse female reproductive tract, and cannabinoid/eCB effects on embryo development and function were differentially executed depending on embryonic stage and ligand levels. In this respect, levels of AEA and CB1 are higher in non-receptive uteri, dormant blastocysts, and interimplantation sites, but decline to lower levels in receptive uteri, activated blastocysts, and implantation sites. This suggests that optimally balanced eCB signaling is critical to synchronizing pre-implantation embryo development and preparing the endometrium for implantation (reviewed in ref. 125). Therefore, site-specific levels of AEA and/or other endogenous ligands of CB1 in the uterus may spatially regulate blastocyst implantation, in addition to effects from embryo-based eCB signaling. Subsequent studies showed that eCBs were important for several aspects of female reproduction, including oviductal embryo transport, placentation and parturition [125], as depicted in Figure 5a.
Figure 5.

a) Schematic diagram showing pregnancy events in mice that are influenced by eCB signaling. Stage-specific eCB signaling during pregnancy is numerically designated (1-11). Implantation and decidualization both are designated by (9), since they are overlapping events. P, placenta; F, fetus. b) Functional activity of ECS elements in human sperm. Far from the oocyte activation of CB1 reduces sperm motility and viability (1), and activation of TRPV1 inhibits a spontaneous acrosome reaction (AR), that would be useless to fertilize the egg (2). Next to the oocyte activation of CB1 inhibits zona pellucida (ZP)-induced AR (3), thus avoiding fertilization of the egg by more than one sperm (polyspermy).
Prior to embryo implantation, oviductal transport of the embryo was also impacted by cannabinoid/eCB exposure. It was shown that a critical balance between AEA synthesis and degradation in mouse embryos and oviducts created a local “AEA tone” that determined normal embryo development and oviductal transport. Defects in oviductal embryo transport under aberrant eCB signaling led to deferred on-time implantation and poor pregnancy outcome. These studies uncovered novel regulation of preimplantation processes by eCB signaling, which could be clinically relevant for fertility regulation in women. For example, CB1 is expressed in low levels within both the Fallopian tube and endometrium of women with ectopic pregnancy, suggesting that aberrant eCB signaling within the Fallopian tubes may lead to this pathology [126-128]. Aberrant eCB signaling was also observed to confer premature trophoblast stem cell differentiation, and defective trophoblast development and invasion. These defects were reflected in retarded fetal development and compromised pregnancy outcome [129]. Thus, a tightly regulated eCB signaling threshold across multiple early pregnancy events is critical for female reproductive success [130]. Independent studies further supported these findings, suggesting a similarly important role for eCB signaling in human pregnancy [125]. Indeed, aberrant eCB signaling may be associated with increased rate of miscarriages and human ectopic pregnancies [126, 131-133].
The effects of eCB signaling on reproductive organs led to the conclusion that also exposure to marijuana during pregnancy may have many adverse effects [125]. With the current legalization of marijuana in the United States and high usage among young adults, there is a critical need for more research into the effects of this drug, and for strong educational efforts to inform the public of the dangers associated with consuming marijuana when pregnant or trying to become pregnant. By contrast, ECS-based drugs have reached the market as treatment for female infertility, and creams containing PEA (N-palmitoylethanolamine; Table 1) appear useful for the treatment of pain-related dysfunctions like chronic vestibulodynia, vulvodynia and vaginismus (Table 2).
Taken together, it can be concluded that: i) eCB signaling is operative in all critical stages of pregnancy; ii) both silenced or amplified eCB signaling affects pregnancy events; and iii) eCB signaling in female reproductive events is primarily mediated by CB1.
On the male side, it should be recalled that nearly 1 in 6 couples suffers from infertility, with male factors accounting for 50% of cases [134]. Although anatomical factors explain a significant number of these, several of them remain unexplained, primarily because factors that are involved in the modulation of spermatogenesis and sperm function are poorly understood. A number of molecules/systems have been suggested as playing a pivotal role in this regard, and one such system is the ECS. The fact that marijuana compromises male fertility indeed provides support for this. Concentrations of AEA, OEA and PEA have been detected at nanomolar levels in seminal plasma [135, 136]. Furthermore, both the receptors (CB1, CB2, TRPV1) and the enzymes (NAPE-PLD, FAAH) regulating these eCB are differentially expressed in human sperm [137]. Indeed, FAAH and NAPE-PLD are localized mainly in the post-acrosomal region and in the middle piece, while CB1 is localised in the plasma membrane over the acrosomal region, the middle piece and tail, and CB2 is localised in the plasma membrane of the head. Finally, TRPV1 is localized in the post-acrosomal region [135]. The localization of these components would suggest that both synthesis and degradation occur within human sperm, and indeed significant evidence is emerging to confirm a functional and physiological role of ECS in the process of fertilization, as summarized in Figure 5b (for a recent review see ref. 137).
Several in vitro studies have shown a concentration-dependent inhibition of mammalian sperm functions by AEA, mediated by CB1 activation. One of these is inhibition of mitochondrial activity that preserves energy and ensures gradual acquisition of the fertilisation capacity during ascent up the female reproductive tract. Sperm fertilizing ability has been shown to be enhanced by the binding of AEA to CB1 that results in the induction of the acrosome reaction pivotal to the fertilization capacity of the sperm. More recently, also a CB2-dependent mechanism has been implicated in this process [138].
Quantification of eCB levels in seminal plasma from an infertility clinics showed AEA, PEA and OEA levels to be significantly lower in men with either asthenozoospermia or oligoasthenozoospermia, compared to normozoospemia [134, 136], and furthermore that supraphysiological levels of methanandamide (a non-hydrolyzable analogue of AEA) decreased sperm motility and viability through CB1-mediated inhibition of mitochondrial activity. Furthermore, PEA and OEA were shown to improve in vitro sperm motility and maintain viability without affecting mitochondrial activity [134]. Taken together, these findings suggest the maintenance of a normal eCB tone is most likely to be necessary for the preservation of normal sperm function, and hence of male fertility. There is indeed some evidence that PEA and OEA, which possess anti-inflammatory, antimicrobial and antioxidant activities, may protect sperm from oxidative damage, inflammation and microbial infections [134]. In men with idiopathic infertility it has been shown that supplementation with PEA and OEA enhances sperm antioxidant activity, and consequently improves sperm kinematics parameters and hyperactivation in vitro. Ultimately this protects sperm from oxidative damage, and could be beneficial in cases of unexplained male infertility [139].
Taken together, it can be concluded that several observations point to ECS involvement in the preservation of normal sperm function, and thus male fertility, and that abnormalities in this system may have implications for adverse reproductive consequences. This is a rapidly evolving domain, and in the future it could be that manipulation of ECS turns out to improve the fertility capacity of men with various forms of sperm dysfunction.
Skin
An increasing body of evidence suggests that ECS is a key player in the regulation of biological processes of the skin, the largest organ of the human body. As was reviewed previously [140], multiple cellular compartments of the skin (i.e., epidermal keratinocytes, hair follicles, sebaceous and sweat glands) produce prototypic eCBs. In addition, several elements of the ECS like CB1, CB2 and metabolic enzymes were identified on most cutaneous cell types; hence, ECS was implicated in a wide array of skin functions in all layers of the skin (summarized in Figure 6).
Figure 6.
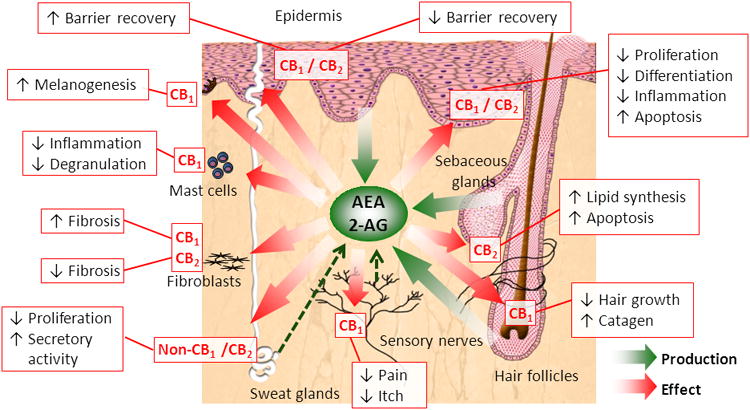
Summary of the effects of cutaneous ECS on multiple skin functions. Adapted from ref. 140.
