

Abstract
G-protein-gated inwardly rectifying K+ (GIRK/Kir3) channel activation underlies key physiological effects of opioids, including analgesia and dependence. GIRK channel activation has also been implicated in the opioid-induced inhibition of midbrain GABA neurons and consequent disinhibition of dopamine (DA) neurons in the ventral tegmental area (VTA). Drug-induced disinhibition of VTA DA neurons has been linked to reward-related behaviors and underlies opioid-induced motor activation. Here, we demonstrate that mouse VTA GABA neurons express a GIRK channel formed by GIRK1 and GIRK2 subunits. Nevertheless, neither constitutive genetic ablation of Girk1 or Girk2, nor the selective ablation of GIRK channels in GABA neurons, diminished morphine-induced motor activity in mice. Moreover, direct activation of GIRK channels in midbrain GABA neurons did not enhance motor activity. In contrast, genetic manipulations that selectively enhanced or suppressed GIRK channel function in midbrain DA neurons correlated with decreased and increased sensitivity, respectively, to the motor-stimulatory effect of systemic morphine. Collectively, these data support the contention that the unique GIRK channel subtype in VTA DA neurons, the GIRK2/GIRK3 heteromer, regulates the sensitivity of the mouse mesolimbic DA system to drugs with addictive potential.
Keywords: conditional knockout, GIRK, Kir3, morphine, rostromedial tegmental area, ventral tegmental area
Introduction
The dopamine (DA) projection from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) is a critical anatomic substrate for drug-induced exploratory/seeking behavior (motor activation) and an associated anticipatory state comparable to “wanting” (Robinson and Berridge, 1993; Di Chiara, 1998; Berridge, 2012). Neuroadaptations within this pathway underlie the development and expression of behavioral sensitization and drug seeking associated with repeated drug administration (Grimm et al., 2001; Berridge, 2012; Salamone and Correa, 2012). Opioids enhance VTA DA neuron activity (Gysling and Wang, 1983), leading to increased DA levels and neuronal activation in the NAc, which in turn augments motor activity (Di Chiara and Imperato, 1988; Kalivas et al., 1990; Garcia et al., 1995; Bontempi and Sharp, 1997). The relevance of the opioid-induced increase in DA neurotransmission is underscored by the inhibitory impact of DA receptor antagonists and intra-NAc DA depletion on opioid-induced motor activity (Stinus et al., 1980; Kalivas et al., 1983; Longoni et al., 1987) and the lack of morphine-induced motor activation in DA-deficient mice (Hnasko et al., 2005).
The motor-stimulatory and associated neurochemical effects of opioids are dependent on the μ-opioid receptor (MOR; Tian et al., 1997; Contarino et al., 2002; Chefer et al., 2003; Yoo et al., 2003). MOR activation has been proposed to inhibit GABA neurons that dampen the output of VTA DA neurons (disinhibition), leading to increased DA neurotransmission (Johnson and North, 1992). Indeed, VTA DA neurons in the rat receive opioid-sensitive input from GABA neurons in the VTA and rostromedial tegmental nucleus (RMTg; Matsui and Williams, 2011; Matsui et al., 2014). Both presynaptic and postsynaptic mechanisms have been proposed to mediate the inhibitory impact of opioids on midbrain GABA neurotransmission (Johnson and North, 1992; Bergevin et al., 2002; Matsui and Williams, 2011; Matsui et al., 2014), though their relative significance to the cellular and behavioral effects of opioids remains unclear.
The postsynaptic inhibitory effect of opioids on GABA neurons is thought to involve activation of G-protein-gated inwardly rectifying K+ (GIRK/Kir3) channels (Johnson and North, 1992; Lüscher and Ungless, 2006). In support of this contention, loss of GIRK1 or GIRK2 correlates with diminished opioid inhibition of locus ceruleus and spinal cord neurons (Torrecilla et al., 2002; Marker et al., 2006) and blunted intrathecal morphine and DAMGO (C26H35N5O6) analgesia (Marker et al., 2004, 2005). Moreover, loss of GIRK2 correlates with diminished systemic morphine-induced analgesia (Mitrovic et al., 2003), and ablation of GIRK channels in the locus ceruleus precludes the induction of opioid dependence (Cruz et al., 2008).
Here, we sought to determine whether and how GIRK channels contribute to opioid-induced motor activation. Our working hypothesis was that opioids inhibit midbrain GABA neurons in a GIRK-dependent manner, leading to disinhibition of VTA DA neurons and an associated increase in motor activity. Using novel mutant mice and pharmacological tools, however, we found that GIRK channel activation in GABA neurons plays little role in opioid-induced motor activity. Instead, our data suggest that the GIRK channel in VTA DA neurons modulates the sensitivity to the motor-stimulatory effects of opioids in mice.
Materials and Methods
Animals.
Animal experimentation was reviewed and approved by the Institutional Animal Care and Use Committee at the University of Minnesota. The generation of constitutive Girk−/− mice was described previously (Signorini et al., 1997; Bettahi et al., 2002; Torrecilla et al., 2002). GAD67- enhanced green fluorescent protein (eGFP) mice (Tamamaki et al., 2003) and Pitx3-eGFP mice (Zhao et al., 2004) were generously provided by Drs. T. Kaneko and Meng Li, respectively. Gad2tm2(cre)Zjh/J (GAD-Cre), B6.SJL-Slc6a3tm1.1\(cre)Bkmn/J (DAT-Cre), and C57BL/6J mice were purchased from The Jackson Laboratory. Girk2flox/flox mice were generated under contract by InGenious Targeting Labs.
Drugs.
DAMGO, naloxone, and baclofen were purchased from Sigma. CGP54626 (C18H28Cl2NO3P.HCl) was purchased from Tocris Bioscience. rTertiapin-Q (TPN) and GIRK2 antibody were purchased from Alomone Labs. Morphine was purchased from Boynton Pharmacy (University of Minnesota). ML297 (C17H14F2N4O) was synthesized in the Vanderbilt Institute of Chemical Biology Chemical Synthesis Core and Vanderbilt Molecular Libraries Probe Production Centers Network Specialized Chemistry Center.
Generation of Girk2flox/flox mice.
The mouse Girk2 gene (also known as Kcnj6; Gene ID 16522) consists of at least seven exons and spans ∼250 kbp on the reverse strand of chromosome 16 (94, 749, 266–94, 997, 696). Several alternative splice variants have been identified that use one of two potential translation start sites (Wei et al., 1998). Exon 4 was targeted because it is present in all known Girk2 splice variants and because it contains the coding sequence for key functional domains of the GIRK2 protein. A C57BL/6 BAC clone (RP23: 155F22) containing Girk2 exon 4 and the surrounding sequence was identified and used to generate the targeting construct. The targeting construct was validated by sequencing, linearized, and electroporated into iTL BA1 (C57BL/6 × 129/SvEv) hybrid embryonic stem cells. Neomycin (NEO)-resistant clones (400) were screened by PCR, which identified five candidate clones that were amplified, reanalyzed by PCR, and evaluated by Southern blotting for proper integration of the targeting construct (including both 5′ and 3′ homology arms); three of these five clones harbored a properly targeted Girk2 allele. Two ES clones (#152 and #364) were then microinjected into C57BL/6 blastocysts, resulting in seven chimeras exhibiting a high percentage of agouti coat color. These mice were mated to C57BL/6N flippase (Flp) deleter mice [C57BL/6-Tg(ACTFLPe)] to remove the NEO cassette. Two F1 offspring (deriving from ES clone #364) were identified that lacked the NEO cassette, but did contain intact 5′ and 3′ homology arms and loxP sites (as assessed by PCR and DNA sequencing). These mice, which were heterozygous for the conditional mutant allele (Girk2flox/wt), were then backcrossed with C57BL/6J mice to amplify the colony. Resulting Girk2flox/wt offspring were then inbred to produce mice homozygous for the Girk2flox allele (Girk2flox/flox mice).
Quantitative RT-PCR.
Punches (2 mm diameter, 2 mm thick) from the cortex, hippocampus, and half of the cerebellum were taken from wild-type and Girk2flox/flox siblings (7–10 weeks old). Punches were frozen on crushed dry ice and stored at −80°C. Total RNA of cortex and hippocampus was isolated using the RNeasy Plus Micro kit (Qiagen). Total RNA from the cerebellum was isolated using TRIzol (Invitrogen) according to the manufacturer's recommendations and treated with recombinant DNase I (Roche). Reverse transcription was performed using an iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed on a LightCycler 480 II system in a final volume of 20 μl with a LightCycler 480 SYBR Green I Master kit (Roche). After preincubation at 95°C for 5 min, amplification consisted of 45 cycles of denaturation for 10 s at 95°C, followed by annealing for 30 s at 60°C and extension for 10 s at 72°C. qRT-PCR oligonucleotide sequences for GIRK2 are as follows: GIRK2, forward, 5′-CGTGGAGTGAATTATTGAATCT-3′; GIRK2, reverse, 5′-GTCATTTCTTCTTTGTGCTTTT-3′. GAPDH mRNA levels were used for normalization purposes using GAPDH QuantiTect oligonucleotides (Qiagen). Samples were analyzed using the 2[ΔCt] method (Livak and Schmittgen, 2001).
