

Abstract
Age-related macular degeneration (AMD) is the leading cause of blindness among older adults in the developed world. Although the pathological mechanisms have not been definitively elucidated, evidence suggests a key role for mitochondrial (mt) dysfunction. The current study used our unique collection of human retinal samples graded for the donor's stage of AMD to address fundamental questions about mtDNA damage in the retina. To evaluate the distribution of mtDNA damage in the diseased retina, damage in the retinal pigment epithelium (RPE) and neural retina from individual donors were compared. To directly test a long-held belief that the macula is selectively damaged with AMD, RPE mtDNA damage was measured in the macula and peripheral sections from individual donors. Small segments of the entire mt genome were examined to determine whether specific regions are preferentially damaged. Our results show that mtDNA damage is limited to the RPE, equivalent mtDNA damage is found in the macular and peripheral RPE, and sites of damage are localized to regions of the mt genome that may impact mt function. These results provide a scientific basis for targeting the RPE mitochondria with therapies that protect and enhance mt function as a strategy for combating AMD.
Keywords: age-related macular degeneration, mitochondria, mitochondrial DNA, oxidative damage, retina, retinal pigment epithelium
Introduction
Age-related macular degeneration (AMD) is the leading cause of blindness in elderly of the developed world (Friedman et al., 2004). This disease affects ∼28% of individuals 75–85 years (Leibowitz et al., 1980). In the US, ∼10 million people are blind or are at substantial risk of vision loss due to AMD (Alliance for Eye and Vision Research, 2013). These numbers are expected to double in the next 20 years when the majority of “baby boomers,” which currently make up ∼25% of the population, will be age 65 or older (Colby and Ortman, 2014).
AMD destroys the macula, which is the part of the retina that supplies high acuity central vision. The loss of central vision affects activities such as reading, driving, and face recognition, and has a significant negative impact on daily function and quality of life. The current estimate for the cost of visual impairment due to retinal disorders in the U.S. is $8.7 billion (Alliance for Eye and Vision Research, 2013). The personal and public costs of AMD, along with the staggering numbers afflicted, create an urgent need to improve AMD treatment strategies, which are currently limited to ∼10% of patients with “wet” AMD. A key feature of the more prevalent “dry” AMD is the loss of retinal pigment epithelial cells (RPE). The RPE are a monolayer of postmitotic cells that perform crucial functions for maintaining retinal homeostasis (Strauss, 2005). The AMD-associated loss of RPE results in photoreceptor death and blindness. Thus, new clinical strategies that effectively protect the RPE from death and target the disease mechanism are desperately needed.
The pathological mechanism(s) for AMD have not been definitively elucidated. However, converging evidence from multiple studies implicate mitochondrial (mt) dysfunction in the AMD disease process. Evidence supporting this hypothesis includes the observed loss in mt mass and intact internal structures, and decreased content of proteins in the electron transport chain in RPE from human donors with AMD (Feher et al., 2006; Nordgaard et al., 2006, 2008). Additionally, our laboratory (Karunadharma et al., 2010) and others (Lin et al., 2011) have recently shown increased levels of mtDNA damage in donor RPE at stages of AMD preceding vision loss. The purpose of this study was to address fundamental questions about mtDNA damage in the retina to gain greater insight into disease mechanism as a critical step in directing development of new treatments. Using our unique collection of human donor samples graded for the stage of AMD, we asked whether mtDNA damage was limited to the RPE or was more globally distributed throughout the diseased retina. To directly test a long-held belief that the macula is selectively damage with AMD, RPE mtDNA damage was compared in the macula and periphery from individual donors. To map regions of mtDNA damage, small segments of the mt genome were examined. Our results provide a scientific basis for targeting the RPE with therapies that protect mt function, suggests that mt damage goes beyond the macula, and provides some indication of potential functional effects associated with mtDNA damage in AMD.
Materials and Methods
Human tissue procurement.