In the epidermis, AEA was shown to inhibit proliferation and reduce differentiation of human epidermal keratinocytes via CB1-coupled signaling (reviewed in ref. 141). Moreover, CB1 also controls melanogenesis in epidermal melanocytes [142]. In addition, further highlighting the role of ECS in epidermal functions, in an animal model using gene-deficient mice, absence of CB1 delayed whereas lack of CB2 accelerated recovery of the epidermal permeability barrier [143], whose establishment is strongly dependent on keratinocyte proliferation and differentiation. Of further importance, CB agonists as well as phytocannabinoids were found to inhibit proliferation of hyperproliferative keratinocytes, whereas various cannabinoids exerted complex anti-tumor activity (anti-proliferative, pro-apoptotic and anti-angiogenic effects together with reduced metastasis formation) in mutiple models of melanoma and non-melanoma skin tumors (for a recent review see ref 140).
ECS is also involved in the biology of skin appendages. CB1-coupled signaling was shown to inhibit human hair shaft elongation and induce premature organ involution (catagen), whereas CB2 was found to “tonically” stimulate the homeostatic lipogenesis in human sebaceous gland-derived sebocytes [140]. Interestingly, AEA inhibited proliferation and induced secretory activity of sweat gland epithelial cells, via non-CB1/CB2-dependent mechanisms [144].
The pioneering mouse study of Meliha Karsak and colleagues [145] has clearly presented that augmentation of the cutaneous eCB tone, as well as certain phytocannabinoids, exerts remarkable anti-inflammatory activities [140]. This effect was recently proven also in human models. “Tonic” activation of CB1 was shown to keep maturation and degranulation of human skin mast cells under constitutive control, and the “pro-inflammatory” degranulation of mast cells was found to be prevented by AEA or CB1 agonists [146]. Of further importance, the prototypic non-psychoactive phytocannabinoid cannabidiol – by modulating a complex signaling network involving TRPV4 ion channels, adenosine A2a receptors, and multiple downstream elements (e.g., P65-NFκB pathway, tribbles homolog 3) – exerted dramatic sebostatic (i.e., suppression of unwanted lipogenesis) and anti-inflammatory effects in a human cellular/organ culture model of acne vulgaris, the most prevalent human skin disease [147].
Opposing roles of CB1 and CB2 were observed in dermal fibroblasts and various fibrosis models. FAAH was found to be down-regulated in dermal fibroblasts (SScDF) of systemic sclerosis (or scleroderma) patients, suggesting that the “eCB tone” might be elevated. Indeed, pharmacological or genetic abrogation of FAAH activity worsened bleomycin-induced fibrosis in mice via a CB1-coupled signaling [148]. Accordingly, CB1 null animals were found to be protected against the pro-fibrotic effects of bleomycin [149]. However, activation of CB2 expressed by leukocytes was able to ameliorate fibrosis in the same animal model; likewise, CB2 stimulation in SScDFs also resulted in significant anti-fibrotic effects [140, 150].
Collectively, the (highly selected) data presented above highlights that appropriate, targeted manipulation of cutaneous ECS and/or administration of certain phytocannabinoids point to completely new therapeutic opportunities in several cutaneous pathologies. Clinical trials are therefore urgently required to assess the putative in vivo efficiency of ECS-oriented treatments in e.g. hyperproliferative skin conditions (tumors, psoriasis), as well as in allergic and inflammatory skin diseases (acne vulgaris, atopic dermatitis). As yet, only PEA has been used as an effective therapeutic for these skin diseases (Table 2). Taken together, it can be concluded that: i) ECS is fundamentally involved is the regulation of a multitude of skin functions such as proliferation, differentiation, cell survival, and immune responses; ii) possibly the most robust function of the ECS is to suppress cutaneous inflammation; and iii) effects of eCBs are chiefly mediated by both CB1 and CB2 expressed by various skin cells.
Other organs
The impact of eCB signaling in other organs, such as respiratory tract and urinary system, still awaits better clarification through more systematic investigations. However, recent studies have proposed an important role of CB1 and CB2 in kidney disease. Indeed, CB1 antagonists (globally acting or peripherally restricted) and selective CB2 agonists protected against type 1 and/or type 2 diabetic nephropathy [151-153], and tubular nephropathy induced by cisplatin [154, 155], so that some of them hold therapeutic potential (Table 2). Such a protection was achieved by attenuating oxidative-nitrosative stress, inflammation, and consequent cell death in glomerular podocytes and/or proximal tubular epithelial cells [151-155].
On a final note, the impact of eCB signaling in the CNS appears immense, and to help to navigate the mare magnum of related data, comprehensive reviews have been recently published [14, 15].
Concluding remarks
To date, the therapeutic exploitation of ECS-based medicines appears still behind our scientific knowledge of eCB signaling, also because there are outstanding questions that still await for an answer (Box 1). A summary that points out the potential applications of ECS-related drugs for peripheral diseases is shown in Table 2. In this context, it should be recalled that peripherally restricted CB1 agonists failed in clinical trials of pain, owing to cardiovascular and metabolic side effects, and synthetic CB1 agonists are damaging to kidneys in young children, in addition to causing serious cardiovascular adverse effects. In keeping with this, over 50 case reports of the cardiovascular side effects of THC/marijuana alone have been reported so far. Additionally, global CB1 antagonist clinical trials were successful in diabetes and obesity, but failed because of unwanted side effects within the central nervous system. Interestingly, in rodent models of obesity and type-2 diabetes, peripherally restricted CB1 antagonists show therapeutic promise, as they retain the full metabolic efficacy of globally acting CB1 antagonists, but are devoid of their neurobehavioral effects. Furthermore, FAAH or MAGL inhibitors appear to be good in some preclinical models of pain, but they promote cardiovascular inflammation and cause metabolic undesirable consequences, and indeed already failed human trials in pain. Finally, there are other cannabis-derived phytocannabinoids with biological activity and therapeutic potential that we chose to exclude from this review due to space limitations. One of them, cannabidiol, is getting more and more approvals by Food and Drug Administration for orphan and other indications (e.g., refractory epilepsy and glioblastoma). Interestingly, a near 1:1 ratio of cannabidiol and THC (nabiximols, trade name Sativex) has beneficial effects for multiple sclerosis patients, who can use it to alleviate neuropathic pain, spasticity, overactive bladder, and other symptoms. Yet, the effects of Sativex cannot be reproduced by THC alone, suggesting that some or many of them could be due to cannabidiol, via molecular mechanisms yet to be disclosed, or to the fact that co-administration of cannabidiol can widen the therapeutic window of THC, thus allowing its administration at higher doses with less side effects. Since overwhelming evidence from preclinical studies also suggests that cannabidiol may be beneficial in myocardial infarction, stroke, diabetic cardiovascular complications, and even in nephropathy, investigations into its mechanism(s) of action appear urgent.
Box 1. Outstanding questions.
When will we have conclusive controlled clinical trials to evaluate the overall safety/toxicity of marijuana and synthetic cannabinoids?
Enhancement of eCB levels is seen in many disease states. Is indeed CB2 activation (and in some cases CB1 activation) a general protective mechanism? What is the role of localization and intracellular trafficking in driving distinct eCB signal cascades in distinct peripheral cells? To what extent can these factors determine efficacy of ECS-oriented therapies?
When will we have proof of the principle clinical trials to evaluate the therapeutic potential of peripherally restricted CB1 antagonists and CB2 agonists in cardiovascular disease? A clear assessment of the importance of the peripheral ECS (especially CB1) and of its therapeutic relevance in humans will have to await the results of clinical studies with novel, peripherally restricted ligands.
Many GI disorders are triggered or exacerbated by stress. How does stress alter the ECS in the gut? What is the balance between CB1 and CB2 activation in GI disorders? CB2 appears to be an important homeostatic regulator in the gut. How is it differentially regulated in GI diseases? How are the enzymes of eCB biosynthesis and degradation regulated in the gut in health and disease? This important question remains to be fully explored.