Quantitative immunoblotting.
Punches (2 mm diameter, 2 mm thick) of defined brain regions were taken from wild-type and Girk2flox/flox siblings (7–10 weeks old). Samples were sonicated in 1% SDS lysis buffer containing a Halt phosphatase and protease inhibitor cocktail (Thermo Fisher Scientific), heated at 85°C for 10 min, and centrifuged at 4°C for 20 min at 16,000 × g. Protein samples (40 μg) were then separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked in 5% milk/PBS, incubated overnight at 4°C with antibodies against GIRK2 (1:200; Alomone Labs) or β-actin (1:10,000; Abcam), and diluted in 5% milk/PBS/0.1% Tween 20. Membranes were washed with PBS/0.1% Tween 20 and incubated with donkey anti-rabbit (926-68072; 1:5000; LI-COR Biosciences) or donkey anti-mouse (926-32212; 1:1000–5000; LI-COR) secondary antibodies. Blots were developed using the Odyssey infrared imaging system (LI-COR), and the integrated density of each band was measured using ImageJ software (NIH).
Immunohistochemistry.
Mice (7–10 weeks old) were given a lethal dose of ketamine/xylazine (250 and 25 mg/kg, i.p., respectively) and perfused transcardially with Ca2+-free Tyrode's solution followed by 4% paraformaldehyde containing 14% picric acid. Brains were postfixed overnight in Sorenson's buffer containing 10% sucrose at 4°C. Tissue was sectioned coronally at 12 μm by cryostat and mounted to slides. Sections containing the VTA were washed in TBS for 10 min and then placed in boiling citric acid (10 mm; 30 min). Slides were then washed in TBS (three times, 5 min each) and incubated for 1 h in TBS blocking solution containing casein, Tween 20, and Triton X-100 (all at 0.2%). Sections were exposed to rabbit anti-GIRK2 antibody (1:1000; Alomone Labs) diluted in TBS solution containing casein and Tween 20 (both 0.2%) overnight at room temperature in a humidity chamber. The following day, sections were washed in TBS (three times, 20 min each) and then incubated for 2 h in donkey anti-rabbit IgG Cy3 secondary antibody (1:500; Jackson ImmunoResearch) diluted in TBS containing casein and Tween 20 (both at 0.2%). Sections were washed in TBS (three times, 20 min each), dehydrated through a series of graded alcohols, cleared in xylene, and coverslipped with DPX (Thermo Fisher Scientific). Fluorescence was visualized with an IX81ZDC2 Olympus microscope with disk-scanning unit and imaged with MetaMorph Advanced software. Digital images were colorized with ImageJ 64 software and prepared for micrographic presentation in Adobe Illustrator and Photoshop 6 (Adobe Systems); no adjustments were made to saturation, brightness, or contrast.
Slice electrophysiology.
Coronal and horizontal slices from wild-type and mutant mice (4–10 weeks old) were prepared as described previously (Arora et al., 2011). VTA neurons found in the lateral aspect of the VTA, near the medial terminal nucleus of the accessory optic tract, were targeted for analysis. Upon achieving whole-cell access, the current response to a 1 s voltage ramp (−60 to −120 mV) was used to measure Ih presence and amplitude, and spontaneous activity (I = 0) was assessed. For rheobase measurements, cells were held in current-clamp mode at 0 pA to obtain resting membrane potential, and then progressively increasing current pulses (1 s duration) were applied until the cell fired an action potential. Changes in somatodendritic holding current evoked by baclofen, DAMGO, or ML297 were measured at a holding potential of −60 mV, as described previously (Arora et al., 2011).
Some studies involved targeted evaluation of VTA DA and GABA neurons, as defined by eGFP expression in VTA neurons from Pitx3-eGFP and GAD67-eGFP mice, respectively (Tamamaki et al., 2003; Zhao et al., 2004). For other studies, we relied on morphological and functional criteria to identify “putative” VTA GABA and DA neurons. Specifically, small (apparent capacitance <40 pF) VTA neurons exhibiting little or no Ih current (<35 pA) were identified as putative GABA neurons, as these properties were typical of eGFP-positive VTA neurons from GAD67-eGFP mice (Labouebe et al., 2007). Larger (apparent capacitance >50 pF) VTA neurons exhibiting substantial Ih current (>100 pA) were identified as putative DA neurons, because these properties correlated well with tyrosine hydroxylase expression (Cruz et al., 2004; Arora et al., 2011), and this was a typical profile for eGFP-positive VTA neurons in slices from Pitx3-eGFP mice (Labouebe et al., 2007).
Morphine-induced motor activity.
Systemic morphine-induced locomotor activity assessments were performed as described previously (Pravetoni and Wickman, 2008). Subjects (6–12 weeks old) were handled and acclimated to the testing room for 1 h, followed by habituation to activity chambers and systemic injections (saline, intraperitoneally), on each of the 3 d preceding testing. Total distance traveled during the 60 min period following saline injection on the last acclimation day was defined as the response to 0 mg/kg morphine. Subjects were then given an ascending series of systemic morphine doses (3, 10, and 30 mg/kg i.p.), with doses separated by 3 d intervals.
Intracranial manipulations.
Mice (6–8 weeks old) were anesthetized with ketamine and xylazine (100 and 10 mg/kg, i.p.) and placed in a stereotaxic frame (David Kopf Instruments). For intracranial pharmacology studies, bilateral stainless-steel guide cannulae (Plastics One) targeting the VTA (AP, −3.08 mm; ML, ±0.5 mm; DV, −3.5 mm) or RMTg (AP, −3.90 mm; ML, ±0.5 mm; DV, −3.4 mm) were implanted 1 mm above the infusion target. Cannulae were held in place by stainless-steel mounting screws (0–80 × 3/32; Plastics One) and dental acrylate (Henry Schein), with stylets inserted following surgery to prevent blockage. Mice were allowed to recover for 7–9 d before behavioral testing.
On day 1 of testing, mice were gently restrained (mimicking the injection procedure) and placed in the open-field chamber. On day 2, mice were restrained, and then dummy cannulae were removed and replaced with injection cannulae attached to the delivery system. On day 3, mice were given infusions (0.5 μl/side/min) of vehicle (saline or saline/DMSO) or drug (DAMGO or ML297) using a KSD230 multichannel syringe pump controller (KD Scientific). Injection cannulae consisted of 20 mm, 33 ga stainless-steel hypodermic tubes inserted into a 9 mm, 26 ga segment to protrude 1 mm beyond the tip of the 10 mm guide cannula. Drug was delivered via catheter lines connected to a 10 μl Hamilton syringe. After infusion, injection cannulae were held in place for an additional 1–2 min to ensure proper drug diffusion. Thereafter, subjects were placed in the motor activity monitoring units for 60 min. Guide cannulae placement was assessed by post hoc histology.
For the viral GIRK3 reconstitution study, cDNAs for yellow fluorescent protein (YFP) fused to mouse GIRK3 or GFP alone as a control were subcloned into a CaMKII-pBOB third-generation self-inactivating lentiviral vector (kindly provided by Dr. Inder Verma, Laboratory of Genetics, Salk Institute for Biological Studies, La Jolla, CA). Lentivirus was produced by the Gene Transfer, Targeting, and Therapeutics Core at The Salk Institute for Biological Studies. Viral titer was ∼8 × 109 TU/ml as determined by real-time PCR of infected 293T cells. GIRK3-YFP or control lentivirus was injected into the VTA (AP, −3.08 mm; ML, ±0.5 mm; DV, −4.4 mm) of male mice (6–7 weeks old) using a 5 μl Hamilton syringe. Following infusion (0.5 μl/site; 0.5 μl/min), the syringe was left in place for 10 min to reduce backflow. Systemic morphine-induced motor activity was evaluated 21 d after viral infusion, and only data from mice with confirmed viral expression in the VTA were analyzed.
Statistical analysis.
Data are presented throughout as the mean ± SEM. Statistical analyses were performed using Prism 6 (GraphPad Software) and SigmaPlot 11.0 (Systat Software). Unless specifically noted, male and female mice were used for all studies. No sex differences were observed for any measured parameter, and thus data from male and female mice were pooled to increase statistical power. Electrophysiological data and motor activity, data were analyzed with a Student's t test, one-way or two-way ANOVA, or two-way repeated measures ANOVA followed by individual comparisons using the Bonferroni post-test method. For all statistical comparisons, differences were considered significant at p < 0.05.
Results
Morphine-induced motor activity in Girk−/− mice
We began by evaluating the impact of constitutive Girk subunit ablation on systemic morphine-induced motor activity. Since ablation of either Girk1 or Girk2 correlated with diminished opioid analgesia (Mitrovic et al., 2003; Marker et al., 2004, 2005), and since Girk2 ablation correlated with decreased opioid-induced hypothermia (Costa et al., 2005), we predicted that mice lacking GIRK1 and/or GIRK2 would exhibit diminished systemic morphine-induced motor activity. Surprisingly, however, the motor-stimulatory effect of systemic morphine was enhanced in Girk2−/− mice at the higher doses tested (10 and 30 mg/kg; Fig. 1). Girk1−/− mice also showed increased morphine-induced activity, but only at the highest morphine dose. Thus, GIRK channels modulate systemic morphine-induced motor activity, but in a manner inconsistent with their predicted role as proximal mediators of the underlying opioid-induced signaling.