Donor eyes were obtained from the Minnesota Lions Eye Bank with the consent of the donor or donor's family for use in medical research in accordance with the tenets of the Declaration of Helsinki. After enucleation, eyes were maintained in a moist chamber at 4°C until photographing and dissection. Dissection included a trephine punch (5 mm) of RPE cells and neural retina centered over the macula. Trephine punches of RPE cells were also obtained from the superior and inferior sections of the retina from a subset of donors, using the fovea, optic nerve, and branch retinal arteries and veins as landmarks. Following dissection, tissues punches were flash frozen in liquid nitrogen and stored at −80°C until further processing.
Evaluation of the donor's stage of AMD was determined by a board-certified ophthalmologist/retina specialist from stereoscopic fundus photographs of the RPE using the criteria established by the Minnesota Grading System (MGS) for eye-bank eyes (Olsen and Feng, 2004; Decanini et al., 2007). MGS 1 represents the control group with no clinically observable eye disease. MGS 2, 3, and 4 are early, intermediate, and advanced stages of AMD, respectively. Advanced AMD (MGS 4) includes both dry AMD (central geographic atrophy) and wet AMD (choroidal neovascularization). Exclusion criteria for the present study include a history of diabetes or glaucoma, clinical symptoms of diabetic retinopathy, advanced glaucoma, and myopic degeneration or atypical debris in the eyes. Records from the Minnesota Lions Eye Bank provided demographics of the donors including age, race, gender, time, and cause of death, and a family report of a limited medical and ocular history (Table 1). Donor tissue from 48 males and 41 females, age 60 and older, were used in this study.
Table 1.
Donor characteristics and clinical informationa
| MGSb grade | Sample (n) | Sex |
Age |
Cause of death (n)c | |
|---|---|---|---|---|---|
| M | F | Mean ± SD | |||
| 1 | 30 | 19 | 11 | 73 ± 9.7d | Cancer (11), COPD (4), CHF (3), sepsis (3), ACE (3), GI bleed (1), ARDS (2), pneumonia (1), CVA (1), organ failure (1) |
| 2 | 25 | 13 | 12 | 79 ± 10.4 | Pulmonary fibrosis (1), CHF (1), organ failure (2), sepsis (2), cancer (4), pneumonia (6), Guillan Barre syndrome (1), ACE (3), GI Bleed (1), CVA (2), bowel obstruction (1), ALS (1) |
| 3 | 21 | 10 | 11 | 82 ± 9.5 | Sepsis (2), metabolic acidosis (1), CHF (2), COPD (1), ACE (3), cancer (1), pneumonia (3), bowel obstruction (1), organ failure (2), ARDS (3), dementia (1), perforated bowel (1) |
| 4 | 13 | 6 | 7 | 85 ± 5.7 | CVA (2), sepsis (4), pneumonia (1), dementia (1), cancer (1), organ failure (1), ACE (2), AAA (1) |
aInformation supplied by Minnesota Lions Eye Bank.
bMinnesota Grading System (MGS) was used to evaluate the stage of AMD in eye-bank eyes (Olsen and Feng, 2004).
cThe number of donors for each cause of death is indicated in parentheses.
dMGS1 is significantly younger than MGS 3 and 4 (p = 0.0003).
ACE, Acute myocardial event; COPD, chronic obstructive pulmonary disease; CHF, congestive heart failure; GI bleed, gastrointestinal bleed; ARDS, acute respiratory disease syndrome; CVA, cerebrovascular accident (stroke); organ failure, kidney and/or multiple organ failure; ALS, amyotrophic lateral sclerosis; AAA, abdominal aortic aneurysm.
DNA isolation and quantification.
Total genomic DNA was isolated from 5 mm trephine punches of the RPE (peripheral and macula) or neural retina (macula) using a QIAamp DNA mini kit (Qiagen) and quantified using Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies), following the manufacturer's protocols. Picogreen dye fluorescence was measured using a multiwell plate reader (CytoFluor, PerSeptive Biosystems) with 485 nm/535 nm excitation/emission filters. Lambda DNA provided with the kit was used to construct a standard curve of known DNA concentrations.
Long-extension PCR.