Do AEA and 2-AG as bioactive lipids act in opposition to each other, in order to maintain a fine-tuned balance in immune functional activities? Since CB2 is found preponderantly on immune cells, can it serve as a selective and specific molecular targets for ablating untoward pathological processes characterized by hyperinflammation?
Insight as to how activation of cannabinoid receptors alters immune functional activities has been obtained through the study of their ligation to phytocannabinoids. Yet, the latter substances and eCBs exhibit differential intracellular fates. How can the assessment of such differential fates provide insights into phytocannabinoid superimposed dysregulation of eCB-mediated activities?
What is the role of the ECS in genetically inherited muscle dystrophies, such as Duchenne's disease? What is the role of TRPV1 channels in eCB regulation of skeletal muscle formation?
What is the mechanism of eCB signaling in bone elongation? How is the hypertrophy of chondrocytes modulated by eCBs? In which processes are CB2 receptors involved?
Evidence shows that both silencing and amplification of eCB signaling can adversely impact female reproductive functions. Is the underlying mechanism the same or are different mechanisms operative? How may manipulation of the ECS influence fertility especially in those with idiopathic male infertility?
Is there a multilateral crosstalk between cutaneous ECS and the complex neuro-immuno-endocrine network (e.g., hormones, cytokines, growth factors, neuropeptides) of the skin?
Against this background, one should admit that the way towards a therapeutic exploitation of ECS-oriented drugs may not be quite close, also because of the apparent complexity of eCB signaling regulation. After 50 years since the discovery of THC, and nearly 20 years since that of AEA (1992) and 2-AG (1995), only recently it is emerging that the correct interaction between the different components of ECS is essential for a proper function of eCB as signaling molecules. For instance, distinct eCB-binding receptors can exchange signals to each other and with other receptors, and to do so they must be properly located on the plasma membrane and/or within the cell [15]. Another emerging concept (already demonstrated for AEA) is that eCBs are transported within the cell by distinct carriers, collectively called eCB intracellular transporters (EITs) [22, and references therein]. The need of a proper localization and the existence of an intracellular trafficking seem to add a new dimension to the already complex regulation of eCB signaling. Thus, in order to understand the many-faceted actions elicited by eCBs (and hence by eCB-related drugs), it appears now crucial to understand how after getting to the right place eCBs can be concentrated to suitable amounts for receptor activation at the right time. In this framework, after 50 years of cannabinoid/eCB research a re-examination of the widely accepted “dogma” issuing that eCBs are exclusively synthesized and released on demand could be proposed, whereby intracellular trafficking, in addition to storage in specific reservoirs like adiposomes [156], could play a key role in determining signal transduction triggered by the same eCB in different cellular contexts. In line with this concept, recent evidence based on pharmacological and genetic manipulation has demonstrated that different EITs may drive AEA signaling at different receptors, and/or AEA to be metabolized by different enzymes [157, 158]. Thus, compounds selectively directed at one or more of these novel EITs could lead to the development of intensely searched ECS-based therapeutics with limited side effects and abuse liability. Chances are that we can take advantage of these novel drugs in the near future.
Highlights.
50 years ago the psychoactive ingredient of Cannabis sativa, Δ9-tetrahydrocannabinol (THC), was isolated.
We critically review current status of endocannabinnoids (eCB) functions in peripheral organs.
We discuss the relevance of the peripheral eCB system for human health and disease pathogenesis
We highlight emerging challenges and therapeutic hopes.
Acknowledgments
MM thanks Professor Alessandro Finazzi Agrò (Campus Bio-Medico University of Rome, Italy) for his continuing interest and support, and Dr. Monica Bari (Tor Vergata University of Rome, Italy) for the male fertility artwork. GC thanks Dr. Melissa Jamerson (Virginia Commonwealth University, Richmond, USA) for her help in writing the immune chapter, and TB thanks Dr. Attila Oláh (University of Debrecen, Hungary) for his help in organizing the skin chapter. MM was supported by the Italian Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN 2010-2011 grant), KAS by the Canadian Institutes of Health Research, SKD by the National Institutes of Health (NIH/NIDA DA06668 grant), and AZ by the Deutsche Forschungsgemeinschaft (Cluster of Excellence “Immunosensation”, FOR926).
Dedication: This paper is dedicated to our dear colleague and friend Itai Bab, sorrowfully missed last October 2014.
Glossary
- Acne vulgaris
one of the commonest human skin diseases, characterized by a pathologically elevated and qualitatively altered sebum production (seborrhoea) and by inflammation of the sebaceous glands (and of the skin in general).
- Acrosome reaction
changes in the sperm head that occur with contact with the egg, allowing it to penetrate and then fertilize the egg.
- Advanced glycation end products
substances that can contribute to development or worsening of many human degenerative diseases.
- Anandamide (AEA)
the amide derivative of arachidonic acid with ethanolamine (N-arachidonoyl-ethanolamine), named after the Sanskrit word for bliss, “ananda”.
- Antigen presentation
an adaptive immune response whereby antigen-specific receptors recognize processed antigens in the form of peptides.
- Arachidonic acid
common name of 5,8,11,14-eicosatetraenoic acid, an essential (diet-derived) ω-6 fatty acid that serves as precursor for eicosanoids and endocannabinoids.
- Asthenozoospermia
reduced sperm motility and sperm count.
- Atherosclerosis
building up of fats, cholesterol and other substances (plaques) in and on artery wall, which can restrict blood flow.
- Autocrine
related to a substance secreted by a cell and acting on surface receptors of the same cell.
- B lymphocyte
an immune cell that makes antibodies against antigens, acts as an antigen-presenting cell, and develops into a memory cell after activation by antigen.
- Blastocyst
an embryonic stage in mammals and is comprised of a fluid-filled cavity (blastocoel) and two cell types – inner cell mass and trophectoderm.
- Cannabinoid receptors (CBs)
G protein-coupled receptors that bind THC, as well as AEA, 2-AG and other endocannabinoids.
- Cannabinoids
natural lipophilic products found in Cannabis sativa extracts (e.g., hashish or marijuana). Among >60 cannabinoids present in this plant, which are all characterized by a terpeno-phenol bicyclic or tricyclic structure, the best known compound is Δ9-tetrahydrocannabinol (THC).
- Catagen
the cessation phase of hair production, that includes also growth (anagen) and rest (telogen) phases.
- Celiac disease
a gastrointestinal autoimmune disorder that results in severe damage to the mucosal lining of the small intestine when foods with gluten are eaten, causing malabsorption, bloating, pain and diarrhea.
- Chemokine
a class of cytokines that functions to attract white blood cells (leukocytes) such as lymphocytes, granulocytes, monocytes, and macrophages to sites of infection.
- Chemotaxis
is the movement of an organism in response to a chemical stimulus.
- Cholecystokinin
a peptide released from “I”-type enteroendocrine cells that are predominantly found in the proximal small intestine of the gastrointestinal tract. Cholecystokinin regulates food intake, and coordinates the digestion of fat and protein.
- Chondrocyte
cartilage cells derived from mesenchymal stem cells that produce extracellular matrix components of cartilage tissue.
- Chondroclast
multinucleated cartilage resorbing cells.
- Cytokine
a category of small proteins (5-20 kDa) that are released by immune cells that play an important role in cell signaling affecting the behavior of other cells; cytokines include chemokines, interferons, and lymphokines.
- Cumulus oophorus
cluster of cells that surround the oocyte in the ovarian follicle.
- Dendritic cell
antigen-presenting cells capable of activating T lymphocytes and stimulating the growth and differentiation of B lymphocytes.
- Embryo development
development of the fertilized egg to the blastocyst stage through several rounds of mitosis.
- Endocannabinoids
family of lipid messengers that behave as endogenous agonists of CBs in animals. AEA and 2-AG are the best characterized compounds described to date, and represent prototype members of fatty acid amides and monoacylglycerols, respectively.
- Endometrium
the inner lining of the uterus and consists of stromal and epithelial cells.
- Endotoxemia
the presence of endotoxins in the blood, which may result in shock.
- Enteric nervous system
The enteric nervous system is the third division of the autonomic nervous system. It consists of nerve cell bodies arranged in two ganglionated plexuses (the myenteric and submucosal plexuses) and an extensive network of nerve fibers that lie in the wall of the gastrointestinal tract. It provides local neural control of the gastrointestinal tract and is required for the coordination of digestive and defensive functions of the gut.
