

Abstract
Oxidative stress contributes to neuronal death in brain ischemia-reperfusion. Tissue levels of the endogenous antioxidant glutathione (GSH) are depleted during ischemia-reperfusion, but it is unknown whether this depletion is a cause or an effect of oxidative stress, and whether it occurs in neurons or other cell types. We used immunohistochemical methods to evaluate glutathione, superoxide, and oxidative stress in mouse hippocampal neurons after transient forebrain ischemia. GSH levels in CA1 pyramidal neurons were normally high relative to surrounding neuropil, and exhibited a time-dependent decrease during the first few hours of reperfusion. Colabeling for superoxide in the neurons showed a concurrent increase in detectable superoxide over this interval. To identify cause–effect relationships between these changes, we independently manipulated superoxide production and GSH metabolism during reperfusion. Mice in which NADPH oxidase activity was blocked to prevent superoxide production showed preservation of neuronal GSH content, thus demonstrating that neuronal GSH depletion is result of oxidative stress. Conversely, mice in which neuronal GSH levels were maintained by N-acetyl cysteine treatment during reperfusion showed less neuronal superoxide signal, oxidative stress, and neuronal death. At 3 d following ischemia, GSH content in reactive astrocytes and microglia was increased in the hippocampal CA1 relative to surviving neurons. Results of these studies demonstrate that neuronal GSH depletion is both a result and a cause of neuronal oxidative stress after ischemia-reperfusion, and that postischemic restoration of neuronal GSH levels can be neuroprotective.
Keywords: glutathione, hyperglycemia, ischemia, N-acetyl cysteine, neuron, oxidative stress
Introduction
Ischemia-reperfusion leads to the production of superoxide and nitric oxide by neurons and other cell types (Chan, 2001; Brennan-Minnella et al., 2015). These oxidants form peroxynitrite and other highly reactive oxygen species (ROS) that rapidly damage lipids, proteins, and DNA. DNA damage in turn triggers the poly(ADP-ribose) polymerase cell death pathway (parthanatosis), which is the primary cause of acute neuronal death in ischemia-reperfusion injury (Moroni, 2008; Andrabi et al., 2011; Baxter et al., 2014).
Glutathione (GSH) plays a crucial role in cell defense against oxidative stress (Dringen, 2000; Maher, 2006). GSH scavenges superoxide and other ROS both directly and, more rapidly, in enzyme-catalyzed reactions. One such reaction class reduces peroxides, as exemplified by the glutathione peroxidase reaction: 2GSH + H2O2 → GSSG + 2H2O. A second class of reactions includes glutaredoxin-mediated repair of proteins that have been oxidatively modified at cysteine residues. These reactions also ultimately generate GSSG from GSH. The GSSG (glutathione disulfide) formed in these processes can either be exported from cells or recycled to GSH in the glutathione reductase reaction: GSSG + NADPH → 2GSH + NADP+. GSSG has intrinsic cytotoxicity, and consequently cells export GSSG when formation exceeds capacity for recycling to GSH (Homolya et al., 2003). GSH moieties lost through GSSG export must be replaced by de novo GSH synthesis, for which cysteine availability is usually the rate liming factor (Jones, 2008). GSH depletion has also been shown to impair mitochondrial ATP production (Vesce et al., 2005) and promote mitochondrially driven apoptosis (Muyderman et al., 2007).
The obligatory role of GSH in these anti-oxidant and repair processes suggests that intracellular GSH levels could be an important factor affecting neuronal survival during ischemia-reperfusion, but there are several gaps to our understanding in this area. It is not known whether ischemia-reperfusion reduces GSH levels specifically in neurons, if so by what mechanism, or if this reduction significantly contributes to neuronal demise. There is also uncertainty as to the relative GSH concentrations in neurons compared with astrocytes. Studies of pure neuronal and astrocyte cultures suggest that neurons contain far less GSH than astrocytes (Makar et al., 1994; Dringen et al., 1999); however, this may be a cell culture artifact because cultured astrocytes display a reactive phenotype in which the GSH biosynthetic pathway is upregulated (Shih et al., 2003), and neuron levels of GSH are artificially depressed when cultured in the absence of astrocytes (Dringen et al., 1999; Dringen, 2000).
To resolve these issues, we used an immunohistochemical method to evaluate GSH content in individual neurons. Results of these studies show that GSH levels in hippocampal pyramidal neurons are normally greater than astrocyte GSH levels, and that neuronal GSH levels fall in a time-dependent manner after ischemia-reperfusion. Blocking superoxide production during reperfusion preserves neuronal GSH levels, and supporting neuronal GSH levels with N-acetyl cysteine reduces oxidative stress and neuronal death.
Materials and Methods
Studies were approved by the SFVAMC animal studies committee. Mice were male, C57BL/6, 3–5 months of age. Wild-type (WT) mice were obtained from Simonsen Laboratories. p47phox−/− Mice were obtained from The Jackson Laboratory, and subsequently back-crossed to WT C57BL/6 mice for >10 generations. Reagents were obtained from Sigma-Aldrich except where otherwise noted.
Transient forebrain ischemia.
Ischemia was induced by transient occlusion of both common carotid arteries, as described previously (Barone et al., 1993; Won et al., 2010). Mice were fasted overnight and anesthetized with 2% isoflurane in a 70% N2O/30% O2 mixture. Body temperature was maintained at 37 ± 0.3°C with a homeothermic blanket and heating lamp (Harvard Apparatus). The common carotid arteries were exposed through a midline neck skin incision and occluded for 12 min with small aneurysm clips. Artery occlusion and reperfusion was confirmed in each case by visual inspection. Anesthetics were discontinued after skin closure. When mice showed spontaneous respiration, they were returned to a recovery chamber maintained at 37°C until ambulatory. Sham-operated animals received neck incisions without artery occlusion.
Where used, N-acetyl cysteine (NAC), apocynin, or their corresponding vehicles were administered as 10 μl/g intraperitoneal injections at the time of reperfusion. NAC was dissolved in physiological saline, and apocynin was dissolved in 1% dimethylsulfoxide. For studies using hyperglycemia, mice were given intraperitoneal injection of 50% glucose in saline, (2.5 μl/g body weight) immediately before reperfusion as described previously (Suh et al., 2008). The corresponding normoglycemic mice received equal volumes of saline only. Blood glucose was measured from tail vein samples 15 min before ischemia onset and 30 min after reperfusion using ACCU-CHEK glucometer (Roche).