The long-extension (LX) PCR assay (Santos et al., 2002; Kovalenko and Santos, 2009) was performed using a GeneAmp XL PCR kit (Life Technologies) for data in Figures 1 and 2. Primer sequences, conditions for each primer set, and PCR amplification profile were performed as described previously (Karunadharma et al., 2010). Half-template samples and no DNA controls were run on each plate to verify linearity and as a negative control, respectively. A small 191 or 222 bp mtDNA fragment in either the 16S rRNA or Cyt b genes, respectively, were amplified to account for total mtDNA copies in each sample. Prior comparison showed that amplicons from these two regions provided comparable results for estimating mtDNA content (Karunadharma et al., 2010).
Figure 1.

RPE mtDNA damage increases with AMD. Average lesion frequency was determined by LX PCR for Regions I–IV of the mt genome for each donor. Data were normalized to the mean for MGS1 (control, n = 27). Sample size for each MGS group is shown in the bar. MGS groups were compared by one-way ANOVA and Tukey's post hoc test. Significance was set at p < 0.05; *p = 0.0005, MGS2 and MGS4 were significantly greater than MGS1. Data are mean ± SEM.
Figure 2.
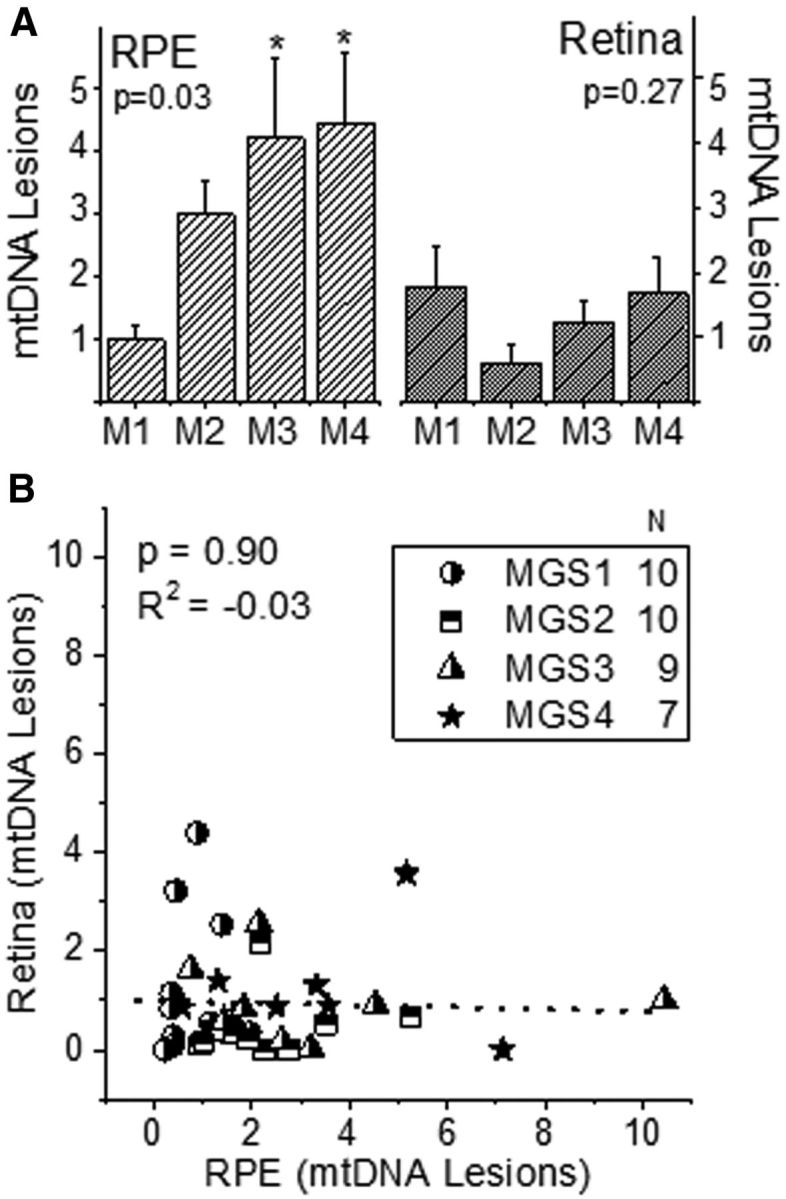
mtDNA damage increase with AMD in the RPE, but not retina. Average lesion frequency was determined for each donor as outlined in Figure 1. Data for both the RPE and retina were normalized to the mean of RPE MGS1 (control, n = 27). A, Plot shows mtDNA lesions increase with AMD progression in the RPE (p = 0.03) but not retina (p = 0.27) in donors with AMD (M2–M4, MGS2–4, n = 26) compared with age-matched controls (M1, MGS1, n = 10). Probability determined by one-way ANOVA and Tukey's post hoc test. Data are mean ± SEM. B, Plot of mtDNA lesions measured in the retina and RPE of individual donors shows no significant relationship (p = 0.90) between the extent of damage in these two ocular tissues. Note that each symbol represents an individual donor. Probability and R2 were determined by linear regression.