Figure 1.

Constitutive Girk subunit ablation and morphine-induced motor activity. The total distance traveled by adult male wild-type (white; n = 26/dose), Girk1−/− (gray; n = 18/dose), and Girk2−/− (black; n = 7/dose) mice during a 60 min test following systemic administration of saline (0) or morphine (3, 10, and 30 mg/kg, i.p.) is shown. Main effects of genotype (F(2,48) = 12.8, p < 0.0001) and dose (F(3,144) = 133.6, p < 0.0001) were observed, as well as a significant interaction between genotype and dose (F(6,144) = 5.4, p < 0.0001). ***p < 0.001 versus wild-type (within dose).
Morphine-induced motor activity in mice lacking GIRK channels in GABA neurons
GIRK channel activation in GABA neurons has been implicated in the opioid-induced disinhibition of VTA DA neurons and associated behavior (Johnson and North, 1992; Lüscher and Ungless, 2006). Global and constitutive GIRK subunit ablation, however, may impact excitability in multiple cell types that exert opposing influences on morphine-induced motor activity (Arora et al., 2010). To evaluate the impact of selective ablation of GIRK channels in GABA neurons on morphine-induced motor activity, we generated conditional Girk2−/− (Girk2flox/flox) mice (Fig. 2A–C). After determining that Girk2flox/flox mice exhibited normal levels of GIRK2 mRNA and protein in multiple brain regions (Fig. 2D,E), we crossed Girk2flox/flox mice with Gad2tm2(cre)Zjh/J (GAD-Cre) mice, which express Cre recombinase under the control of the Gad2 promoter. Cre recombinase-driven reporter expression in GAD-Cre mice is observed throughout the brain, but is restricted to GABA neurons (Taniguchi et al., 2011).
Figure 2.

Generation of Girk2flox/flox mice. A, Depiction of the mouse Girk2/Kcnj6 gene, highlighting the seven known exons (rectangles with corresponding numbers below; Wei et al., 1998). Although several alternative splice variants have been identified, using two distinct translation initiation codons (ATG) and four distinct translation stop codons (TGA), all known variants contain exon 4. Shaded regions of exons denote protein-coding sequences. Some exons (1, 4, and 6) contain internal splice acceptor sites, denoted by vertical lines. B, Schematic depictions of a GIRK2 subunit. The image on the left shows the membrane topology of a GIRK2 subunit. Each subunit contains intracellular N- and C-terminal domains, two membrane-spanning domains (M1 and M2), two extracellular loops (×1 and ×2), and a pore domain (P). The schematic on the right is a linear depiction of the GIRK2a subunit, a prominent isoform in the brain (Wei et al., 1998). The region encoded by exon 4, which includes most of the N terminus (NT), both membrane-spanning domains and extracellular loops, the pore domain, and much of the C terminus (CT), is denoted by arrows. C, The Girk2 targeting strategy included engineering a loxP site just upstream (214 bp) of exon 4 and incorporating a NEO resistance cassette containing Flp recognition target (FRT) sites and a second loxP site; this cassette was positioned 795 bp downstream from exon 4. In total, the target region spanned 1.93 Kbp and was flanked by a 5′ homology arm (extending ∼5.4 Kbp upstream from exon 4) and a 3′ homology arm (an ∼7 Kbp fragment found immediately downstream from the target region). The NEO cassette was removed by crossing selected chimeras with C57BL/6N Flp deleter mice, yielding the Girk2flox allele. D, Tissue from the cortex (n = 9–10/genotype; t(17) = 0.09, p = 0.9), hippocampus (n = 6–7/genotype; t11 = 0.4, p = 0.7), and cerebellum (n = 7–8/genotype; t(13) = 1.3, p = 0.2) of wild-type and Girk2flox/flox (f/f) mice was evaluated for GIRK2 mRNA levels using quantitative RT-PCR. E, GIRK2 protein immunoreactivity (ir) relative to β-actin in samples from the cortex (n = 6–7/genotype; t(11) = 0.4, p = 0.7), hippocampus (n = 5–7/genotype; t(10) = 0.05, p = 1.0), and cerebellum (n = 4–7/genotype; t(9) = 0.6, p = 0.6) of wild-type and Girk2flox/flox (f/f) mice was assessed using quantitative immunoblotting. The level of GIRK2, which is seen as a doublet (top), was compared to the level of β-actin control (bottom) in each sample. Gel images of GIRK2 and β-actin were cropped and aligned with a molecular weight scale showing the markers for 52 and 38 kDA.
To assess the functional impact and cell-type specificity of GIRK channel ablation in GAD-Cre:Girk2flox/flox mice, we measured somatodendritic currents evoked by the GABAB receptor agonist baclofen in putative GABA and DA neurons of the VTA. Previous work has shown that baclofen-induced currents in these two neuron populations are mediated in large part by GIRK2-containing channels (Cruz et al., 2004; Labouebe et al., 2007). As predicted, baclofen-induced currents in putative VTA DA neurons were comparable in slices from GAD-Cre(+):Girk2flox/flox and GAD-Cre(−):Girk2flox/flox mice (Fig. 3A,B). In contrast, currents in putative VTA GABA neurons were significantly smaller in slices from GAD-Cre(+):Girk2flox/flox mice compared to GAD-Cre(−):Girk2flox/flox controls (Fig. 3C,D). Despite the loss of GIRK channels in putative GABA neurons in GAD-Cre(+):Girk2flox/flox mice, no difference in systemic morphine-induced motor activity was observed between these mice and GAD-Cre(−):Girk2flox/flox controls (Fig. 3E).
Figure 3.

GABA neuron-specific ablation of GIRK channels and morphine-induced motor activity. A, Representative traces of currents evoked by baclofen (Bac; 200 μm) in putative VTA DA neurons from GAD-Cre(−):Girk2flox/flox (black) and GAD-Cre(+):Girk2flox/flox (gray) mice. Currents were reversed by the GABAB receptor antagonist CGP54626 (CGP; 2 μm). B, Summary plot of baclofen-induced currents in putative VTA DA neurons from GAD-Cre(−):Girk2flox/flox (white; n = 7) and GAD-Cre(+):Girk2flox/flox (gray; n = 8) mice. No genotype-dependent difference was observed (t(13) = 0.09, p = 0.9). C, Representative traces of currents evoked by baclofen (200 μm) in putative VTA GABA neurons from GAD-Cre(−):Girk2flox/flox (left, black) and GAD-Cre(+):Girk2flox/flox (right, gray) mice. D, Summary plot of baclofen-induced currents in putative VTA GABA neurons from GAD-Cre(−):Girk2flox/flox (white; n = 6) and GAD-Cre(+):Girk2flox/flox (gray; n = 5) mice. A significant genotype-dependent difference was observed (t(9) = 4.1, **p < 0.01). E, Total distance traveled (in meters) by GAD-Cre(−):Girk2flox/flox (white; n = 14) and GAD-Cre(+):Girk2flox/flox (gray; n = 7) mice during a 60 min period following systemic (intraperitoneal) administration of saline (0) or morphine (3, 10, and 30 mg/kg). A main effect of dose (F(3,57) = 76.6, p < 0.0001) was observed, but there was no main effect of genotype (F(1,19) = 0.02, p = 0.9), nor was there an interaction between genotype and dose (F(3,57) = 0.3, p = 0.8). Calibrations: A, 60 pA, 200 s; C, 20 pA, 200 s.
Impact of GIRK channel manipulations on intracranial DAMGO-induced activity
VTA DA neurons express a unique GIRK channel subtype (GIRK2/GIRK3 heteromer), whereas single-cell RT-PCR analysis suggests that VTA GABA neurons may express GIRK1, GIRK2, and GIRK3 (Fig. 4A; Cruz et al., 2004; Labouebe et al., 2007). To test this proposal directly, we characterized GIRK channel activity in VTA GABA neurons. In slices from wild-type mice, baclofen (200 μm) reliably evoked an outward (inhibitory) current in putative VTA GABA neurons (Fig. 4B). Bath application of the nonselective K+ channel blocker Ba2+ (0.3 mm) or the GIRK channel blocker TPN (200 nm) blunted baclofen-induced currents (50–60%) in these neurons (Fig. 4C). Baclofen-induced currents in putative VTA GABA neurons from Girk1−/− or Girk2−/− mice (but not Girk3−/− mice) were attenuated relative to controls and were comparable in magnitude to currents measured in wild-type neurons in the presence of GIRK channel blockers (Fig. 4C).
Figure 4.