Immunohistochemistry.
Mice were anesthetized and transcardially perfused with cold saline followed by 4% formaldehyde solution. Brains were removed and postfixed in 4% formaldehyde overnight. Brains used for 4-hydroxynonenal (4HNE) and nitrotyrosine (NT) immunostaining were embedded in paraffin for collection of 7 μm coronal sections. The sections were deparaffinized with xylene, rehydrated, and then boiled in 10 mm sodium citrate, pH 7.0, for 10 min to achieve antigen retrieval (Shi et al., 1991). Brains used for other outcome measures were cryoprotected in sucrose and cryostat sectioned at either 25 μm thickness for Fluoro-Jade B staining, or 40 μm thickness for all other measures.
GSH was detected in sections that were preincubated with 10 mm N-ethylmaleimide (NEM) for 4 h at 4°C, using a mouse antibody to GSH-NEM (clone 8.1GSH, Millipore) as described previously (Miller et al., 2009; Escartin et al., 2011). The sections were subsequently incubated with rabbit antibodies to 4HNE (Abcam), NT (Millipore), MAP2 (Millipore), GFAP (Millipore), or Iba1 (Waco). Antibody binding was visualized using AlexaFluor 488-conjugated goat anti-mouse IgG or AlexaFluor 594-conjugated goat anti-rabbit IgG (Invitrogen).
For detection of superoxide-derived ROS, 3 mg/kg dihydroethidium (Invitrogen) dissolved in 1% dimethylsulfoxide was intraperitoneally injected 15 min before artery occlusion and brains were harvested at the designated time points following reperfusion. Cryostat sections were photographed with a fluorescent microscope using excitation at 510–550 nm and emission >580 nm to detect oxidized ethidium species (Eth) (Murakami et al., 1998; Won et al., 2010).
Quantification of fluorescent labeling was performed using four evenly spaced sections collected through the hippocampus of each mouse. Each section was photographed with a fluorescence microscope using uniform conditions. Antibody or Eth fluorescence intensity was measured in the bilateral CA1 pyramidal layer of each of section, and the values obtained in the four sections were averaged for each brain. To assess colocalization of glutathione and superoxide formation at the single-cell level, brain sections from dihydroethidium-injected mice were immunostained with anti-GS-NEM and photographed with a confocal fluorescence microscope. Eth and anti-GS-NEM fluorescence were measured in each neuronal soma of hippocampal CA1, with >70 cells analyzed in each section.
Neuron death.
Degenerating neurons were evaluated by Fluoro-Jade B staining as described previously (Schmued and Hopkins, 2000; Won et al., 2010), using five evenly spaced sections through the hippocampus. In brief, 25 μm sections mounted on slides were immersed in a basic alcohol solution followed by 0.06% potassium permanganate for 15 min, placed in 0.0004% Fluoro-Jade B (Millipore) for 20 min. The number of degenerating neurons in CA1 pyramidal layer was counted bilaterally and averaged over the five sections from each brain.
Acute brain-slice cultures.
Mouse brains were vibratome sectioned into 300 μm coronal slices. The slices were placed in ice-cold artificial CSF (130 mm NaCl, 3.5 mm KCl, 1.25 mm NaH2PO4, 2 mm MgSO4, 2 mm CaCl2, 20 mm NaHCO3 and 10 mm glucose, pH7.2) while equilibrated with 95% oxygen and 5% CO2. After 30 min, the incubation temperature was raised to 30°C and one-half of the slices were treated with 0.5 mm 3-morpholinosydnonimine (SIN-1) plus 1 mm buthionine sulfoxamine (BSO) to reduce glutathione content (SIN-1 generates peroxynitrite, which avidly reacts with GSH, and BSO prevents de novo GSH synthesis; Griffith and Meister, 1979; Zhang et al., 1997). After 6 h, slices were either frozen for biochemical GSH determination or fixed in 4% formaldehyde for GSH-NEM immunohistochemistry.
GSH assay.
Brain slices were sonicated with 0.5 ml of 5% sulfosalicylic acid and centrifuged at 10,000 × g for 10 min at 4°C. The supernatant was mixed with 1 mm dithiobis-2-nitrobenzoic acid and 1 mm EDTA in 100 mm sodium phosphate buffer, pH 7.5, and 1 mm NADPH and 200 U/ml of glutathione reductase were added (Baker et al., 1990). GSH standards were treated identically, and optical absorbance of samples and standards was measured at 405 nm. Values were normalized to protein content as determined with a BCA protein assay kit (Thermo Scientific).
Statistical analyses.
Quantified data are presented as box-and-whisker plots, with the boxes showing the median and the upper and lower quartiles, and the whiskers showing the highest and lowest values in each the dataset. Statistical significance was assessed with the Mann–Whitney U test for two-group comparisons, and with the Kruskal–Wallis nonparametric one-way ANOVA test followed by Dunn's test for multiple group comparisons. P values <0.05 were considered significant. The number of mice in each experimental group is displayed in each figure.
Results
Ischemia reduces neuronal GSH content
To evaluate cell-type-specific changes in glutathione content, we adapted an immunohistochemical approach that uses antibody to GSH-NEM adducts. This method specifically identifies GSH in NEM-treated tissues, and thereby overcomes the more limited specificity of antibodies directed to native GSH (Miller et al., 2009). Hippocampal sections evaluated using this approach showed a strong GSH signal in the CA1 pyramidal neuron soma, with lesser signal in the adjacent neuropil and astrocyte cell bodies (Fig. 1A). The CA1 pyramidal layer was chosen for analysis of neuronal GSH changes because this region is relatively poor in astrocyte processes (Ouyang et al., 2007). This was confirmed here by staining for the astrocyte marker, GFAP (Fig. 1B; see also Fig. 7A). GSH-NEM staining was absent in sections that had not been treated with NEM, and in sections treated with iodoacetamide to render sulfhydryl groups unreactive to NEM (Fig. 1B). To further evaluate the specificity of this histochemical approach, we compared the GSH-NEM signal to biochemical measurements of GSH in acutely isolated hippocampal slices. In one-half of the slices GSH levels were lowered by 6 h incubations in medium containing 3-morpholinosydnonimine and l-buthionine sulfoxamine. Slices treated this way showed a ∼50% reduction in total slice GSH-NEM signal, and a comparable reduction in biochemically measured slice GSH content (Fig. 1C–E).