Due to technical issues with the enzyme (rTth) in the GeneAmp XL PCR kit, we used AccuPrime Taq Polymerase High Fidelity (Life Technologies) for the LX PCR analysis of four mtDNA regions (Regions I–IV) for punches from retinal sections (macula, inferior, superior; data shown in Fig. 3) and nine primer sets (A–I; data shown in Fig. 4). Primer sequences for amplification of nine regions of the human mtDNA genome were as reported previously (Taylor et al., 2001).
Figure 3.
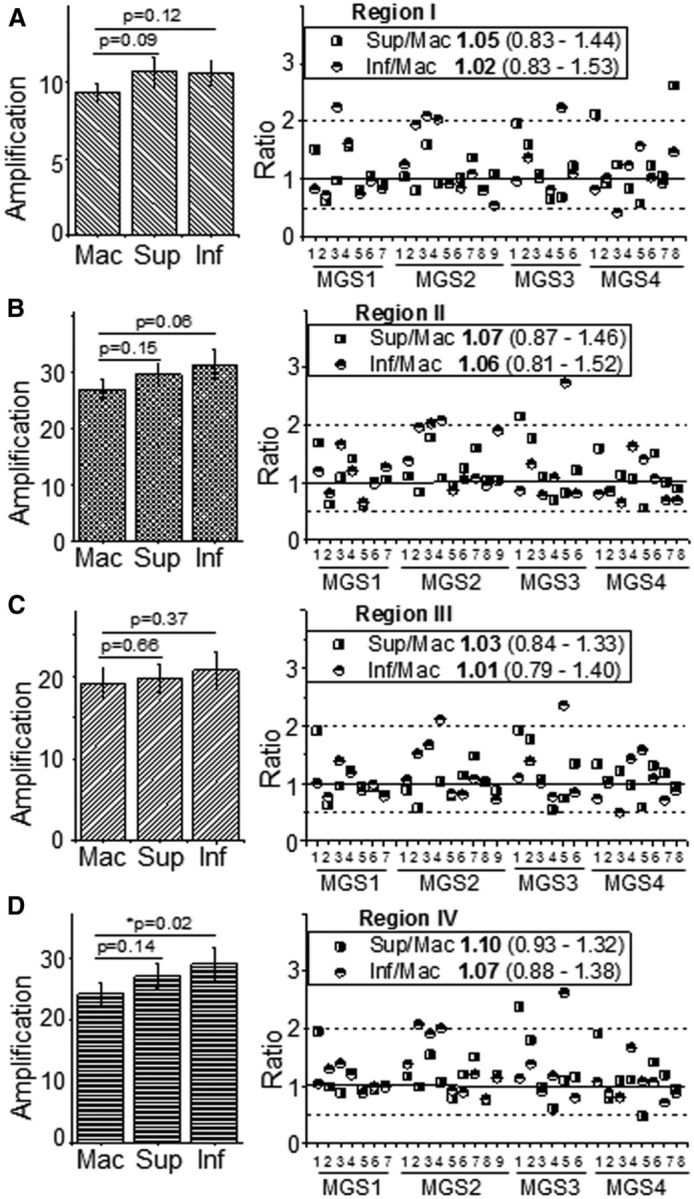
Comparison of RPE mtDNA damage in the macula and periphery. Graphs show results for mtDNA amplification for mt Regions I (A), II (B), III (C), and IV (D). The plots on the left report the mean relative amplification (±SEM) for punches from the macula, superior and inferior sections (n = 30/section). Relative amplification was compared between punches from the macula and periphery (inferior or superior) for individual donors using a paired t test. Probability values per comparison are reported above the graphs. Statistical significance was set at 0.025 based on a Bonferroni correction to control for the familywise error rate associated with multiple comparisons. Right, The ratio (peripheral section/macula) from individual donors from MGS1–4 (n = 6–9 per group) for each mtDNA region. Median and the interquartile range for the superior/macula or inferior/macula are also provided in the plot legends. Solid line indicates a ratio of 1; dashed lines demarcate the boundaries of a twofold increase or decrease in ratio.