Characterization of the GIRK channel in VTA GABA neurons. A, Proposed GIRK subunit and receptor expression patterns in VTA DA and GABA neurons. B, Typical current induced by baclofen (200 μm) and reversed by CGP54626 (CGP; 2 μm) in a putative VTA GABA neuron from a wild-type mouse. C, The impact of pharmacologic and genetic ablation of GIRK channels on baclofen-induced currents in putative VTA GABA neurons (F(5,58) = 12.5; p < 0.0001). Currents are plotted for putative VTA GABA neurons from wild-type mice in the absence (control; n = 21) or presence of external Ba2+ (0.3 mm; n = 8) or TPN (200 nm; n = 4), and in neurons from Girk1−/− (red; n = 9), Girk2−/− (gray; n = 14), and Girk3−/− mice (gray; n = 8). *p < 0.05, ***p < 0.001 versus control. D, Responses of two VTA GABA neurons (eGFP-positive VTA neurons in slices from GAD67-eGFP mice) to a saturating concentration of DAMGO (3 μm). The response observed in the neuron shown on the left was reversed by naloxone (Nal; 10 μm). E, Summary of DAMGO-induced currents in putative VTA GABA neurons from wild-type (4 of 15 responders; 28.6 ± 2.7 pA) and Girk1−/− (0 of 13 responders) mice. Approximately one-third (4 of 15) of putative VTA GABA neurons evaluated exhibited a response to DAMGO (defined as a naloxone (Nal)-reversible current of >15 pA); none of the neurons in slices from Girk1−/− mice exhibited a response to DAMGO. Calibrations: B, D, 5 pA, 120 s.
To test whether GIRK1-containing channels mediate the MOR-dependent inhibition of VTA GABA neurons, we measured somatodendritic currents evoked by the MOR agonist DAMGO. Outward currents evoked by DAMGO (3 μm) and reversed by naloxone (10 μm) were observed in only a subset (4 of 15) of VTA GABA neurons (Fig. 4D,E). None of the putative VTA GABA neurons from Girk1−/− mice (0 of 13) displayed an evident DAMGO-induced current (Fig. 4E), supporting the contention that GIRK1-containing GIRK channels mediate the direct inhibitory effect of MOR agonists on a subset of VTA GABA neurons.
Since Girk1 ablation should alter GIRK channel activity in VTA GABA but not VTA DA neurons, we next compared the motor activity evoked by infusion of DAMGO into the VTA of wild-type and Girk1−/− mice. Intra-VTA DAMGO increased motor activity in wild-type mice, with the maximal response observed at 1 nmol (Fig. 5A,B). The motor-stimulatory effect of intra-VTA DAMGO was completely preserved in Girk1−/− mice. Administration of DAMGO to the adjacent RMTg also evoked increased motor activity in wild-type mice, in a dose-dependent manner, with peak response observed at the 0.1 nmol dose (Fig. 5C,D). Intra-RMTg opioid-induced motor activity was not reduced in Girk1−/− mice. Rather, elevated motor activity was seen in Girk1−/− mice at the two highest doses tested. These findings argue that GIRK channel activation in midbrain GABA neurons is not required for opioid-induced motor activity.
Figure 5.
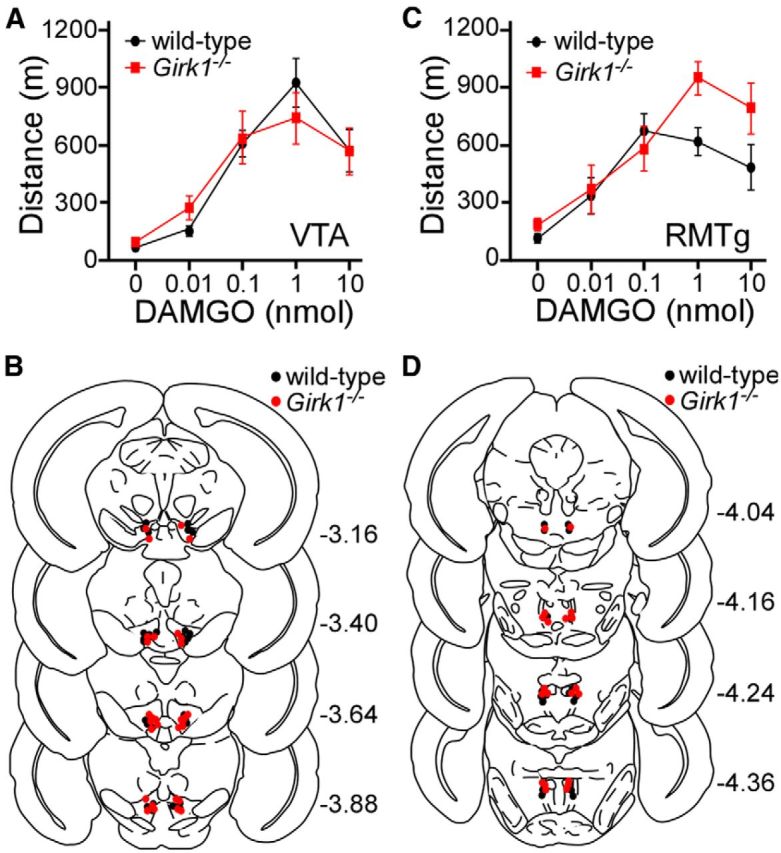
Intracranial DAMGO-induced motor activity. A, Open-field motor activity of adult male wild-type (black; n = 15) and congenic Girk1−/− (red; n = 15) mice during a 60 min period following bilateral intra-VTA infusion of 0 nmol (saline; n = 15/genotype), 0.01 nmol (n = 5–6/genotype), 0.1 nmol (6–7/genotype), 1 nmol (5–8/genotype), and 10 nmol (5–8/genotype) of DAMGO. A main effect of DAMGO dose was observed (F(4,70) = 36.2, p < 0.001), but there was no effect of genotype (F(1,70) = 0.005, p = 1.0) or genotype/concentration interaction (F(4,70) = 0.8, p = 0.5). B, Schematic of coronal sections (from bregma) depicting cannulae placements in mice used for intra-VTA experiments. C, Open-field motor activity of male wild-type (black; n = 11) and congenic Girk1−/− (red; n = 10) mice during a 60 min test following bilateral intra-RMTg infusion of 0 nmol (saline; n = 10–11/genotype), 0.01 nmol (n = 5–7/genotype), 0.1 nmol (5–7/genotype), 1 nmol (6–10/genotype, and 10 nmol (6–10/genotype) of DAMGO. A main effect of DAMGO dose (F(4,67) = 18.2, p < 0.001) and genotype (F(1,67) = 5.2, p < 0.05) was observed, but there was no genotype/concentration interaction (F(4,67) = 2.0, p = 0.1). D, Schematic of cannulae placements in mice evaluated in the intra-RMTg DAMGO study.
To test whether GIRK channel activation in GABA neurons is sufficient to enhance motor activity, we employed a novel direct-acting agonist (ML297) of GIRK1-containing GIRK channels (Kaufmann et al., 2013). In VTA slices, ML297 induced an outward current in putative GABA neurons from wild-type mice, but not in putative GABA neurons from Girk1−/− mice (Fig. 6A,B). ML297 also evoked no response in VTA DA neurons (Fig. 6A), suggesting that intracranial infusion of ML297 should selectively inhibit neurons expressing GIRK1-containing channels (e.g., VTA GABA neurons) without affecting neurons expressing GIRK1-lacking channels (e.g., VTA DA neurons). When administered directly to the VTA or RMTg of wild-type mice, ML297 failed to increase motor activity (Fig. 6C–E). Thus, direct activation of GIRK1-containing channels in the VTA and RMTg is insufficient to trigger motor activation.
Figure 6.

Direct activation of GIRK1-containing GIRK channels and motor activity. A, ML297 (10 μm) evoked a Ba2+-sensitive (0.3 mm) outward current in putative VTA GABA neurons from wild-type (left, black) but not Girk1−/− (middle, gray) mice. ML297 did not evoke an outward current in VTA DA neurons (right, black; n = 6), as shown in this recording from an eGFP-positive neuron from a Pitx3-eGFP mouse. Calibrations: left and middle, 15 pA, 180 s; right, 30 pA, 200 s. B, Summary of ML297-induced currents in putative VTA GABA neurons from wild-type (n = 6) and Girk1−/− (n = 5) mice (t(9) = 2.9). *p < 0.05). C, Open-field motor activity of male wild-type mice during a 60 min test following bilateral infusion of vehicle (0.1% DMSO; white) or ML297 (10 nmol; black) into the VTA (n = 10/group, t(18) = 0.4, p = 0.7) or RMTg (n = 9/group, t(16) = 0.3, p = 0.7). D, E, Schematic of coronal sections (from bregma) depicting cannulae placements in mice used for the intra-VTA and intra-RMTg ML297 studies.
Morphine-induced activity in mice lacking GIRK channels in DA neurons
Collectively, the data presented above argue that GIRK channel activation in GABA neurons is not required for opioid-induced motor activation. To test whether loss of GIRK channels in DA neurons modulates opioid-induced motor activity, Girk2flox/flox mice were crossed with B6.SJL-Slc6a3tm1.1c\(cre)Bkmn/J (DAT-Cre) mice, which express Cre recombinase under the control of the DA transporter (DAT) promoter. Cre recombinase-driven reporter expression in DAT-Cre mice is restricted to DA neurons (Bäckman et al., 2006). Immunohistochemical analysis revealed a striking reduction in GIRK2 immunoreactivity in the VTA from DAT-Cre(+):Girk2flox/flox mice (Fig. 7A).
Figure 7.