Figure 1.

Immunohistochemical quantification of neuronal GSH content. A, Confocal images of mouse hippocampal sections treated with NEM to generate NEM-GSH adducts and immunostained with antibody for GSH-NEM (green). Sections were counterstained (red) with either anti-MAP2 to identify neuronal processes or GFAP to identify astrocyte processes. Specificity was validated by lack of GSH-NEM signal in sections not treated with NEM, and in sections pretreated with iodoacetamide to render sulhydryl groups unreactive with NEM (B). Scale bar, 20 μm. C, In ex vivo brain slices, neuronal GSH-NEM signal was reduced after incubation with 1 mm BSO plus 500 μm SIN1 for 6 h. Scale bar, 20 μm. D, Immunohistochemical quantification of neuronal GSH in ex vivo brain slices. E, Biochemical quantification of GSH in the same ex vivo brain slices (*p < 0.05).
Figure 7.
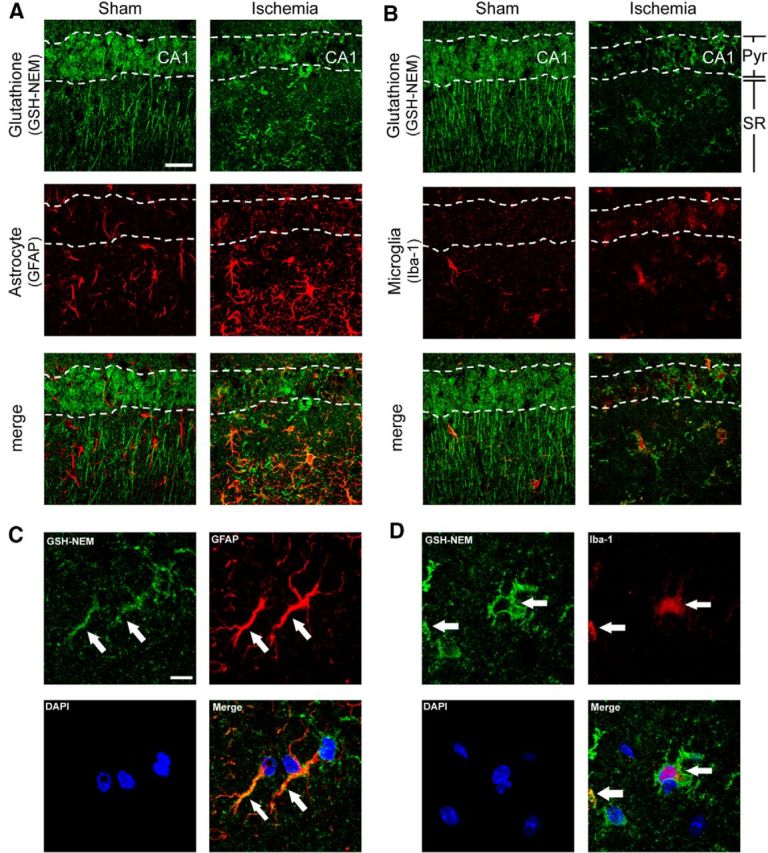
GSH content is increased in glia at 3 d after ischemia-reperfusion. Confocal images show fewer pyramidal neurons in the postischemic CA1 region, but increased numbers of astrocytes (A) and microglial (B) in both the CA1 pyramidal cell layer and underlying stratum radiatum. Colabeling for GSH shows increased signal in astrocytes (GFAP) and microglia (Iba-1) in the postischemic CA1. Pyr, Pyramidal layer; SR, stratum radiatum. Scale bar, 20 μm. C, D, High-magnification images with glial processes marked with arrows and cell nuclei stained with DAPI (blue). Scale bar, 10 μm. Images are representative of n = 4 sham and ischemic mice.
We then used the GSH-NEM method to evaluate changes in the GSH content of hippocampal CA1 pyramidal neurons at serial time points after transient forebrain ischemia. GSH levels were not detectably altered at 30 min after reperfusion, but were significantly decreased at 3 h and nearly absent at 24 h after reperfusion (Fig. 2A,B). To determine whether these decreases were simply indicative of neuronal death, degenerating neurons were identified by Fluoro-Jade B staining in adjacent sections. The Fluoro-Jade B staining showed only scattered dead neurons at 24 h and no signal at the earlier time points (Fig. 2C), consistent with delayed neuronal death in this stroke model (Barone et al., 1993; Bennett et al., 1996; Lee et al., 2004; Won et al., 2010).
Figure 2.
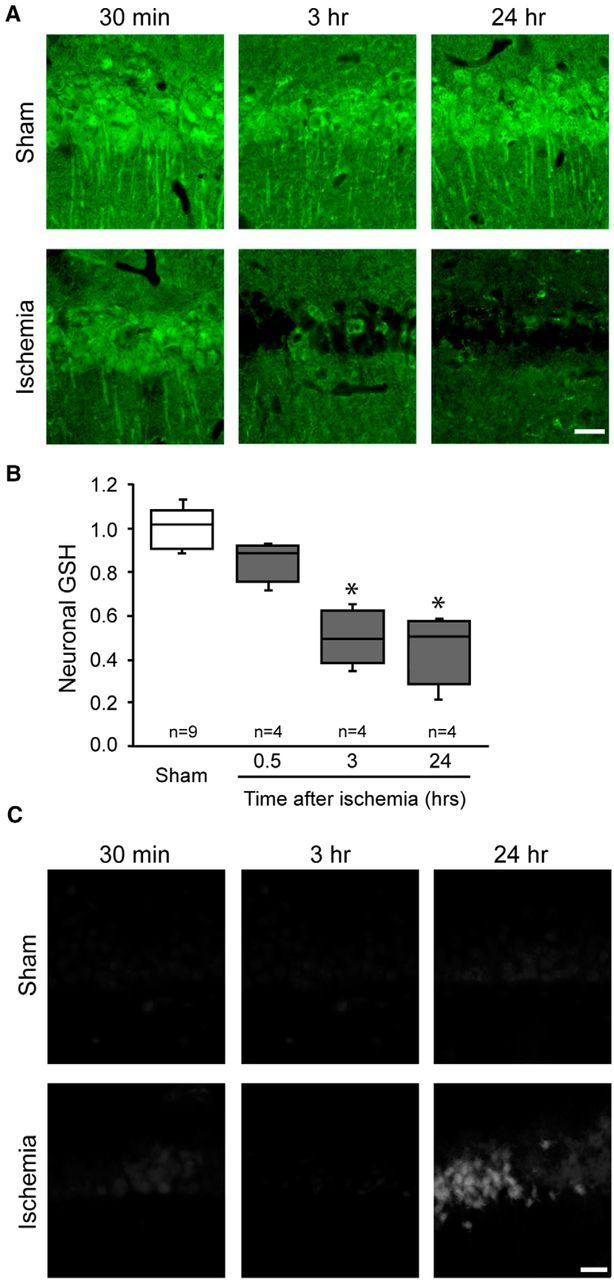
Transient forebrain ischemic injury produces a delayed depletion of GSH in hippocampal CA1 pyramidal neurons. A, Hippocampal brain sections harvested at the designated times after ischemia-reperfusion or sham surgery and immunostained for GSH. Scale bar, 20 μm. B, Quantification of neuronal GSH immunoreactivity (*p < 0.05 vs sham). C, Neuronal death as detected with Fluoro-Jade B under conditions as in A. Scale bar, 20 μm.