Figure 4.

Localization of mt DNA damage. LX PCR was performed on donor RPE samples from the macular region using nine primer sets (A–I) encompassing the entire mitochondrial genome, including the D Loop. Lesion frequency (lesions/10 kb/double-strand) was normalized to the average relative amplification of MGS1 samples (n = 12–15) for each region. A two sample Student's t test was performed for each region, with significance set at p ≤ 0.05; *MGS3 was significantly higher than MGS1 for primer sets B (p = 0.02), G (p = 0.04), H (p = 0.03), and I (p = 0.001). Data are mean ± SEM.
The PCR amplification profile for Regions I–IV using AccuPrime Taq Polymerase included an initial denaturation for 30 s at 94°C, followed by 21 cycles of 94°C denaturation for 30 s for Region I, and 25 cycles for Regions II, III, and IV, and annealing/extension at 57°C for 30 s for all regions. The final extension was performed at 68°C for 4 min for Regions I and II, 6 min for Region III, and 2 min and 30 s for Region IV. The amplification profile for 191 bp mtDNA fragment included an initial denaturation for 30 s at 94°C, followed by 19 cycles of 94°C denaturation for 30 s, and annealing/extension at 55°C for 30 s and final extension at 30 s. The cycling parameters for the nine primer sets A–I included an initial denaturation for 30 s at 94°C, followed by 25 cycles of 94°C denaturation for 30 s, annealing/extension at 55.1°C for 30 s for primers A, B, E, F, G, and I; 59.4°C for primers C, D, and H. The final extension was performed at 68°C for 2 min for all primer sets. Half-template samples and no DNA controls were run on each plate to verify linearity and as a negative control, respectively. Each 25 μl PCR mixture contained sample genomic DNA (3 ng), sense and antisense primer (0.6 μm), AccuPrime Taq Polymerase (1 U), 10× Accuprime PCR Buffer II (0.25 μl), MgSO4 (1 mm), bovine serum albumin (2.5 μg), and DMSO (0.05%) and water. Quant-iT PicoGreen dsDNA Assay Kit was used to measure the total DNA amplified and was performed in triplicate for each reaction. All reactions were run on agarose gels (1% and 2%) and photographed under UV illumination to confirm that the amplicon size was correct.
Calculation of mtDNA damage.
MtDNA lesion frequency was calculated according to the Poisson equation assuming a random distribution of lesions (Ayala-Torres et al., 2000). Lesion frequency per 10 kb per genome (both strands) of the mtDNA was calculated by dividing sample amplification (relative to mtDNA content) by the average relative amplification of donors in MGS1 (no AMD, n = 27 donors). Graphs show data that is normalized to lesion frequency in MGS1.
For comparison of mtDNA damage between 5 mm trephine punches from the macula, superior, and inferior sections of individual donors, amplification of each primer set relative to mtDNA content was used as an estimate of mtDNA damage and compared between retinal sections. Data are presented as the ratio of relative amplification for inferior:macula and superior:macula for individual donors.
Statistics.