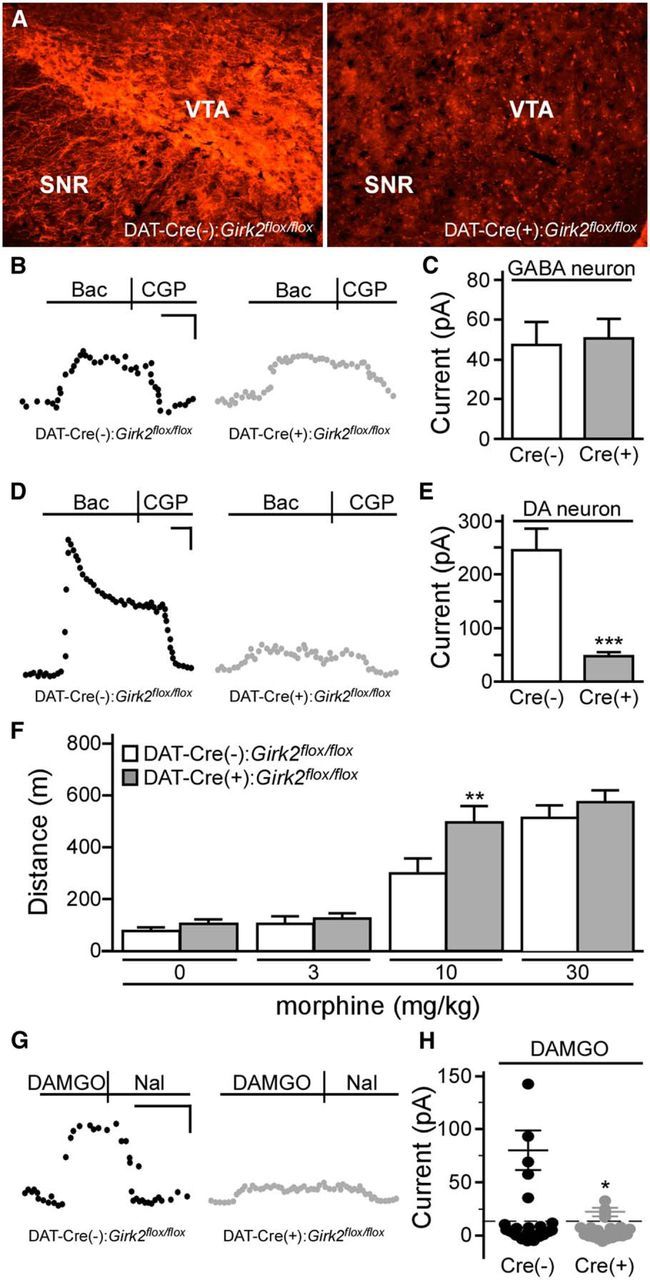
DA neuron-specific ablation of GIRK channels and systemic morphine-induced motor activity. A, GIRK2 immunolabeling in midbrain sections from DAT-Cre(−):Girk2flox/flox and DAT-Cre(+):Girk2flox/flox mice. Images shown were taken from slices −2.98 mm relative to bregma. SNR, Substantia nigra pars reticulata. B, Representative traces evoked by baclofen (Bac, 200 μm) in putative VTA GABA (eGFP-negative) neurons from Pitx3-eGFP(+)/DAT-Cre(−):Girk2flox/flox (left, black) and Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox (right, gray) mice. C, Summary of baclofen-induced currents in putative VTA GABA (eGFP-negative) neurons from Pitx3-eGFP(+)/DAT-Cre(−):Girk2flox/flox (white; n = 10) and Pitx3-eGFP(+)/GADCre(+):Girk2flox/flox (gray; n = 7) mice (t(15) = 0.2, p = 0.8). D, Representative currents evoked by baclofen (200 μm) in VTA DA (eGFP-positive) neurons from Pitx3-eGFP(+)/DAT-Cre(−):Girk2flox/flox (left, black) and Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox (right, gray) mice. E, Summary of baclofen-induced currents in VTA DA (eGFP-positive) neurons from Pitx3-eGFP(+)/DAT-Cre(−):Girk2flox/flox (white; n = 10) and Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox (gray; n = 12) mice (t(20) = 5.2, ***p < 0.0001). F, Open-field motor activity of DAT-Cre(−):Girk2flox/flox (white; n = 6/dose) and DAT-Cre(+):Girk2flox/flox (gray; n = 12/dose) mice during a 60 min period following systemic administration of saline (0) or morphine (3, 10, and 30 mg/kg). Main effects of dose (F(3,48) = 85.3, p < 0.0001) and genotype (F(1,16) = 2.9, p = 0.1) were observed, along with a dose/genotype interaction (F(3,48) = 2.9, p < 0.05). G, Responses of DA neurons from DAT-Cre(−):Girk2flox/flox and DAT-Cre(+):Girk2flox/flox mice to a saturating concentration of DAMGO (3 μm). DAMGO-induced currents were reversed by naloxone (Nal, 10 μm). H, Summary of DAMGO-induced currents in VTA DA neurons from DAT-Cre(−):Girk2flox/flox (5/25 responders; 80.1 ± 18.4 pA) and DAT-Cre(+):Girk2flox/flox (4/20 responders; 22.7 ± 3.4 pA) mice. Calibrations: B, G, 40 pA, 200 s; D, 60 pA, 200 s. *p < 0.05 versus DAT-Cre(−):Girk2flox/flox mice; **p < 0.01 versus DAT-Cre(−):Girk2flox/flox mice; ***p < 0.001 versus DAT-Cre(-):Girk2flox/flox mice.
To assess the functional impact and cell-type specificity of GIRK channel ablation in DAT-Cre(+):Girk2flox/flox mice, we measured baclofen-induced GIRK currents in VTA DA neurons. To permit direct electrophysiological characterization of DA and non-DA VTA neurons, DAT-Cre:Girk2flox/flox mice were crossed with mice expressing eGFP under the control of the promoter for the DA neuron-specific transcription factor Pitx3 (Zhao et al., 2004). As predicted, baclofen-induced currents in small eGFP-negative neurons lacking Ih current (putative VTA GABA neurons) were comparable in slices from Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox and Pitx3-eGFP(+)/DAT-Cre(−):Girk2flox/flox mice (Fig. 7B,C). In contrast, baclofen-induced currents in eGFP-positive (DA) VTA neurons were dramatically reduced in slices from Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox mice compared to controls (Fig. 7D,E). Importantly, like constitutive Girk2−/− mice, DAT-Cre(+):Girk2flox/flox mice were more sensitive to the motor-stimulatory effect of systemic morphine than DAT-Cre(−):Girk2flox/flox controls (Fig. 7F).
Since some VTA DA neurons are directly inhibited by MOR agonists (Cameron et al., 1997; Margolis et al., 2003, 2014; Ford et al., 2006), we evaluated the impact of the DA neuron-specific loss of GIRK2 on DAMGO-induced currents in VTA DA neurons. DAMGO (3 μm) evoked naloxone-sensitive outward currents in a subset (5 of 25) of VTA DA neurons from Pitx3-eGFP(+)/DAT-Cre(−):Girk2flox/flox mice (Fig. 7G,H). Though a similar fraction of VTA DA neurons from Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox mice exhibited DAMGO-induced outward currents, current amplitudes were significantly blunted relative to controls, indicating that activation of GIRK2-containing channels contributes to the direct inhibitory effect of DAMGO in this subset of VTA DA neurons.
We also asked whether there were differences in the excitability of VTA DA neurons from Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox and Pitx3-eGFP(+)/DAT-Cre(−):Girk2flox/flox mice. VTA DA neurons from both genotypes exhibited tonic spontaneous firing patterns (data not shown), with no difference in firing rate [Cre(+), 2.2 ± 0.3 Hz vs Cre(−), 2.0 ± 0.2 Hz; n = 31–33/group; p = 0.6]. Similarly, there was no difference in rheobase [Cre(+), −12 ± 5.0 pA vs Cre(−), −13.9 ± 3.6 pA; n = 26–30/group; p = 0.8] or holding current [Vhold = −60 mV; Cre(+), 10.82 ± 17.9 pA vs Cre(−), 44.35 ± 20.6 pA; n = 26–27/group; p = 0.1] between Pitx3-eGFP(+)/DAT-Cre(+):Girk2flox/flox and control mice. Thus, the cell-specific loss of GIRK2 did not significantly alter key measures of excitability in VTA DA neurons.
Morphine-induced activity in Girk3−/− mice
Data from DAT-Cre(+):Girk2flox/flox mice suggest that loss of GIRK channels in VTA DA neurons contributes in part to the increased sensitivity of constitutive Girk2−/− mice to systemic morphine-induced motor activity, and that enhanced GIRK channel activity in these neurons might yield diminished opioid sensitivity. Since prior work has shown that the residual GIRK channel in VTA DA neurons from Girk3−/− mice (GIRK2 homomer) is more sensitive to GABABR-dependent stimulation than its wild-type counterpart (GIRK2/GIRK3 heteromer) (Cruz et al., 2004), we next evaluated the impact of Girk3 ablation on systemic morphine-induced motor activity. Constitutive Girk3−/− mice were significantly less sensitive to morphine-induced motor activity than wild-type counterparts (Fig. 8A). To determine whether the loss of GIRK3 in the VTA underlies the diminished sensitivity of Girk3−/− mice to systemic morphine-induced motor activity, we restored GIRK3 expression in the VTA of Girk3−/− mice using a recombinant lentivirus. Viral expression of GIRK3 in the VTA of Girk3−/− mice restored normal sensitivity to the motor-stimulatory effect of morphine (Fig. 8A–C).