Reduced neuronal GSH content is accompanied by increased superoxide levels
Superoxide and certain superoxide-derived ROS produced during ischemia-reperfusion can be detected by evaluating oxidation of dihydroethidium to fluorescent oxidized Eth (Murakami et al., 1998; Peshavariya et al., 2007; Won et al., 2010). We prepared confocal images from sections double-labeled for Eth formation and GSH-NEM to evaluate the relationships between superoxide and GSH levels in individual neurons. Eth formation in hippocampal sections harvested at serial time points after ischemia-reperfusion showed increased signal in the CA1 pyramidal neurons at 0.5, 3, and 24 h after reperfusion (Fig. 3A). Focusing on the 3 h time-point, preceding neuronal death, the colabeling showed a corresponding reduction in neuronal GSH levels (Fig. 3A–D).
Figure 3.

Inverse relationship between superoxide and GSH levels in postischemic hippocampal neurons. A, Confocal images showing glutathione (GSH-NEM, green) and superoxide (Eth, red) in neurons of hippocampal sections harvested after ischemia-reperfusion or sham surgery. Scale bar, 20 μm. B, Scatterplot shows relative levels of GSH and superoxide measured in individual neurons harvested 3 h after ischemia or sham surgery (sham n = 4,744 cells; ischemia n = 4,783 cells). C, Histograms show the distribution of data plotted in B. D, Box-and-whisker plots show the median values from n = 4 animals in each treatment group (*p < 0.05).
Superoxide production causes neuronal GSH depletion after ischemia
GSH is consumed in the process of scavenging superoxide and superoxide-derived reactive oxygen species. As a consequence, the reciprocal changes in GSH and superoxide levels in postischemic neurons could result either from increased GSH consumption in neurons with increased superoxide formation, or from reduced superoxide-scavenging capacity in neurons with reduced GSH content. To test these possibilities, we first evaluated the effects of increased and decreased superoxide production on neuronal GSH levels. Superoxide formation was increased by rendering mice hyperglycemic during reperfusion (Muranyi and Li, 2006; Kamada et al., 2007; Suh et al., 2008; Won et al., 2011). The hyperglycemia-induced increase in superoxide production produced a corresponding decrease in neuronal GSH content (Fig. 4A). Next, we suppressed superoxide formation by treating mice with the NADPH oxidase inhibitor apocynin (Stolk et al., 1994; Suh et al., 2008). The reduced superoxide signal in neurons of these mice was accompanied by a corresponding increase in neuronal GSH content (Fig. 4B). We also evaluated this relationship in p47phox−/− mice, which cannot assemble a functional NADPH oxidase-2 complex (Bedard and Krause, 2007). Ischemia-reperfusion in these mice produced no significant increase in neuronal superoxide signal, and no reduction in neuronal GSH content (Fig. 4C). Together, these findings support the idea that accelerated superoxide production is the primary cause of GSH depletion in postischemic neurons. The absence of ischemia-induced neuronal Eth formation in mice lacking NADPH oxidase-2 activity also confirms that this signal is attributable to superoxide or superoxide-derived ROS.
Figure 4.