- Enterocyte
absorptive epithelial cells that line the lumen of the gastrointestinal tract.
- Enteroendocrine cells
specialized hormone producing epithelial cells of the gastrointestinal tract and pancreas. Enteroendocrine cells of the intestine are the most numerous endocrine cells of the body.
- Epidermal barrier
the barrier – consisting of physico-chemical, immunological and microbial aspects – formed chiefly by the outermost (i.e., epidermal) layer of the skin, that aims at protecting the body against various external challenges and at maintaining cutaneous homeostasis.
- Fatty acid amide hydrolase (FAAH)
endomembrane-bound serine hydrolase that cleaves AEA into arachidonic acid and ethanolamine.
- Fibrosis
excessive formation of connective tissue, due to the over-activity of dermal fibroblasts.
- Idiopathic infertility
infertility for which there is no obvious cause.
- Implantation site
the site of embryo attachment in the uterus (implantation chamber).
- Inflammatory bowel disease
a group of chronic inflammatory conditions of the gastrointestinal tract that are characterized by abdominal pain, diarrhea, rectal bleeding, severe internal cramping and weight loss. The two main forms of inflammatory bowel disease are Crohn's disease and ulcerative colitis.
- Inotropy
alteration of the force or energy of muscular contractions.
- Interleukin
a class of glycoproteins that is produced by leukocytes for regulating immune responses.
- Lipogenesis
the process by which acetyl-CoA is converted to fatty acids.
- Macrophage
a type of leukocyte found in tissues that engulfs and digests cell debris, foreign substances, microbes, and cancer cells; macrophages process foreign antigens into peptides and present them to T lymphocytes.
- Microglia
resident macrophages of the brain and spinal cord.
- Monocyte
a type of leukocyte that replenishes resident macrophages and moves to localized sites of infection in tissues where they divide into macrophages and dendritic cells to elicit an immune response.
- Muscular dystrophy
a pathological condition that weakens the musculoskeletal system thereby impairing locomotion. Muscular dystrophies of genetic origin are often due to defective skeletal muscle formation and characterized by progressive skeletal muscle weakness, defects in muscle proteins, muscle inflammation and death of muscle cells and tissues.
- Muscular hypertrophy
a condition that involves an increase in size of skeletal muscle through an increase in the size of its component cells, often as a consequence of both sarcoplasmic hypertrophy, with increased muscle glycogen storage, and myofibrillar hypertrophy, with increased myofibril size.
- Natural killer (NK) cell
a lymphocyte that binds to select tumor cells and virus-infected cells with antigen stimulation; NK cells kill target cells by inserting granules containing perforin.
- Neutrophil
a type of leukocyte that constitutes one of the first responders to inflammation, particularly due to bacterial infection; neutrophils are a type of phagocyte normally found in the bloodstream.
- (Non) receptive uterus
the uterus which is (in) capable for supporting blastocyst growth and attachment.
- Normozoospemia
normal sperm count, morphology and motility.
- Oligoasthenozoospermia
reduced sperm motility and count.
- Oligozoospermia
reduced sperm count.
- Osteoblast
bone-forming cells derived from mesenchymal stem cells.
- Osteoclast
multinucleated myeloid cells resorbing bone.
- Osteoporosis
a progressive degenerative bone disorder, characterized by reduced bone mass and a concomitantly increased fracture risk.
- Ovariectomy
surgical removal of the ovaries.
- Oviductal embryo transport
the journey of fertilized eggs through the oviduct and ultimately into the uterus.
- Paracrine
related to a substance secreted by a cell and acting on surface receptors of nearby cells.
- Parturition
the action or process of giving birth to offspring.
- Perforin
a protein released by NK cells that destroys target cells by inducing pores in their membranes.
- Peroxisome proliferator-activated receptors (PPARs)
superfamily of nuclear receptor transcription factors, that include three members: α, γ and δ. PPAR-α and -γ are targets of AEA and its congeners.
- Placentation
role of the placenta which is comprised of trophoblast cells and establish vascular connection between the conceptus and the mother.
- Polymorphism
natural variations in a DNA sequence that have been identified in a population.
- Restenosis
literally means the recurrence of stenosis, a narrowing of a blood vessel, leading to restricted blood flow.
- Sperm kinematics parameters
these refer to the parameters of movement in a forward direction and at an acceptable pace.
- Steatosis
the process that describes the abnormal retention of lipids within a cell.
- Sympathetic and parasympathetic projections
nerve endings of the autonomic nervous system originating from cranial or sacral nerves belonging to the sympathetic nervous system or the parasympathetic nervous system.
- Systemic sclerosis (also known as “scleroderma”)
a systemic autoimmune disease, which can be characterized by the excessive accumulation of collagen fibers in the skin and in the internal organs.
- T lymphocyte
immune cells that can directly kill infected cells or cancer cells; these cells assist in B lymphocyte maturation and produce cytokines.
- Tight junction proteins
families of proteins, including occludins, zonula occludens and claudins, which together form an intercellular tight junction between adjacent gastrointestinal epithelial cells. The tight junction lies just beneath the apical surface and consists of interlinked rows of integral membrane proteins, limiting the passage of water and other molecules.
- Transient receptor potential vanilloid-1 (TRPV1)
a six-transmembrane receptor-channel that is activated by physical and chemical stimuli, as well as by AEA.
- Trophectoderm
the outer layer of the blastocyst and is the progenitor of future placenta.
- Δ9-Tetrahydrocannabinol (THC)
psychoactive ingredient of cannabis (Cannabis sativa) extracts like hashish and marijuana.
- Zona pellucida
glycoprotein layer surrounding the plasma membrane of mammalian oocytes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. J Amer Chem Soc. 1964;86:1646–1647. [Google Scholar]
- 2.Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 1984;26:532–538. [PubMed] [Google Scholar]
- 3.Mechoulam R, et al. Enantiomeric cannabinoids: stereospecificity of psychotropic activity. Experientia. 1988;44:762–764. doi: 10.1007/BF01959156. [DOI] [PubMed] [Google Scholar]
- 4.Devane WA, et al. Determination and characterization of a cannabinoireceptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 5.Matsuda LA, et al. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 6.Munro S, et al. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 7.Devane WA, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 8.Mechoulam R, et al. Identification of an endogenous 2 monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 9.Sugiura T, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 10.Tan B, et al. Targeted lipidomics: discovery of new fatty acyl amides. AAPS J. 2006;8:E461–E465. doi: 10.1208/aapsj080354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanus LE, et al. N-Acyl amino acids and their impact on biological processes. BioFactors. 2014;40:381–388. doi: 10.1002/biof.1166. [DOI] [PubMed] [Google Scholar]
- 12.Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 13.Tantimonaco M, et al. Physical activity and the endocannabinoid system: an overview. Cell Mol Life Sci. 2014;71:2681–2698. doi: 10.1007/s00018-014-1575-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mechoulam R, et al. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nature Rev Neurosci. 2014;15:757–776. doi: 10.1038/nrn3811. [DOI] [PubMed] [Google Scholar]