Figure 8.

Girk3 ablation and rescue: morphine-induced motor activity. A, Total distance traveled by wild-type (white; n = 6) and Girk3−/− (gray; n = 6) mice, as well as Girk3−/− mice given intra-VTA GIRK3 (red; n = 5) or control (black; n = 5) lentivirus, during a 60 min period following systemic administration of saline (0) or morphine (3, 10, and 30 mg/kg). A main effect of dose (F(3,54) = 103.4, p < 0.0001) and genotype (F(3,18) = 3.2, p < 0.05) was observed, but there was no dose/genotype interaction (F(9,54) = 1.8, p = 0.1). **p < 0.01 versus wild-type; ++p < 0.01 versus Girk3−/−; #p < 0.05 versus Girk3−/− + control. B, Representative image showing lentiviral-driven GIRK3-YFP (YFP expression shown in red) expression in the VTA of a Girk3−/− mouse. SN, Substantia nigra; PBP, parabrachial pigmented nucleus; IF, interfascicular nucleus; MM, medial mammillary nucleus. C, Schematic of coronal sections (from bregma) depicting viral expression in mice included in this study.
Discussion
Opioid-induced motor-activation is an unconditioned behavioral response that involves increased DA neurotransmission in the mesolimbic (VTA–NAc) pathway (Stinus et al., 1980; Kalivas et al., 1983). GIRK channels have been proposed to mediate the direct inhibitory effects of opioids on midbrain GABA neurons that suppress VTA DA neuron output, and thus we predicted that loss of GIRK signaling would suppress opioid-induced motor activity. Instead, we found that GIRK channel activation in GABA neurons is not required for opioid-induced motor activation in mice, nor is it sufficient to evoke an increase in motor activity. GIRK channels in VTA DA neurons are, however, a key determinant of sensitivity to the motor-stimulatory effect of opioids.
The opioid-induced disinhibition of VTA DA neurons was proposed initially to involve the direct inhibition of local (VTA) GABA neurons (Johnson and North, 1992). In support of this model, MOR expression within the VTA is most prominent in GABA neurons (Garzón and Pickel, 2001), VTA GABA neurons synapse onto VTA DA neurons (Omelchenko and Sesack, 2009; Tan et al., 2012; Bocklisch et al., 2013; Matsui et al., 2014), opioids hyperpolarize and/or inhibit the spontaneous activity of VTA GABA neurons (Johnson and North, 1992; Cameron et al., 1997; Steffensen et al., 2006; Chieng et al., 2011), and input to VTA DA neurons from VTA GABA neurons is opioid sensitive (Matsui et al., 2014). Moreover, inhibition of VTA GABA neurons leads to activation of VTA DA neurons (Bocklisch et al., 2013). Previous evidence, however, suggests that the disinhibition of VTA DA neurons reflects the opioid-induced inhibition of GABAergic input from the RMTg (Barrot et al., 2012; Bourdy and Barrot, 2012). Indeed, most midbrain (VTA) DA neurons in rodents and nonhuman primates are innervated and inhibited by RMTg GABA projections (Hong et al., 2011; Barrot et al., 2012), and this comparably dense input may be better poised to regulate the excitability of VTA DA neurons than the more sparse input from VTA GABA neurons (Omelchenko and Sesack, 2009; Jalabert et al., 2011; Lecca et al., 2011).
Whether the opioid-induced disinhibition of VTA DA neurons is attributable to the inhibition of VTA or RMTg GABA neurons or reflects a broad suppression of GABA input to VTA DA neurons from these and other structures, including the NAc and ventral pallidum (Kalivas et al., 1993; Watabe-Uchida et al., 2012), our data argue that the opioid-induced inhibition of GABAergic input to VTA DA neurons involves a presynaptic mechanism and/or non-GIRK somatodendritic mechanism(s), at least in the mouse. Consistent with the former possibility, MOR labeling in the VTA is found on axon terminals of GABA neurons (Garzón and Pickel, 2001), and opioid inhibition of GABAergic neurotransmission in several brain regions, including the VTA, is mediated by a reduced probability of presynaptic neurotransmitter release (Bergevin et al., 2002; Matsui and Williams, 2011; Matsui et al., 2014). Furthermore, pharmacological inactivation of the RMTg blocked the ability of intra-VTA morphine to activate VTA DA neurons (Jalabert et al., 2011), arguing that the disinhibitory effect of intra-VTA opioids on VTA DA neurons is due to inhibition of RMTg GABA afferents.
Non-GIRK postsynaptic mechanisms may also contribute to the opioid-induced disinhibition of VTA DA neurons. Notably, the opioid-induced hyperpolarization and inhibition of spontaneous activity in VTA and RMTg GABA neurons has been observed by multiple groups (Chieng et al., 2011; Lecca et al., 2011; Matsui and Williams, 2011). Indeed, DAMGO suppressed the spontaneous activity of 92% of eGFP-positive (GABA) VTA neurons from GAD67-eGFP mice (Chieng et al., 2011). Using this same mouse line, we show here that most VTA GABA neurons lack an evident DAMGO-induced inhibitory current, reminiscent of findings in the rat (Margolis et al., 2012). Similarly, only one-third of the RMTg neurons projecting to the ventral midbrain (VTA and susbstantia nigra) were hyperpolarized by MOR agonists (Matsui and Williams, 2011). Our finding that intra-RMTg DAMGO stimulated motor activity, however, argues that the direct inhibition of RMTg neurons is sufficient to stimulate motor activity. Interestingly, morphine was reported to suppress excitatory input to RMTg neurons via a GIRK-independent postsynaptic mechanism (Lecca et al., 2011). Thus, MOR may couple preferentially to a non-GIRK somatodendritic effector(s) in midbrain GABA neurons. At present, the behavioral relevance of the opioid-induced, GIRK-dependent, and GIRK-independent inhibition of midbrain GABA neurons is unclear.
Some VTA DA neurons express MOR, and both direct excitatory and inhibitory effects of MOR activation on VTA DA neurons have been reported (Margolis et al., 2014). Consistent with previous reports (Cameron et al., 1997; Ford et al., 2006; Margolis et al., 2014), we found that ∼20% of mouse VTA DA neurons were inhibited by DAMGO. Moreover, we show that GIRK channel activation mediates the direct inhibitory effect of DAMGO on this subset of VTA DA neurons. Disruption of MOR-GIRK signaling in these neurons, however, seems unlikely to explain the enhanced morphine-induced motor activity observed in Girk2−/− or DAT-Cre(+):Girk2flox/flox mice. Indeed, if MOR-GIRK signaling in these neurons underlies the motor-stimulatory effect of systemic or intra-VTA opioids, then the loss of GIRK channels should diminish and not enhance the opioid-induced behavior. It is possible, however, that this VTA DA neuron subpopulation normally tempers opioid-induced motor stimulation, perhaps akin to a negative feedback pathway. In this scenario, diminished MOR-GIRK signaling could result in enhanced opioid-induced motor stimulation.
The output of VTA DA neurons is tempered by “long-loop” (GABABR-dependent) and D2 DA autoreceptor (D2R)-mediated negative feedback mechanisms that are engaged by in vivo administration of drugs of abuse (Einhorn et al., 1988; Sugita et al., 1992). The GIRK2/GIRK3 channel mediates the inhibitory effect of GABABR and D2R activation on VTA DA neurons (Beckstead et al., 2004; Cruz et al., 2004). The enhanced morphine-induced motor activity seen in DAT-Cre(+):Girk2flox/flox mice may reflect, therefore, the loss of these feedback mechanisms. In support of this contention, key excitability measures in the absence of drug were comparable in VTA DA neurons from DAT-Cre(+):Girk2flox/flox and DAT-Cre(−):Girk2flox/flox mice. Furthermore, our observations with Girk3−/− mice and viral reconstitution of GIRK3 in the VTA, together with previous work showing that Girk3 ablation correlates with enhanced sensitivity of the residual GIRK channel to GPCR-dependent activation (Labouebe et al., 2007), argues that the sensitivity of the VTA DA neuron GIRK channel to GPCR stimulation is a critical determinant of morphine-induced motor activity.
Acute and repeated psychostimulant exposure suppresses GABAB and D2 DA receptor-dependent inhibitory signaling in VTA DA neurons (Hearing et al., 2012). A single injection of cocaine, for example, transiently suppressed GABABR-GIRK signaling in VTA DA neurons by triggering a subcellular redistribution of GIRK channels from the membrane to intracellular sites (Arora et al., 2011). As decreased GIRK-dependent signaling in VTA DA neurons should correlate with enhanced opioid-induced motor activity, our findings suggest a plausible explanation for behavioral cross-sensitization observed between opioids and psychostimulants (Stewart and Vezina, 1987; DuMars et al., 1988; Lett, 1989; Vezina and Stewart, 1990; Cador et al., 1995; McDaid et al., 2005).