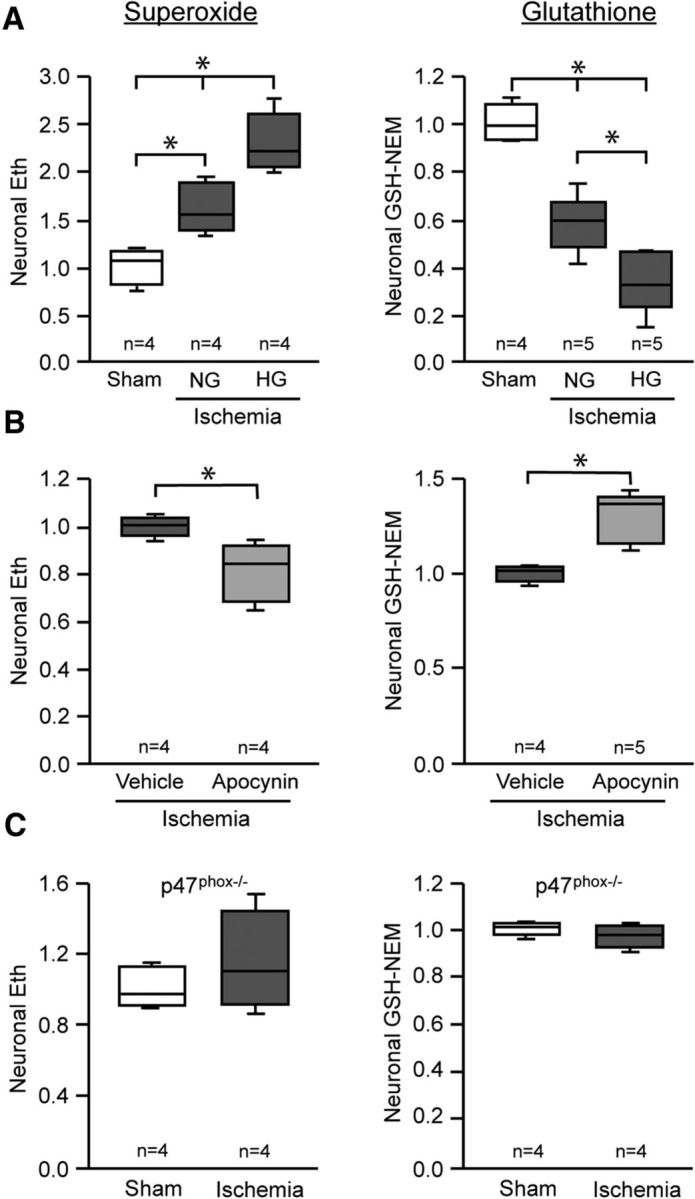
Neuronal GSH levels are influenced by postischemic superoxide production. Neuronal superoxide and GSH levels were assessed in hippocampal CA1 pyramidal neurons fixed 3 h after ischemia-reperfusion or sham surgery. A, Superoxide levels were increased by normoglycemic ischemia-reperfusion (NG), and further increased in mice rendered hyperglycemic during reperfusion (HG; *p < 0.05 vs sham). Neuronal GSH exhibited reciprocal changes. Blood glucose levels were 4.61 ± 0.82 mmol/L in normoglycemic mice and 14.89 ± 2.93 mmol/L in the hyperglycemic mice. B, Apocynin (15 mg/kg) reduced superoxide production in postischemic neurons, and prevented GSH depletion (*p < 0.05). C, Ischemia-reperfusion did not increase superoxide levels in p47phox−/− mice, and neurons in these mice showed no significant fall in GSH levels.
Increased GSH content suppresses superoxide levels after ischemia
The above findings do not exclude the alternative possibility, that elevated superoxide levels result from decreased neuronal GSH content. We evaluated this possibility by treating mice with NAC immediately after reperfusion. NAC serves as a source of cysteine, the rate-limiting step for de novo GSH synthesis (Aoyama et al., 2008; Samuni et al., 2013). Mice treated with NAC after ischemia had normal neuronal GSH levels and less Eth formation than vehicle-treated mice (Fig. 5), suggesting that the normalized GSH content prevents elevated superoxide levels during reperfusion. This suggestion was supported by measures of 4-hydroxynonenal and nitrotyrosine, which are oxidative modifications of lipids and proteins that are produced in part by superoxide (Reiter et al., 2000; Awasthi et al., 2004). Both 4-hydroxynonenal and nitrotyrosine were formed in CA1 neurons after ischemia-reperfusion (Fig. 6A), and their formation was attenuated in mice treated with NAC (Fig. 6A,B). NAC administration also reduced CA1 neuronal death, as measured by Fluoro-Jade B in brains harvested 3 d after ischemia-reperfusion (Fig. 6C,D). Together, these findings results indicate that neuronal GSH levels contribute to the ability of neurons to scavenge superoxide and suppress oxidative damage after ischemia-reperfusion.
Figure 5.
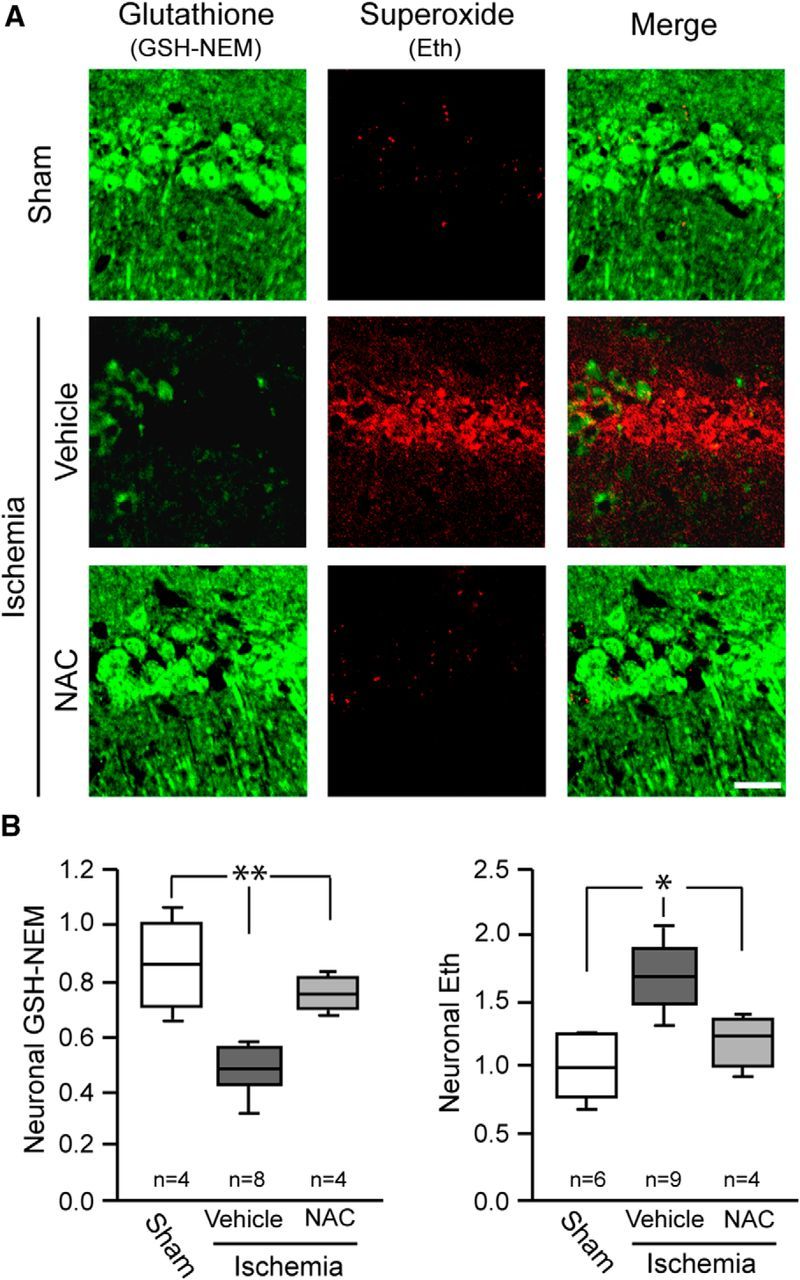
N-acetyl cysteine administered after ischemia-reperfusion restores neuronal GSH and reduces detectable superoxide. A, Representative photomicrographs showing relative levels of GSH and superoxide detection in hippocampal CA1 neurons. NAC or saline vehicle was administered at reperfusion, and tissues were harvested 3 h later. Scale bar, 20 μm. B, Quantified results (*p < 0.05, **p < 0.01 vs ischemia/vehicle).