- 15.Maccarrone M, et al. Programming of neural cells by (endo) cannabinoids: from physiological rules to emerging therapies. Nat Rev Neurosci. 2014;15:786–801. doi: 10.1038/nrn3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International Union of Basic and Clinical Pharmacology. LXXIX Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiPatrizio NV, Piomelli D. The thrifty lipids: endocannabinoids and the neural control of energy conservation. Trends Neurosci. 2012;35:403–411. doi: 10.1016/j.tins.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galve-Roperh I, et al. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res. 2013;52:633–650. doi: 10.1016/j.plipres.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Ross RA. The enigmatic pharmacology of GPR55. Trends Pharmacol Sci. 2009;30:156–163. doi: 10.1016/j.tips.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Di Marzo V, De Petrocellis L. Endocannabinoids as regulators of transient receptor potential (TRP) channels: A further opportunity to develop new endocannabinoid-based therapeutic drugs. Curr Med Chem. 2010;17:1430–1449. doi: 10.2174/092986710790980078. [DOI] [PubMed] [Google Scholar]
- 21.Pistis M, Melis M. From surface to nuclear receptors: the endocannabinoid family extends its assets. Curr Med Chem. 2010;17:1450–1467. doi: 10.2174/092986710790980014. [DOI] [PubMed] [Google Scholar]
- 22.Maccarrone M, et al. Intracellular trafficking of anandamide: new concepts for signaling. Trends Biochem Sci. 2010;35:601–608. doi: 10.1016/j.tibs.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Rahman IA, et al. New players in the fatty acyl ethanolamide metabolism. Pharmacol Res. 2014;86:1–10. doi: 10.1016/j.phrs.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Cravatt BF, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Marzo V, Maccarrone M. FAAH and anandamide: is 2-AG really the odd one out? Trends Pharmacol Sci. 2008;29:229–233. doi: 10.1016/j.tips.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 26.Fezza F, et al. Endocannabinoids, related compounds and their metabolic routes. Molecules. 2014;19:17078–17106. doi: 10.3390/molecules191117078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pacher P, et al. Theendocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steffens S, Pacher P. Targeting cannabinoid receptor CB2 in cardiovascular disorders: promises and controversies. Br J Pharmacol. 2012;167:313–323. doi: 10.1111/j.1476-5381.2012.02042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steffens S, Pacher P. The activated endocannabinoid system in atherosclerosis: driving force or protective mechanism? Curr Drug Targets. 2014 doi: 10.2174/1389450115666141202113225. in press. [DOI] [PubMed] [Google Scholar]
- 30.Rajesh M, et al. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br J Pharmacol. 2010;160:688–700. doi: 10.1111/j.1476-5381.2010.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhopadhyay P, et al. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc Res. 2010;85:773–784. doi: 10.1093/cvr/cvp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajesh M, et al. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes. 2012;61:716–727. doi: 10.2337/db11-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slavic S, et al. Cannabinoid receptor 1 inhibition improves cardiac function and remodelling after myocardial infarction and in experimental metabolic syndrome. J Mol Med. 2013;91:811–823. doi: 10.1007/s00109-013-1034-0. [DOI] [PubMed] [Google Scholar]
- 34.Schaich CL, et al. Acute and chronic systemic CB1 cannabinoid receptor blockade improves blood pressure regulation and metabolic profile in hypertensive (mRen2) 27 rats. Physiol Rep. 2014;2 doi: 10.14814/phy2.12108. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease--successes and failures. FEBS J. 2013;280:1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jouanjus E, et al. Cannabis use: signal of increasing risk of serious cardiovascular disorders. J Am Heart Assoc. 2014;3:e000638. doi: 10.1161/JAHA.113.000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126:21–38. doi: 10.1016/j.pharmthera.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Hons IM, et al. Plasticity of mouse enteric synapses mediated through endocannabinoid and purinergic signaling. Neurogastroenterol Motil. 2012;24:e113–124. doi: 10.1111/j.1365-2982.2011.01860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sykaras AG, et al. Duodenal enteroendocrine I-cells contain mRNA transcripts encoding key endocannabinoid and fatty acid receptors. PLoS One. 2012;7:e42373. doi: 10.1371/journal.pone.0042373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muccioli GG, et al. The endocannabinoid system links gut microbiota to adipogenesis. Mol Syst Biol. 2010;6:392. doi: 10.1038/msb.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piomelli D. A fatty gut feeling. Trends Endocrinol Metab. 2013;24:332–341. doi: 10.1016/j.tem.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tellez LA, et al. A gut lipid messenger links excess dietary fat to dopamine deficiency. Science. 2013;341:800–802. doi: 10.1126/science.1239275. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz GJ, et al. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. 2008;8:281–288. doi: 10.1016/j.cmet.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duncan M, et al. Cannabinoid CB2 receptors in the enteric nervous system modulate gastrointestinal contractility in lipopolysaccharide-treated rats. Am J Physiol Gastrointest Liver Physiol. 2008;295:G78–G87. doi: 10.1152/ajpgi.90285.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bashashati M, et al. Inhibiting fatty acid amide hydrolase normalizes endotoxin-induced enhanced gastrointestinal motility in mice. Br J Pharmacol. 2012;165:1556–1571. doi: 10.1111/j.1476-5381.2011.01644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fichna J, et al. Cannabinoids alleviate experimentally induced intestinal inflammation by acting at central and peripheral receptors. PLoS One. 2014;9:e109115. doi: 10.1371/journal.pone.0109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kinsey SG, et al. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Battista N, et al. Altered expression of type-1 and type-2 cannabinoid receptors in celiac disease. PLoS One. 2013;8:e62078. doi: 10.1371/journal.pone.0062078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D'Argenio G, et al. Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB J. 2006;20:568–570. doi: 10.1096/fj.05-4943fje. [DOI] [PubMed] [Google Scholar]
- 50.Kunos G, et al. Should peripheral CB1 cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol Sci. 2009;30:1–7. doi: 10.1016/j.tips.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Naftali T, et al. Cannabis induces a clinical response in patients with Crohn's disease: a prospective placebo-controlled study. Clin Gastroenterol Hepatol. 2013;11:1276–1280 e1271. doi: 10.1016/j.cgh.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 52.Storr M, et al. Cannabis use provides symptom relief in patients with inflammatory bowel disease but is associated with worse disease prognosis in patients with Crohn's disease. Inflamm Bowel Dis. 2014;20:472–480. doi: 10.1097/01.MIB.0000440982.79036.d6. [DOI] [PubMed] [Google Scholar]
- 53.Osei-Hyiaman D, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu HM, et al. Rimonabant, a cannabinoid receptor type 1 inverse agonist, inhibits hepatocyte lipogenesis by activating liver kinase B1 and AMP-activated protein kinase axis downstream of Galpha i/o inhibition. Mol Pharmacol. 2011;80:859–869. doi: 10.1124/mol.111.072769. [DOI] [PubMed] [Google Scholar]
- 55.Jourdan T, et al. Antagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: evidence from cultured explants. Hepatology. 2012;55:790–799. doi: 10.1002/hep.24733. [DOI] [PubMed] [Google Scholar]
- 56.Osei-Hyiaman D, et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118:3160–3169. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, et al. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology. 2012;142:1218–1228. doi: 10.1053/j.gastro.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chanda D, et al. Activation of cannabinoid receptor type 1 (Cb1r) disrupts hepatic insulin receptor signaling via cyclic AMP-response element-binding protein H (Crebh)-mediated induction of Lipin1 gene. J Biol Chem. 2012;287:38041–38049. doi: 10.1074/jbc.M112.377978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeong WI, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 60.Jourdan T, et al. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese mice. Diabetes. 2010;59:926–934. doi: 10.2337/db09-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tam J, et al. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–179. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teixeira-Clerc F, et al. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 63.Batkai S, et al. Endocannabinoids acting at vascular CB1 receptors mediate the vasodilated state in advanced liver cirrhosis. Nat Med. 2001;7(7):827–832. doi: 10.1038/89953. [DOI] [PubMed] [Google Scholar]