Although the vast majority of neurons in the mouse VTA are dopaminergic or GABAergic (Chieng et al., 2011), some glutamatergic neurons are present as well (Yamaguchi et al., 2007; Nair-Roberts et al., 2008). Most glutamatergic neurons are found in the medial aspect of the VTA (Yamaguchi et al., 2007), including some that coexpress VGlut2 and TH (Yamaguchi et al., 2011; Morales and Root, 2014). We targeted the lateral aspect of the VTA to minimize the potential impact of this small neuron population on our study. Whereas many of our experiments used transgenic mice with fluorescently tagged GABA and DA neurons, some experiments relied on a combination of cell size (apparent capacitance) and Ih current size to identify putative VTA GABA and DA neurons. This combination of features, as well as the amplitude of the baclofen-induced somatodendritic current, has proven useful for distinguishing between DA and GABA neurons in the mouse VTA (Cruz et al., 2004; Labouebe et al., 2007; Arora et al., 2011). We cannot exclude the possibility, however, that some putative VTA DA and GABA neurons evaluated in this study were glutamatergic neurons. At present, the expression of GIRK channels and MOR in VTA glutamatergic neurons and their relevance to the motor-stimulatory effects of systemic and intracranial opioids are unclear.
Opioids such as morphine are the most commonly administered drugs for pain, despite possessing an unfavorable side-effect profile and significant addictive liability (Inturrisi, 2002). Given the increase in abuse of prescription opioids (Maxwell, 2011), it is particularly important that we understand the circuitry, cell types, and mechanisms mediating both the beneficial and problematic effects of opioids. These insights may suggest novel approaches to eliciting the beneficial outcome associated with opioids (analgesia) while avoiding undesirable side effects (addiction). Data described herein suggest that the unique GIRK channel subtype (GIRK2/GIRK3 heteromer) in VTA DA neurons sets the sensitivity of the mesolimbic circuit to morphine. Given that previous work has implicated this channel in circuit-level and behavioral effects of other drugs of abuse (Cruz et al., 2004; Munoz and Slesinger, 2014), these findings suggest that therapeutic approaches targeting the GIRK2/GIRK3 channel might prove useful for treating some aspects of drug addiction.
Footnotes
This work was supported by National Institute of Health Grants DA007097 (L.K., M.H.), GM008471 and DA007234 (N.M.M.), DA07234 (N.C.V.), CA068485 (C.D.W.), and DA029343, DA011806, DA034696, and MH061933 (K.W.). We thank Desirae Haluk for assistance with intracranial experiments and Wickman lab staff for exceptional care of the mouse colony. In addition, we thank Ian Romaine, the Chemical Synthesis Core in the Vanderbilt Institute of Chemical Biology, and the Vanderbilt MLPCN Specialized Chemistry Center for synthesis of ML297.
C.D.W. receives royalties from the sale of the thallium-sensitive dye, Thallos, through a licensing agreement between Vanderbilt University and TEFlabs. The remaining authors declare no competing financial interests.
References
- Arora D, Haluk DM, Kourrich S, Pravetoni M, Fernández-Alacid L, Nicolau JC, Luján R, Wickman K. Altered neurotransmission in the mesolimbic reward system of Girk mice. J Neurochem. 2010;114:1487–1497. doi: 10.1111/j.1471-4159.2010.06864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora D, Hearing M, Haluk DM, Mirkovic K, Fajardo-Serrano A, Wessendorf MW, Watanabe M, Luján R, Wickman K. Acute cocaine exposure weakens GABA(B) receptor-dependent G-protein-gated inwardly rectifying K+ signaling in dopamine neurons of the ventral tegmental area. J Neurosci. 2011;31:12251–12257. doi: 10.1523/JNEUROSCI.0494-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, Tomac AC. Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis. 2006;44:383–390. doi: 10.1002/dvg.20228. [DOI] [PubMed] [Google Scholar]
- Barrot M, Sesack SR, Georges F, Pistis M, Hong S, Jhou TC. Braking dopamine systems: a new GABA master structure for mesolimbic and nigrostriatal functions. J Neurosci. 2012;32:14094–14101. doi: 10.1523/JNEUROSCI.3370-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–946. doi: 10.1016/j.neuron.2004.05.019. [DOI] [PubMed] [Google Scholar]
- Bergevin A, Girardot D, Bourque MJ, Trudeau LE. Presynaptic mu-opioid receptors regulate a late step of the secretory process in rat ventral tegmental area GABAergic neurons. Neuropharmacology. 2002;42:1065–1078. doi: 10.1016/S0028-3908(02)00061-8. [DOI] [PubMed] [Google Scholar]
- Berridge KC. From prediction error to incentive salience: mesolimbic computation of reward motivation. Eur J Neurosci. 2012;35:1124–1143. doi: 10.1111/j.1460-9568.2012.07990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettahi I, Marker CL, Roman MI, Wickman K. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem. 2002;277:48282–48288. doi: 10.1074/jbc.M209599200. [DOI] [PubMed] [Google Scholar]
- Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, Tan KR, Lüscher C. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science. 2013;341:1521–1525. doi: 10.1126/science.1237059. [DOI] [PubMed] [Google Scholar]
- Bontempi B, Sharp FR. Systemic morphine-induced Fos protein in the rat striatum and nucleus accumbens is regulated by mu opioid receptors in the substantia nigra and ventral tegmental area. J Neurosci. 1997;17:8596–8612. doi: 10.1523/JNEUROSCI.17-21-08596.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdy R, Barrot M. A new control center for dopaminergic systems: pulling the VTA by the tail. Trends Neurosci. 2012;35:681–690. doi: 10.1016/j.tins.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Cador M, Bjijou Y, Stinus L. Evidence of a complete independence of the neurobiological substrates for the induction and expression of behavioral sensitization to amphetamine. Neuroscience. 1995;65:385–395. doi: 10.1016/0306-4522(94)00524-9. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Wessendorf MW, Williams JT. A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience. 1997;77:155–166. doi: 10.1016/S0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Kieffer BL, Shippenberg TS. Basal and morphine-evoked dopaminergic neurotransmission in the nucleus accumbens of MOR- and DOR-knockout mice. Eur J Neurosci. 2003;18:1915–1922. doi: 10.1046/j.1460-9568.2003.02912.x. [DOI] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ. Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol. 2011;589:3775–3787. doi: 10.1113/jphysiol.2011.210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contarino A, Picetti R, Matthes HW, Koob GF, Kieffer BL, Gold LH. Lack of reward and locomotor stimulation induced by heroin in mu-opioid receptor-deficient mice. Eur J Pharmacol. 2002;446:103–109. doi: 10.1016/S0014-2999(02)01812-5. [DOI] [PubMed] [Google Scholar]
- Costa AC, Stasko MR, Stoffel M, Scott-McKean JJ. G-protein-gated potassium (GIRK) channels containing the GIRK2 subunit are control hubs for pharmacologically induced hypothermic responses. J Neurosci. 2005;25:7801–7804. doi: 10.1523/JNEUROSCI.1699-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C. Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- Cruz HG, Berton F, Sollini M, Blanchet C, Pravetoni M, Wickman K, Lüscher C. Absence and rescue of morphine withdrawal in GIRK/Kir3 knock-out mice. J Neurosci. 2008;28:4069–4077. doi: 10.1523/JNEUROSCI.0267-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G. A motivational learning hypothesis of the role of mesolimbic dopamine in compulsive drug use. J Psychopharmacol. 1998;12:54–67. doi: 10.1177/026988119801200108. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMars LA, Rodger LD, Kalivas PW. Behavioral cross-sensitization between cocaine and enkephalin in the A10 dopamine region. Behav Brain Res. 1988;27:87–91. doi: 10.1016/0166-4328(88)90111-8. [DOI] [PubMed] [Google Scholar]
- Einhorn LC, Johansen PA, White FJ. Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: studies in the ventral tegmental area. J Neurosci. 1988;8:100–112. doi: 10.1523/JNEUROSCI.08-01-00100.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MM, Brown HE, Harlan RE. Alterations in immediate-early gene proteins in the rat forebrain induced by acute morphine injection. Brain Res. 1995;692:23–40. doi: 10.1016/0006-8993(95)00625-Z. [DOI] [PubMed] [Google Scholar]
- Garzón M, Pickel VM. Plasmalemmal mu-opioid receptor distribution mainly in nondopaminergic neurons in the rat ventral tegmental area. Synapse. 2001;41:311–328. doi: 10.1002/syn.1088. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, Shaham Y. Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature. 2001;412:141–142. doi: 10.1038/35084134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res. 1983;277:119–127. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- Hearing MC, Zink AN, Wickman K. Cocaine-induced adaptations in metabotropic inhibitory signaling in the mesocorticolimbic system. Rev Neurosci. 2012;23:325–351. doi: 10.1515/revneuro-2012-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Sotak BN, Palmiter RD. Morphine reward in dopamine-deficient mice. Nature. 2005;438:854–857. doi: 10.1038/nature04172. [DOI] [PubMed] [Google Scholar]
- Hong S, Jhou TC, Smith M, Saleem KS, Hikosaka O. Negative reward signals from the lateral habenula to dopamine neurons are mediated by rostromedial tegmental nucleus in primates. J Neurosci. 2011;31:11457–11471. doi: 10.1523/JNEUROSCI.1384-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inturrisi CE. Clinical pharmacology of opioids for pain. Clin J Pain. 2002;18:S3–S13. doi: 10.1097/00002508-200207001-00002. [DOI] [PubMed] [Google Scholar]
- Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, Georges F. Neuronal circuits underlying acute morphine action on dopamine neurons. Proc Natl Acad Sci U S A. 2011;108:16446–16450. doi: 10.1073/pnas.1105418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Widerlöv E, Stanley D, Breese G, Prange AJ., Jr Enkephalin action on the mesolimbic system: a dopamine-dependent and a dopamine-independent increase in locomotor activity. J Pharmacol Exp Ther. 1983;227:229–237. [PubMed] [Google Scholar]
- Kalivas PW, Duffy P, Eberhardt H. Modulation of A10 dopamine neurons by gamma-aminobutyric acid agonists. J Pharmacol Exp Ther. 1990;253:858–866. [PubMed] [Google Scholar]
- Kalivas PW, Churchill L, Klitenick MA. GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience. 1993;57:1047–1060. doi: 10.1016/0306-4522(93)90048-K. [DOI] [PubMed] [Google Scholar]
- Kaufmann K, Romaine I, Days E, Pascual C, Malik A, Yang L, Zou B, Du Y, Sliwoski G, Morrison RD, Denton J, Niswender CM, Daniels JS, Sulikowski GA, Xie XS, Lindsley CW, Weaver CD. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem Neurosci. 2013;4:1278–1286. doi: 10.1021/cn400062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouèbe G, Lomazzi M, Cruz HG, Creton C, Luján R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, Boyer SB, Slesinger PA, Lüscher C. RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci. 2007;10:1559–1568. doi: 10.1038/nn2006. [DOI] [PubMed] [Google Scholar]
- Lecca S, Melis M, Luchicchi A, Ennas MG, Castelli MP, Muntoni AL, Pistis M. Effects of drugs of abuse on putative rostromedial tegmental neurons, inhibitory afferents to midbrain dopamine cells. Neuropsychopharmacology. 2011;36:589–602. doi: 10.1038/npp.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett BT. Repeated exposures intensify rather than diminish the rewarding effects of amphetamine, morphine, and cocaine. Psychopharmacology (Berl) 1989;98:357–362. doi: 10.1007/BF00451687. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Longoni R, Spina L, Di Chiara G. Dopaminergic D-1 receptors: essential role in morphine-induced hypermotility. Psychopharmacology (Berl) 1987;93:401–402. doi: 10.1007/BF00187265. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Ungless MA. The mechanistic classification of addictive drugs. PLoS Med. 2006;3:e437. doi: 10.1371/journal.pmed.0030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Toy B, Himmels P, Morales M, Fields HL. Identification of rat ventral tegmental area GABAergic neurons. PLoS One. 2012;7:e42365. doi: 10.1371/journal.pone.0042365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Fujita W, Fields HL. Direct bidirectional μ-opioid control of midbrain dopamine neurons. J Neurosci. 2014;34:14707–14716. doi: 10.1523/JNEUROSCI.2144-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker CL, Stoffel M, Wickman K. Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J Neurosci. 2004;24:2806–2812. doi: 10.1523/JNEUROSCI.5251-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker CL, Luján R, Loh HH, Wickman K. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids. J Neurosci. 2005;25:3551–3559. doi: 10.1523/JNEUROSCI.4899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker CL, Luján R, Colón J, Wickman K. Distinct populations of spinal cord lamina II interneurons expressing G-protein-gated potassium channels. J Neurosci. 2006;26:12251–12259. doi: 10.1523/JNEUROSCI.3693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui A, Williams JT. Opioid-sensitive GABA inputs from rostromedial tegmental nucleus synapse onto midbrain dopamine neurons. J Neurosci. 2011;31:17729–17735. doi: 10.1523/JNEUROSCI.4570-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui A, Jarvie BC, Robinson BG, Hentges ST, Williams JT. Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron. 2014;82:1346–1356. doi: 10.1016/j.neuron.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell JC. The prescription drug epidemic in the United States: a perfect storm. Drug Alcohol Rev. 2011;30:264–270. doi: 10.1111/j.1465-3362.2011.00291.x. [DOI] [PubMed] [Google Scholar]
- McDaid J, Dallimore JE, Mackie AR, Mickiewicz AL, Napier TC. Cross-sensitization to morphine in cocaine-sensitized rats: behavioral assessments correlate with enhanced responding of ventral pallidal neurons to morphine and glutamate, with diminished effects of GABA. J Pharmacol Exp Ther. 2005;313:1182–1193. doi: 10.1124/jpet.105.084038. [DOI] [PubMed] [Google Scholar]
- Mitrovic I, Margeta-Mitrovic M, Bader S, Stoffel M, Jan LY, Basbaum AI. Contribution of GIRK2-mediated postsynaptic signaling to opiate and alpha 2-adrenergic analgesia and analgesic sex differences. Proc Natl Acad Sci U S A. 2003;100:271–276. doi: 10.1073/pnas.0136822100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Root DH. Glutamate neurons within the midbrain dopamine regions. Neuroscience. 2014;282C:60–68. doi: 10.1016/j.neuroscience.2014.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz MB, Slesinger PA. Sorting nexin 27 regulation of G protein-gated inwardly rectifying K(+) channels attenuates in vivo cocaine response. Neuron. 2014;82:659–669. doi: 10.1016/j.neuron.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA. Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience. 2008;152:1024–1031. doi: 10.1016/j.neuroscience.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omelchenko N, Sesack SR. Ultrastructural analysis of local collaterals of rat ventral tegmental area neurons: GABA phenotype and synapses onto dopamine and GABA cells. Synapse. 2009;63:895–906. doi: 10.1002/syn.20668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravetoni M, Wickman K. Behavioral characterization of mice lacking GIRK/Kir3 channel subunits. Genes Brain Behav. 2008;7:523–531. doi: 10.1111/j.1601-183X.2008.00388.x. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-P. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76:470–485. doi: 10.1016/j.neuron.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci U S A. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Stobbs SH, Colago EE, Lee RS, Koob GF, Gallegos RA, Henriksen SJ. Contingent and non-contingent effects of heroin on mu-opioid receptor-containing ventral tegmental area GABA neurons. Exp Neurol. 2006;202:139–151. doi: 10.1016/j.expneurol.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Stewart J, Vezina P. Environment-specific enhancement of the hyperactivity induced by systemic or intra-VTA morphine injections in rats preexposed to amphetamine. Psychobiology. 1987;15:144–153. doi: 10.3758/BF03333103. [DOI] [Google Scholar]
- Stinus L, Koob GF, Ling N, Bloom FE, Le Moal M. Locomotor activation induced by infusion of endorphins into the ventral tegmental area: evidence for opiate-dopamine interactions. Proc Natl Acad Sci U S A. 1980;77:2323–2327. doi: 10.1073/pnas.77.4.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Johnson SW, North RA. Synaptic inputs to GABAA and GABAB receptors originate from discrete afferent neurons. Neurosci Lett. 1992;134:207–211. doi: 10.1016/0304-3940(92)90518-C. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012;73:1173–1183. doi: 10.1016/j.neuron.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Broxmeyer HE, Fan Y, Lai Z, Zhang S, Aronica S, Cooper S, Bigsby RM, Steinmetz R, Engle SJ, Mestek A, Pollock JD, Lehman MN, Jansen HT, Ying M, Stambrook PJ, Tischfield JA, Yu L. Altered hematopoiesis, behavior, and sexual function in mu opioid receptor-deficient mice. J Exp Med. 1997;185:1517–1522. doi: 10.1084/jem.185.8.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K. G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J Neurosci. 2002;22:4328–4334. doi: 10.1523/JNEUROSCI.22-11-04328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezina P, Stewart J. Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Res. 1990;516:99–106. doi: 10.1016/0006-8993(90)90902-N. [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron. 2012;74:858–873. doi: 10.1016/j.neuron.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Wei J, Hodes ME, Piva R, Feng Y, Wang Y, Ghetti B, Dlouhy SR. Characterization of murine Girk2 transcript isoforms: structure and differential expression. Genomics. 1998;51:379–390. doi: 10.1006/geno.1998.5369. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Sheen W, Morales M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 2007;25:106–118. doi: 10.1111/j.1460-9568.2006.05263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Wang HL, Li X, Ng TH, Morales M. Mesocorticolimbic glutamatergic pathway. J Neurosci. 2011;31:8476–8490. doi: 10.1523/JNEUROSCI.1598-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JH, Yang EM, Lee SY, Loh HH, Ho IK, Jang CG. Differential effects of morphine and cocaine on locomotor activity and sensitization in mu-opioid receptor knockout mice. Neurosci Lett. 2003;344:37–40. doi: 10.1016/S0304-3940(03)00410-5. [DOI] [PubMed] [Google Scholar]
- Zhao S, Maxwell S, Jimenez-Beristain A, Vives J, Kuehner E, Zhao J, O'Brien C, de Felipe C, Semina E, Li M. Generation of embryonic stem cells and transgenic mice expressing green fluorescence protein in midbrain dopaminergic neurons. Eur J Neurosci. 2004;19:1133–1140. doi: 10.1111/j.1460-9568.2004.03206.x. [DOI] [PubMed] [Google Scholar]