Figure 6.

N-acetyl cysteine administered after ischemia-reperfusion attenuates oxidative stress and cell death. A, Hippocampal sections immunostained for 4HNE and NT. Mice received 7.5 mg/kg NAC or saline vehicle at reperfusion, and tissues were harvested 3 h later. Scale bar, 20 μm. B, Quantification of 4HNE and NT immunofluorescence. C, Fluoro-Jade B staining identifies degenerating hippocampal CA1 neurons in mouse brain 3 d after ischemia-reperfusion. Scale bar, 50 μm. D, Quantified cell death (*p < 0.05 vs ischemia/vehicle).
GSH content is increased in glia at 3 d after ischemia-reperfusion
Although our findings show a fall in neuronal GSH content over the first few hours after ischemia-reperfusion, prior studies using biochemical GSH determinations have identified an increase in bulk tissue GSH content at later time points (days) after ischemia-reperfusion (Uemura et al., 1991; Ningaraj and Rao, 1998; Namba et al., 2001). To determine whether the later increase in GSH content might be in non-neuronal cells, we performed double-labeling for GSH-NEM with astrocyte and microglial markers in brains harvested 3 d after ischemia reperfusion. These studies showed reduced GSH signal in the CA1 pyramidal layer, corresponding to a reduced number of surviving neurons. However, these studies also showed increased GSH content in both astrocytes and microglia in the underling stratum radiatum (Fig. 7). High-power views (Fig. 7C,D) suggested an increase in GSH concentration in the glial processes, in addition to increased numbers of cells and processes.
Discussion
These findings demonstrate that ischemia-reperfusion causes both a fall in GSH and an increase in superoxide levels in pyramidal CA1 neurons. Pharmacological or genetic blockade of superoxide production preserved neuronal GSH content, whereas conversely, N-acetyl cysteine treatment supported GSH levels and reduced superoxide levels, oxidative stress, and neuronal death. These results indicate that GSH depletion is both a result and a cause of neuronal oxidative stress after ischemia-reperfusion, and that restoration of neuronal GSH levels during reperfusion can be neuroprotective.
It can be difficult to reliably distinguish between GSH and other thiols by histochemical methods. Initial studies of brain tissue using mercury orange as a label found a relatively low GSH signal in neuronal somata (Slivka et al., 1987), but this agent was later found to react with protein sulfhydryls in addition to GSH (Thomas et al., 1995). Studies using monochlorobimane to detect GSH also suggested a relative paucity of GSH in neurons (Bragin et al., 2010). However, the reaction between monochlorobimane and GSH requires glutathione S-transferase. The GSH-bimane signal is therefore influenced by cell-type differences in this enzyme, as well as by differing rates of GSH-bimane excretion (Ublacker et al., 1991). In contrast to these histochemical methods, immunostaining with antibodies to GSH showed abundant labeling in neuronal soma and processes (Ong et al., 2000), in-line with the present results. Nonenzymatic GSH labeling with fluorescently labeled C5-maleimide also shows strongest signal in neurons (Aoyama et al., 2006; Escartin et al., 2011). The NEM-GSH antibody method used here uses antibodies to N-ethylmaleimide-glutathione adducts to further improve specificity for GSH (Miller et al., 2009).
In the present studies, comparisons of GSH-NEM staining with biochemical GSH measures in acute hippocampal slices confirmed that the GSH-NEM method provides a quantitative measure of brain tissue GSH content. The GSH-NEM approach has limitations, however, as with other immunohistochemical methods a linear relationship between neuronal GSH content and the GSH-NEM signal cannot be assumed to hold over the full range of signal detected. Additionally, the inability to discriminate between neuronal and non-neuronal processes in the neuropil restricted our analysis to neuronal cell bodies, and it is possible that these do not uniformly reflect GSH metabolism in the neuronal processes.
When used in studies of ischemia-reperfusion, GSH-NEM staining showed GSH depletion in hippocampal pyramidal neurons over the first few hours of ischemia-reperfusion. This depletion occurs long before neuronal death, as evidenced both by Fluoro-Jade B staining (Fig. 2C) and by prior work showing delayed CA1 neuronal death in this transient ischemia model (Barone et al., 1993; Bennett et al., 1996; Lee et al., 2004; Won et al., 2010). Biochemical measurements of GSH in brain tissue homogenates have shown a decrease in bulk tissue GSH content over this same time period (Uemura et al., 1991; Shivakumar et al., 1995), and a subsequent normalization or supranormalization after 72 h (Ningaraj and Rao, 1998; Namba et al., 2001). Our studies using GSH-NEM staining together with cell-type-specific markers indicates that this later recovery of GSH levels is attributable to GSH accumulation in reactive astrocytes and microglia, rather than to a rebound increase in surviving neurons. The mechanism and functional effects of this delayed GSH accumulation are not known, but may be attributable to increased NRF2 signaling in reactive astrocytes (Hamby and Sofroniew, 2010).
Although Eth formation is specific for superoxide under cell-free conditions, the presence of metals and peroxidases in cells allows H2O2 and other peroxides to oxidize dihydroethidium to Eth species (Fernandes et al., 2007). Elevated Eth signal is generally considered an indicator of increased ROS production, but in the present studies, the high Eth signal observed in neurons with low GSH content more likely reflects impaired superoxide scavenging. Superoxide is scavenged in part by direct reaction with GSH, and superoxide-derived peroxides and protein oxidation products are reduced primarily by enzymatic processes coupled to GSH (Dringen, 2000; Maher, 2006).