- 64.Julien B, et al. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology. 2005;128:742–755. doi: 10.1053/j.gastro.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 65.Mallat A, et al. The endocannabinoid system as a key mediator during liver diseases: new insights and therapeutic openings. Br J Pharmacol. 2011;163:1432–1440. doi: 10.1111/j.1476-5381.2011.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Munoz-Luque J, et al. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther. 2008;324:475–483. doi: 10.1124/jpet.107.131896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guillot A, et al. Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology. 2014;59:296–306. doi: 10.1002/hep.26598. [DOI] [PubMed] [Google Scholar]
- 68.Louvet A, et al. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology. 2011;54:1217–1226. doi: 10.1002/hep.24524. [DOI] [PubMed] [Google Scholar]
- 69.Batkai S, et al. Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J. 2007;21:1788–1800. doi: 10.1096/fj.06-7451com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao Z, et al. Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology. 2013;144:808–817 e815. doi: 10.1053/j.gastro.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mallat A, et al. Environmental factors as disease accelerators during chronic hepatitis C. J Hepatol. 2008;48:657–665. doi: 10.1016/j.jhep.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 72.Tanasescu R, Constantinescu CS. Cannabinoids and the immune system: an overview. Immunobiol. 2010;215:588–597. doi: 10.1016/j.imbio.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 73.Chiurchiu V, et al. Endocannabinoid signalling in innate and adaptive immunity. Immunology. 2015 doi: 10.1111/imm.12441. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hasko J, et al. CB2 receptor activation inhibits melanoma cell transmigration through the blood-brain barrier. Int J Mol Sci. 2014;15:8063–8074. doi: 10.3390/ijms15058063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu YJ, et al. Cannabinoid receptor 2 suppresses leukocyte inflammatory migration by modulating the JNK/c-Jun/Alox5 pathway. J Biol Chem. 2013;288:13551–13562. doi: 10.1074/jbc.M113.453811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sugiura T, et al. Evidence that 2-arachidonoylglycerol but not N-palmitoyl-ethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J Biol Chem. 2000;275:605–612. doi: 10.1074/jbc.275.1.605. [DOI] [PubMed] [Google Scholar]
- 77.Berdyshev EV, et al. Influence of fatty acid ethanolamides and delta9-tetrahydrocannabinol on cytokine and arachidonate release by mononuclear cells. Eur J Pharmacol. 1997;330:231–240. doi: 10.1016/s0014-2999(97)01007-8. [DOI] [PubMed] [Google Scholar]
- 78.Cencioni MT, et al. Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS ONE. 2010;5:e8688. doi: 10.1371/journal.pone.0008688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Derocq JM, et al. The endogenous cannabinoid anandamide is a lipid messenger activating cell growth via a cannabinoid receptor-independent pathway in hematopoietic cell lines. FEBS Lett. 1998;425:419–425. doi: 10.1016/s0014-5793(98)00275-0. [DOI] [PubMed] [Google Scholar]
- 80.Facci L, et al. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci USA. 1995;92:3376–3380. doi: 10.1073/pnas.92.8.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Puffenbarger RA, et al. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia. 2000;29:58–69. [PubMed] [Google Scholar]
- 82.Cabral GA, Staab A. Effects on the immune system. Handb Exp Pharmacol. 2005;168:385–423. doi: 10.1007/3-540-26573-2_13. [DOI] [PubMed] [Google Scholar]
- 83.Cavuoto P, et al. Effects of cannabinoid receptors on skeletal muscle oxidative pathways. Mol Cell Endocrinol. 2007;267:63–69. doi: 10.1016/j.mce.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 84.Cavuoto P, et al. The expression of receptors for endocannabinoids in human and rodent skeletal muscle. Biochem Biophys Res Commun. 2007;364:105–110. doi: 10.1016/j.bbrc.2007.09.099. [DOI] [PubMed] [Google Scholar]
- 85.Crespillo A, et al. Expression of the cannabinoid system in muscle: effects of a high-fat diet and CB1 receptor blockade. Biochem J. 2011;433:175–185. doi: 10.1042/BJ20100751. [DOI] [PubMed] [Google Scholar]
- 86.Eckardt K, et al. Cannabinoid type 1 receptors in human skeletal muscle cells participate in the negative crosstalk between fat and muscle. Diabetologia. 2009;52:664–674. doi: 10.1007/s00125-008-1240-4. [DOI] [PubMed] [Google Scholar]
- 87.Esposito I, et al. The cannabinoid CB1 receptor antagonist rimonabant stimulates 2-deoxyglucose uptake in skeletal muscle cells by regulating the expression of phosphatidylinositol-3-kinase. Mol Pharmacol. 2008;74:1678–1686. doi: 10.1124/mol.108.049205. [DOI] [PubMed] [Google Scholar]
- 88.Lipina C, et al. Regulation of MAP kinase-directed mitogenic and protein kinase B-mediated signaling by cannabinoid receptor type 1 in skeletal muscle cells. Diabetes. 2010;59:375–385. doi: 10.2337/db09-0979. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Lindborg KA, et al. Effects of in vitro antagonism of endocannabinoid-1 receptors on the glucose transport system in normal and insulin-resistant rat skeletal muscle. Diabetes Obes Metab. 2010;12:722–730. doi: 10.1111/j.1463-1326.2010.01227.x. [DOI] [PubMed] [Google Scholar]
- 90.Iannotti FA, et al. Analysis of the “endocannabinoidome” in peripheral tissues of obese Zucker rats. Prostaglandins Leukot Essent Fatty Acids. 2013;89:127–135. doi: 10.1016/j.plefa.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 91.Pagotto U, et al. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27:73–100. doi: 10.1210/er.2005-0009. [DOI] [PubMed] [Google Scholar]
- 92.Matias I, et al. Dysregulation of peripheral endocannabinoid levels in hyperglycemia and obesity: Effect of high fat diets. Mol Cell Endocrinol. 2008;286:S66–S78. doi: 10.1016/j.mce.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 93.Addy C, et al. The acyclic CB1R inverse agonist taranabant mediates weight loss by increasing energy expenditure and decreasing caloric intake. Cell Metab. 2008;7:68–78. doi: 10.1016/j.cmet.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 94.Herling AW, et al. Increased energy expenditure contributes more to the body weight-reducing effect of rimonabant than reduced food intake in candy-fed wistar rats. Endocrinology. 2008;149:2557–2566. doi: 10.1210/en.2007-1515. [DOI] [PubMed] [Google Scholar]
- 95.Kunz I, et al. Effects of rimonabant, a cannabinoid CB1 receptor ligand, on energy expenditure in lean rats. Int J Obes. 2008;32:863–870. doi: 10.1038/ijo.2008.3. [DOI] [PubMed] [Google Scholar]
- 96.Liu YL, et al. Effects of the cannabinoid CB1 receptor antagonist SR141716 on oxygen consumption and soleus muscle glucose uptake in Lep (ob)/Lep (ob) mice. Int J Obes. 2005;29:183–187. doi: 10.1038/sj.ijo.0802847. [DOI] [PubMed] [Google Scholar]
- 97.Tedesco L, et al. Cannabinoid receptor stimulation impairs mitochondrial biogenesis in mouse white adipose tissue, muscle, and liver: the role of eNOS, p38 MAPK, and AMPK pathways. Diabetes. 2010;59:2826–2836. doi: 10.2337/db09-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lindborg KA, et al. Effects of chronic antagonism of endocannabinoid-1 receptors on glucose tolerance and insulin action in skeletal muscles of lean and obese Zucker rats. Cardiorenal Med. 2011;1:31–44. doi: 10.1159/000322826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song D, et al. Acute cannabinoid receptor type 1 (CB1R) modulation influences insulin sensitivity by an effect outside the central nervous system in mice. Diabetologia. 2011;54:1181–1189. doi: 10.1007/s00125-011-2082-z. [DOI] [PubMed] [Google Scholar]
- 100.Iannotti FA, et al. The endocannabinoid 2-AG controls skeletal muscle cell differentiation via CB1 receptor-dependent inhibition of Kv7 channels. Proc Natl Acad Sci USA. 2014;117:2472–2481. doi: 10.1073/pnas.1406728111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luo Z, et al. TRPV1 activation improves exercise endurance and energy metabolism through PGC-1a upregulation in mice. Cell Res. 2012;22:551–564. doi: 10.1038/cr.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ito N, et al. Capsaicin mimics mechanical load-induced intracellular signaling events: involvement of TRPV1-mediated calcium signaling in induction of skeletal muscle hypertrophy. Channels. 2013;7:221–224. doi: 10.4161/chan.24583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bab I, Zimmer A. Cannabinoid receptors and the regulation of bone mass. Br J Pharmacol. 2008;153:182–188. doi: 10.1038/sj.bjp.0707593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bab I, et al. Endocannabinoids and the regulation of bone metabolism. J Neuroendocrinol. 2008;20:69–74. doi: 10.1111/j.1365-2826.2008.01675.x. [DOI] [PubMed] [Google Scholar]
- 105.Bab I. Regulation of skeletal remodeling by the endocannabinoid system. Ann NY Acad Sci. 2007;1116:414–422. doi: 10.1196/annals.1402.014. [DOI] [PubMed] [Google Scholar]
- 106.Bab I, et al. Cannabinoids and the skeleton: from marijuana a treversal of bone loss. Ann Med. 2009;41:560–567. doi: 10.1080/07853890903121025. [DOI] [PubMed] [Google Scholar]
- 107.Idris AI, Ralston SH. Role of cannabinoids in the regulation of bone remodeling. Front Endocrinol. 2012;3:136. doi: 10.3389/fendo.2012.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Idris AI, et al. Cannabinoid receptor type 1 protects against age-related osteoporosis by regulating osteoblast and adipocyte differentiation in marrow stromal cells. Cell Metab. 2009;10:139–147. doi: 10.1016/j.cmet.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 109.Idris AI, et al. Regulation of bone mass, osteoclast function, and ovariectomy-induced bone loss by the type 2 cannabinoid receptor. Endocrinology. 2008;149:5619–5626. doi: 10.1210/en.2008-0150. [DOI] [PubMed] [Google Scholar]
- 110.Idris AI, et al. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat Med. 2005;11:774–779. doi: 10.1038/nm1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ko JY, et al. Cannabinoid receptor 1 mediates glucocorticoid-induced bone loss in rats by perturbing bone mineral acquisition and marrow adipogenesis. Arthritis Rheum. 2012;64:1204–1214. doi: 10.1002/art.33457. [DOI] [PubMed] [Google Scholar]
- 112.Ofek O, et al. CB2 cannabinoid receptor targets mitogenic Gi protein-cyclin D1 axis in osteoblasts. J Bone Miner Res. 2011;26:308–316. doi: 10.1002/jbmr.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ofek O, et al. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA. 2006;103:696–701. doi: 10.1073/pnas.0504187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tam J, et al. Involvement of neuronal cannabinoid receptor CB1 in regulation of bone mass and bone remodeling. Mol Pharmacol. 2006;70:786–792. doi: 10.1124/mol.106.026435. [DOI] [PubMed] [Google Scholar]
- 115.Tam J, et al. The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J. 2008;22:285–294. doi: 10.1096/fj.06-7957com. [DOI] [PubMed] [Google Scholar]
- 116.Rossi F, et al. The genetic ablation or pharmacological inhibition of TRPV1 signalling is beneficial for the restoration of quiescent osteoclast activity in ovariectomized mice. Br J Pharmacol. 2014;171:2621–2630. doi: 10.1111/bph.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Karsak M, et al. The cannabinoid receptor type 2 (CNR2) gene is associated with hand bone strength phenotypes in an ethnically homogeneous family sample. Hum Genet. 2009;126:629–636. doi: 10.1007/s00439-009-0708-8. [DOI] [PubMed] [Google Scholar]
- 118.Yamada Y, et al. Association of candidate gene polymorphisms with bone mineral density in community-dwelling Japanese women and men. Int J Mol Med. 2007;19:791–801. [PubMed] [Google Scholar]
- 119.Elefteriou F, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–520. doi: 10.1038/nature03398. [DOI] [PubMed] [Google Scholar]
- 120.Yirmiya R, et al. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci USA. 2006;103:16876–16881. doi: 10.1073/pnas.0604234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bajayo A, et al. Skeletal parasympathetic innervations communicate central IL-1 signals regulating bone mass accrual. Proc Natl Acad Sci USA. 2012;109:15455–15460. doi: 10.1073/pnas.1206061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wasserman E, et al. CB1 cannabinoid receptors mediate endochondral skeletal growth attenuation by Δ9-tetrahydrocannabinol. Marrow. 2014 doi: 10.1111/nyas.12642. in press. [DOI] [PubMed] [Google Scholar]
- 123.Marroun EH, et al. Intrauterine cannabis exposure affects fetal growth trajectories: the Generation R Study. J Am Acad Child Adolesc Psychiatry. 2009;48:1173–1181. doi: 10.1097/CHI.0b013e3181bfa8ee. [DOI] [PubMed] [Google Scholar]
- 124.Zuckerman B, et al. Effects of maternal marijuana and cocaine use on fetal growth. N Engl J Med. 1989;320:762–768. doi: 10.1056/NEJM198903233201203. [DOI] [PubMed] [Google Scholar]
- 125.Wang H, et al. Jekyll and hyde: two faces of cannabinoid signaling in male and female fertility. Endocrine Rev. 2006;27:427–448. doi: 10.1210/er.2006-0006. [DOI] [PubMed] [Google Scholar]
- 126.Horne AW, et al. CB1 expression is attenuated in Fallopian tube and decidua of women with ectopic pregnancy. PLoS ONE. 2008;3:e3969. doi: 10.1371/journal.pone.0003969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang H, et al. Aberrant cannabinoid signaling impairs oviductal transport of embryos. Nat Med. 2004;10:1074–1080. doi: 10.1038/nm1104. [DOI] [PubMed] [Google Scholar]
- 128.Wang H, et al. Fatty acid amide hydrolase deficiency limits early pregnancy events. J Clin Invest. 2006;116:2122–2131. doi: 10.1172/JCI28621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sun X, et al. Endocannabinoid signaling directs differentiation of trophoblast cell lineages and placentation. Proc Nat Acad Sci USA. 2010;107:16887–16892. doi: 10.1073/pnas.1010892107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xie H, et al. Silencing or amplification of endocannabinoid signaling in blastocysts via CB1 compromises trophoblast cell migration. J Biol Chem. 2012;287:32288–32297. doi: 10.1074/jbc.M112.381145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maccarrone M, et al. Relation between decreased anandamide hydrolase concentrations in human lymphocytes and miscarriage. Lancet. 2000;355:1326–1329. doi: 10.1016/S0140-6736(00)02115-2. [DOI] [PubMed] [Google Scholar]
- 132.Chamley LW, et al. Nuclear localisation of the endocannabinoid metabolizing enzyme fatty acid amide hydrolase (FAAH) in invasive trophoblasts and an association with recurrent miscarriage. Placenta. 2008;29:970–975. doi: 10.1016/j.placenta.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 133.Gebeh AK, et al. Ectopic pregnancy is associated with high anandamide levels and aberrant expression of FAAH and CB1 in fallopian tubes. J Clin Endocrinol Metab. 2012;97:2827–2835. doi: 10.1210/jc.2012-1780. [DOI] [PubMed] [Google Scholar]
- 134.Amoako AA, et al. Relationship between seminal plasma levels of anandamide congeners palmitoylethanolamide and oleolyethanolamide and semen quality. Fertil Steril. 2014;102:1260–1267. doi: 10.1016/j.fertnstert.2014.07.767. [DOI] [PubMed] [Google Scholar]
- 135.Francavilla F, et al. Characterisation of the endocannabinoid system in human spermatozoa and involvement of the transient receptor potential vanilloid 1 receptor in their fertilization ability. Endocrinology. 2009;150:4692–4700. doi: 10.1210/en.2009-0057. [DOI] [PubMed] [Google Scholar]
- 136.Amoako AA, et al. Anandamide modulates human sperm motility: implications for men with asthenozoosperma and oligoasthenozoosperma. Hum Reprod. 2013;28:2058–2066. doi: 10.1093/humrep/det232. [DOI] [PubMed] [Google Scholar]
- 137.Rapino C, et al. Endocannabinoid as biomarkers of human reproduction. Hum Reprod Update. 2014;20:501–516. doi: 10.1093/humupd/dmu004. [DOI] [PubMed] [Google Scholar]
- 138.Meccariello R, et al. Update in reproduction coming from the endocannabinoid system. Int J Endocrinol. 2014;2014:412354. doi: 10.1155/2014/412354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ambrosini A, et al. Idiopathic infertility, effect of palmitoylethanolamide (a homologue of anandamide) on activated sperm cell motility and Ca2+ influx. J Androl. 2005;26:429–436. doi: 10.2164/jandrol.04141. [DOI] [PubMed] [Google Scholar]
- 140.Bíró T, et al. The endocannabinoid system of the skin in health and disease: novel perspectives and therapeutic opportunities. Trends Pharmacol Sci. 2009;30:411–420. doi: 10.1016/j.tips.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pucci M, et al. Endocannabinoid signaling and epidermal differentiation. Eur J Dermatol. 2011;21:29–34. doi: 10.1684/ejd.2011.1266. [DOI] [PubMed] [Google Scholar]