The mechanism by which ischemia-reperfusion causes GSH depletion has not been identified, but our results suggest that superoxide production is a major factor. We found that neuronal GSH depletion was attenuated when superoxide production by NADPH oxidase-2 was blocked, and increased in when superoxide production was accelerated by hyperglycemia (Fig. 4A). GSH is oxidized to GSSG in the process of scavenging superoxide and superoxide-derived peroxides, and exported from cells when GSSG formation outstrips its reduction back to GSH (Homolya et al., 2003).
GSH that is lost in the form of exported GSSG must be replaced by de novo synthesis from cysteine, glycine, and glutamate, of which cysteine availability is the rate-limiting factor. NAC is membrane-permeable form of cysteine that can be de-acetylated and used for GSH synthesis by neurons (Berman et al., 2011; Samuni et al., 2013). Prior studies have shown that NAC administered before brain ischemia can be neuroprotective (Knuckey et al., 1995; Carroll et al., 1998; Cuzzocrea et al., 2000). Our results additionally show that NAC administered during reperfusion following ischemia can restore or maintain intracellular GSH content in neurons, and thereby limit oxidative neuronal damage and death. It is possible that the effects of NAC are due in part to direct actions of this thiol with superoxide or other ROS; however, this is likely a minor factor because the reaction between NAC and superoxide is slow (Samuni et al., 2013), and because unlike GSH, NAC cannot serve as a substrate for either the reduction of peroxides by glutathione peroxidase or the repair of oxidized proteins by glutaredoxins. Accordingly, prior studies show that the antioxidant effects of NAC during acute oxidative stress are negated if de novo GSH synthesis is prevented with buthionine sulfoximine (Wong and Corcoran, 1987; Yang et al., 2003; Aoyama et al., 2006). This control was not feasible in the present ischemia studies because inhibitors of GSH synthesis downregulate NMDA receptor activity and thus also affect superoxide production (Ryu et al., 2003; Brennan et al., 2009).
The observation that both GSH depletion and neuronal demise were attenuated in mice treated with NAC during reperfusion after ischemia also suggests that this approach could have therapeutic potential. NAC is inexpensive and safe, and it is already approved by the US Federal Drug Administration for other indications (Miller and Rumack, 1983). Our findings suggest that administration of NAC to stroke patients during the initial period of reperfusion could improve outcomes, but the “therapeutic window of opportunity” remains to be established.
Footnotes
This work was supported by the NIH (NS081149 to R.A.S.) and the US Department of Veterans Affairs.
The authors declare no competing financial interests.
References
- Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi Z, West AB, Koehler RC, Poirier GG, Dawson TM, Dawson VL. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med. 2011;17:692–699. doi: 10.1038/nm.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Watabe M, Nakaki T. Regulation of neuronal glutathione synthesis. J Pharmacol Sci. 2008;108:227–238. doi: 10.1254/jphs.08R01CR. [DOI] [PubMed] [Google Scholar]
- Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic Biol Med. 2004;37:607–619. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- Baker MA, Cerniglia GJ, Zaman A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem. 1990;190:360–365. doi: 10.1016/0003-2697(90)90208-Q. [DOI] [PubMed] [Google Scholar]
- Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab. 1993;13:683–692. doi: 10.1038/jcbfm.1993.87. [DOI] [PubMed] [Google Scholar]
- Baxter P, Chen Y, Xu Y, Swanson RA. Mitochondrial dysfunction induced by nuclear poly(ADP-ribose) polymerase-1: a treatable cause of cell death in stroke. Transl Stroke Res. 2014;5:136–144. doi: 10.1007/s12975-013-0283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Pellegrini-Giampietro DE, Gorter JA, Aronica E, Connor JA, Zukin RS. The GluR2 hypothesis: Ca(++)-permeable AMPA receptors in delayed neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:373–384. doi: 10.1101/SQB.1996.061.01.040. [DOI] [PubMed] [Google Scholar]
- Berman AE, Chan WY, Brennan AM, Reyes RC, Adler BL, Suh SW, Kauppinen TM, Edling Y, Swanson RA. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1−/− mouse. Ann Neurol. 2011;69:509–520. doi: 10.1002/ana.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin DE, Zhou B, Ramamoorthy P, Müller WS, Connor JA, Shi H. Differential changes of glutathione levels in astrocytes and neurons in ischemic brains by two-photon imaging. J Cereb Blood Flow Metab. 2010;30:734–738. doi: 10.1038/jcbfm.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan-Minnella AM, Won SJ, Swanson RA. NADPH oxidase-2: linking glucose, acidosis, and excitotoxicity in stroke. Antioxid Redox Signal. 2015;22:161–174. doi: 10.1089/ars.2013.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JE, Howard EF, Hess DC, Wakade CG, Chen Q, Cheng C. Nuclear factor-kappa B activation during cerebral reperfusion: effect of attenuation with N-acetylcysteine treatment. Brain Res Mol Brain Res. 1998;56:186–191. doi: 10.1016/S0169-328X(98)00045-X. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Costantino G, Serraino I, Dugo L, Calabrò G, Cucinotta G, De Sarro A, Caputi AP. Beneficial effects of N-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130:1219–1226. doi: 10.1038/sj.bjp.0703421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/S0301-0082(99)00060-X. [DOI] [PubMed] [Google Scholar]
- Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C, Won SJ, Malgorn C, Auregan G, Berman AE, Chen PC, Déglon N, Johnson JA, Suh SW, Swanson RA. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J Neurosci. 2011;31:7392–7401. doi: 10.1523/JNEUROSCI.6577-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes DC, Wosniak J, Jr, Pescatore LA, Bertoline MA, Liberman M, Laurindo FR, Santos CX. Analysis of DHE-derived oxidation products by HPLC in the assessment of superoxide production and NADPH oxidase activity in vascular systems. Am J Physiol Cell Physiol. 2007;292:C413–C422. doi: 10.1152/ajpcell.00188.2006. [DOI] [PubMed] [Google Scholar]
- Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J Biol Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7:494–506. doi: 10.1016/j.nurt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolya L, Váradi A, Sarkadi B. Multidrug resistance-associated proteins: export pumps for conjugates with glutathione, glucuronate or sulfate. BioFactors. 2003;17:103–114. doi: 10.1002/biof.5520170111. [DOI] [PubMed] [Google Scholar]
- Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood–brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckey NW, Palm D, Primiano M, Epstein MH, Johanson CE. N-acetylcysteine enhances hippocampal neuronal survival after transient forebrain ischemia in rats. Stroke. 1995;26:305–310. doi: 10.1161/01.STR.26.2.305. discussion 311. [DOI] [PubMed] [Google Scholar]
- Lee SR, Tsuji K, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P. Redox control of neural function: background, mechanisms, and significance. Antioxid Redox Signal. 2006;8:1941–1970. doi: 10.1089/ars.2006.8.1941. [DOI] [PubMed] [Google Scholar]
- Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem. 1994;62:45–53. doi: 10.1046/j.1471-4159.1994.62010045.x. [DOI] [PubMed] [Google Scholar]
- Miller LF, Rumack BH. Clinical safety of high oral doses of acetylcysteine. Semin Oncol. 1983;10:76–85. [PubMed] [Google Scholar]
- Miller VM, Lawrence DA, Mondal TK, Seegal RF. Reduced glutathione is highly expressed in white matter and neurons in the unperturbed mouse brain: implications for oxidative stress associated with neurodegeneration. Brain Res. 2009;1276:22–30. doi: 10.1016/j.brainres.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni F. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8:96–103. doi: 10.1016/j.coph.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranyi M, Li PA. Hyperglycemia increases superoxide production in the CA1 pyramidal neurons after global cerebral ischemia. Neurosci Lett. 2006;393:119–121. doi: 10.1016/j.neulet.2005.09.079. [DOI] [PubMed] [Google Scholar]
- Muyderman H, Wadey AL, Nilsson M, Sims NR. Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J Neurochem. 2007;102:1369–1382. doi: 10.1111/j.1471-4159.2007.04641.x. [DOI] [PubMed] [Google Scholar]
- Namba K, Takeda Y, Sunami K, Hirakawa M. Temporal profiles of the levels of endogenous antioxidants after four-vessel occlusion in rats. J Neurosurg Anesthesiol. 2001;13:131–137. doi: 10.1097/00008506-200104000-00010. [DOI] [PubMed] [Google Scholar]
- Ningaraj NS, Rao MK. Disulfiram augments oxidative stress in rat brain following bilateral carotid artery occlusion. J Biomed Sci. 1998;5:226–230. doi: 10.1007/BF02253473. [DOI] [PubMed] [Google Scholar]
- Ong WY, Hu CY, Hjelle OP, Ottersen OP, Halliwell B. Changes in glutathione in the hippocampus of rats injected with kainate: depletion in neurons and upregulation in glia. Exp Brain Res. 2000;132:510–516. doi: 10.1007/s002210000347. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshavariya HM, Dusting GJ, Selemidis S. Analysis of dihydroethidium fluorescence for the detection of intracellular and extracellular superoxide produced by NADPH oxidase. Free Radic Res. 2007;41:699–712. doi: 10.1080/10715760701297354. [DOI] [PubMed] [Google Scholar]
- Reiter CD, Teng RJ, Beckman JS. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J Biol Chem. 2000;275:32460–32466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- Ryu BR, Lee YA, Won SJ, Noh JH, Chang SY, Chung JM, Choi JS, Joo CK, Yoon SH, Gwag BJ. The novel neuroprotective action of sulfasalazine through blockade of NMDA receptors. J Pharmacol Exp Ther. 2003;305:48–56. doi: 10.1124/jpet.102.042606. [DOI] [PubMed] [Google Scholar]
- Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830:4117–4129. doi: 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/S0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Shi SR, Key ME, Kalra KL. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J Histochem Cytochem. 1991;39:741–748. doi: 10.1177/39.6.1709656. [DOI] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar BR, Kolluri SV, Ravindranath V. Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. J Pharmacol Exp Ther. 1995;274:1167–1173. [PubMed] [Google Scholar]
- Slivka A, Mytilineou C, Cohen G. Histochemical evaluation of glutathione in brain. Brain Res. 1987;409:275–284. doi: 10.1016/0006-8993(87)90712-8. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol. 2008;64:654–663. doi: 10.1002/ana.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Nicklee T, Hedley DW. Differential effects of depleting agents on cytoplasmic and nuclear non-protein sulphydryls: a fluorescence image cytometry study. Br J Cancer. 1995;72:45–50. doi: 10.1038/bjc.1995.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ublacker GA, Johnson JA, Siegel FL, Mulcahy RT. Influence of glutathione S-transferases on cellular glutathione determination by flow cytometry using monochlorobimane. Cancer Res. 1991;51:1783–1788. [PubMed] [Google Scholar]
- Uemura Y, Miller JM, Matson WR, Beal MF. Neurochemical analysis of focal ischemia in rats. Stroke. 1991;22:1548–1553. doi: 10.1161/01.STR.22.12.1548. [DOI] [PubMed] [Google Scholar]
- Vesce S, Jekabsons MB, Johnson-Cadwell LI, Nicholls DG. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. J Biol Chem. 2005;280:38720–38728. doi: 10.1074/jbc.M506575200. [DOI] [PubMed] [Google Scholar]
- Won SJ, Yoo BH, Brennan AM, Shin BS, Kauppinen TM, Berman AE, Swanson RA, Suh SW. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J Neurosci. 2010;30:15409–15418. doi: 10.1523/JNEUROSCI.2084-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann Neurol. 2011;70:583–590. doi: 10.1002/ana.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BK, Corcoran GB. Effects of esterase inhibitors and buthionine sulfoximine on the prevention of acetaminophen hepatotoxicity by N-acetylcysteine. Res Commun Chem Pathol Pharmacol. 1987;55:397–408. [PubMed] [Google Scholar]
- Yang B, Keshelava N, Anderson CP, Reynolds CP. Antagonism of buthionine sulfoximine cytotoxicity for human neuroblastoma cell lines by hypoxia is reversed by the bioreductive agent tirapazamine. Cancer Res. 2003;63:1520–1526. [PubMed] [Google Scholar]
- Zhang H, Squadrito GL, Uppu RM, Lemercier JN, Cueto R, Pryor WA. Inhibition of peroxynitrite-mediated oxidation of glutathione by carbon dioxide. Arch Biochem Biophys. 1997;339:183–189. doi: 10.1006/abbi.1996.9863. [DOI] [PubMed] [Google Scholar]