- 142.Pucci M, et al. Endocannabinoids stimulate human melanogenesis via type-1 cannabinoid receptor. J Biol Chem. 2012;287:15466–15478. doi: 10.1074/jbc.M111.314880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Roelandt T, et al. Cannabinoid receptors 1 and 2 oppositely regulate epidermal permeability barrier status and differentiation. Exp Dermatol. 2012;21:688–693. doi: 10.1111/j.1600-0625.2012.01561.x. [DOI] [PubMed] [Google Scholar]
- 144.Czifra G, et al. Endocannabinoids regulate growth and survival of human eccrine sweat gland-derived epithelial cells. J Invest Dermatol. 2012;132:1967–1976. doi: 10.1038/jid.2012.118. [DOI] [PubMed] [Google Scholar]
- 145.Karsak M, et al. Attenuation of allergic contact dermatitis through the endocannabinoid system. Science. 2007;316:1494–1497. doi: 10.1126/science.1142265. [DOI] [PubMed] [Google Scholar]
- 146.Sugawara K, et al. Endocannabinoids limit excessive mast cell maturation and activation in human skin. J Allergy Clin Immunol. 2012;129:726–738. doi: 10.1016/j.jaci.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 147.Oláh A, et al. Cannabidiol exerts sebostatic and antiinflammatory effects on human sebocytes. J Clin Invest. 2014;124:3713–3724. doi: 10.1172/JCI64628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Palumbo-Zerr K, et al. Inactivation of fatty acid amide hydrolase exacerbates experimental fibrosis by enhanced endocannabinoid-mediated activation of CB1. Ann Rheum Dis. 2012;71:2051–2054. doi: 10.1136/annrheumdis-2012-201823. [DOI] [PubMed] [Google Scholar]
- 149.Marquart S, et al. Inactivation of the cannabinoid receptor CB1 prevents leukocyte infiltration and experimental fibrosis. Arthritis Rheum. 2010;62:3467–3476. doi: 10.1002/art.27642. [DOI] [PubMed] [Google Scholar]
- 150.Lazzerini PE, et al. Adenosine A2A receptor activation stimulates collagen production in sclerodermic dermal fibroblasts either directly and through a cross-talk with the cannabinoid system. J Mol Med. 2012;90:331–342. doi: 10.1007/s00109-011-0824-5. [DOI] [PubMed] [Google Scholar]
- 151.Barutta F, et al. Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes. 2010;59:1046–1054. doi: 10.2337/db09-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jourdan T, et al. Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc Natl Acad Sci USA. 2014;111:E5420–E5428. doi: 10.1073/pnas.1419901111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Barutta F, et al. Protective role of cannabinoid receptor type 2 in a mouse model of diabetic nephropathy. Diabetes. 2011;60:2386–2396. doi: 10.2337/db10-1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mukhopadhyay P, et al. CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol. 2010;160:657–668. doi: 10.1111/j.1476-5381.2010.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mukhopadhyay P, et al. Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic Biol Med. 2010;48:457–467. doi: 10.1016/j.freeradbiomed.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Oddi S, et al. Evidence for the intracellular accumulation of anandamide in adiposomes. Cell Mol Life Sci. 2008;65:840–850. doi: 10.1007/s00018-008-7494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kaczocha M, et al. Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS One. 2014;9:e94200. doi: 10.1371/journal.pone.0094200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yu S, et al. Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor β/δ (PPARβ/δ) in the brain. J Biol Chem. 2014;289:12748–12758. doi: 10.1074/jbc.M114.559062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mukhopadhyay P, et al. Pharmacological inhibition of CB1 cannabinoid receptor protects against doxorubicin-induced cardiotoxicity. J Am Coll Cardiol. 2007;50:528–536. doi: 10.1016/j.jacc.2007.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pacher P, Mechoulam R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res. 2011;50:193–211. doi: 10.1016/j.plipres.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Beaumont H, et al. Effect of delta9-tetrahydrocannabinol, a cannabinoid receptor agonist, on the triggering of transient lower oesophageal sphincter relaxations in dogs and humans. Br J Pharmacol. 2009;156:153–162. doi: 10.1111/j.1476-5381.2008.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lehmann A, et al. Cannabinoid receptor agonism inhibits transient lower esophageal sphincter relaxations and reflux in dogs. Gastroenterology. 2002;123:1129–1134. doi: 10.1053/gast.2002.36025. [DOI] [PubMed] [Google Scholar]
- 163.Cluny NL, et al. Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl) methanone (SAB378), a peripherally restricted cannabinoid CB1/CB2 receptor agonist, inhibits gastrointestinal motility but has no effect on experimental colitis in mice. J Pharmacol Exp Ther. 2010;334:973–980. doi: 10.1124/jpet.110.169946. [DOI] [PubMed] [Google Scholar]
- 164.Izzo AA, et al. Cannabinoid CB1-receptor mediated regulation of gastrointestinal motility in mice in a model of intestinal inflammation. Br J Pharmacol. 2001;134:563–570. doi: 10.1038/sj.bjp.0704293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Izzo AA, et al. Defaecation, intestinal fluid accumulation and motility in rodents: implications of cannabinoid CB1 receptors. Naunyn Schmied Arch Pharmacol. 1999;359:65–70. doi: 10.1007/pl00005325. [DOI] [PubMed] [Google Scholar]
- 166.Massa F, et al. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113:1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Germano MP, et al. Cannabinoid CB1-mediated inhibition of stress-induced gastric ulcers in rats. Naunyn Schmied Arch Pharmacol. 2001;363:241–244. doi: 10.1007/s002100000360. [DOI] [PubMed] [Google Scholar]
- 168.Li YY, et al. Involvement of cannabinoid-1 and cannabinoid-2 receptors in septic ileus. Neurogastroenterol Motil. 2010;22:350–e388. doi: 10.1111/j.1365-2982.2009.01419.x. [DOI] [PubMed] [Google Scholar]
- 169.Mascolo N, et al. The endocannabinoid system and the molecular basis of paralytic ileus in mice. FASEB J. 2002;16:1973–1975. doi: 10.1096/fj.02-0338fje. [DOI] [PubMed] [Google Scholar]
- 170.Kimball ES, et al. Agonists of cannabinoid receptor 1 and 2 inhibit experimental colitis induced by oil of mustard and by dextran sulfate sodium. Am J Physiol GI Liver Physiol. 2006;291:G364–371. doi: 10.1152/ajpgi.00407.2005. [DOI] [PubMed] [Google Scholar]
- 171.Capasso R, et al. Fatty acid amide hydrolase controls mouse intestinal motility in vivo. Gastroenterology. 2005;129:941–951. doi: 10.1053/j.gastro.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 172.Fichna J, et al. Selective inhibition of FAAH produces antidiarrheal and antinociceptive effect mediated by endocannabinoids and cannabinoid-like fatty acid amides. Neurogastroenterol Motil. 2014;26:470–481. doi: 10.1111/nmo.12272. [DOI] [PubMed] [Google Scholar]
- 173.Sasso O, et al. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res. 2012;65:553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Alhouayek M, et al. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J. 2011;25:2711–2721. doi: 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- 175.Duncan M, et al. Distribution and function of monoacylglycerol lipase in the gastrointestinal tract. Am J Physiol GI Liver Physiol. 2008;295:G1255–1265. doi: 10.1152/ajpgi.90500.2008. [DOI] [PubMed] [Google Scholar]
- 176.Kinsey SG, et al. Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther. 2013;345:492–501. doi: 10.1124/jpet.112.201426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tam J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953–2966. doi: 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jourdan T, et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med. 2013;19:1132–1140. doi: 10.1038/nm.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Murina F, et al. Vestibulodynia: synergy between palmitoylethanolamide + transpolydatin and transcutaneous electrical nerve stimulation. J Low Genit Tract Dis. 2013;17:111–116. doi: 10.1097/LGT.0b013e3182652316. [DOI] [PubMed] [Google Scholar]
- 180.Keppel Hesselink JM, et al. Vulvodynia and proctodynia treated with topical baclofen 5% and palmitoylethanolamide. Arch Gynecol Obstet. 2014;290:389–393. doi: 10.1007/s00404-014-3218-4. [DOI] [PubMed] [Google Scholar]