Statistical analysis included one-way ANOVA and Tukey's post hoc test to determine whether there was a significant difference in donor characteristics (Table 1) or mtDNA damage with AMD progression (Figs. 1, 2A). Linear Regression analysis compared the extent of damage in the RPE and retina for each donor to determine whether there was an association between damage in these two ocular tissues (Fig. 2B). A Student's t test was used to compare mtDNA damage in younger and older donors in MGS1 (Table 1), and for MGS1 and MGS3 for nine regions of the mt genome (Fig. 4). Significance was set at p < 0.05 for the above statistical tests. A paired t test, which allows for data from individual donors to remain linked during the analysis, was used to determine whether there were significant differences in the extent of mtDNA damage in peripheral regions (inferior and superior) versus the macula (Fig. 3). Statistical significance was set at 0.025 based on a Bonferroni correction to control for the familywise error rate associated with multiple comparisons. Analyses were performed using the statistical software in Origin 9.1 (Originlab). Data are reported as mean ± SEM for each group.
Results
Donor characteristics
Eyes from 89 donors, age 60 years and older, were graded for the stage of AMD by a board-certified ophthalmologist/retina specialist using the MGS (Olsen and Feng, 2004). Donors in MGS1 (n = 30) represent the age-matched control group with no clinically observable eye disease. Donors in MGS2 (n = 25), MGS3 (n = 21), and MGS4 (n = 13) represent progressively more severe stages of the disease. A summary of donor demographics and clinical information for each MGS group used in this study is provided in Table 1. The Minnesota Lions Eye Bank supplied all of the information about the donors, except for the MGS stage. The number of donors experiencing a specific “cause of death” is indicated for each group.
Average age for MGS1 (73 ± 9.7; mean ± SD) was not different from MGS2 (79 ± 10.4), but was significantly younger compared with MGS 3 (82 ± 9.5) and MGS 4 (85 ± 5.7; p = 0.0003). To determine whether there was an age-dependent difference in mtDNA damage, we compared damage in younger MGS1 donors (age 60–72; average 65 ± 3.8, n = 14) versus older MGS1 donors (age 75–91; average 82 ± 5.3, n = 13). There was no statistically significant difference between younger and older donors (p = 0.39), so the slight difference in age between MGS1 and MGS3/4 should not bias our results. The average time from death to enucleation for donors used in this study was 5.2 ± 2.6 h and was not different between MGS groups (p > 0.48). The average time from death to tissue cryopreservation was 18.2 ± 4.2 h and was not different between MGS groups (p > 0.08).
Increased mtDNA damage with AMD is limited to the RPE
The 16.6 kb mt genome encodes for 13 proteins in the electron transport chain and the machinery required for translation of these proteins within the mitochondria. Unlike nuclear DNA, mtDNA does not contain introns. The mt genome does contain one noncoding control region that regulates mtDNA replication and translation. Thus, ROS-induced mtDNA damage, which includes abasic sites, as well as single- and double-strand breaks (Demple and Harrison, 1994), can produce somatic mutations that have detrimental consequences to mt-encoded proteins participating in production of ATP. The net result of these changes includes reduced bioenergetics, increased generation of mt ROS, mt dysfunction, and eventual cell death (Richter et al., 1988; Bohr, 2002; Santos et al., 2004).
Our laboratory and others have previously shown that LX PCR is an effective method for determining whether the mt genome is preferentially damaged with AMD (Udar et al., 2009; Karunadharma et al., 2010; Lin et al., 2011). This assay is based on the principle that DNA lesions (i.e., abasic sites, strand breaks, and thymine dimers) can slow down or block the progression of a thermostable DNA polymerase and prevent complete product synthesis (Santos et al., 2002). Thus, lower-amplification results when the mt genome is damaged. To account for potential differences in mtDNA content, amplification of each mitochondrial region is normalized to total mtDNA content, which is determined by amplifying a small region (100–200 bases) of the mt genome. Since the probability of lesion occurrence in a very small region is low, this measurement can be used to estimate the total relative number of mtDNA molecules in each sample.
We had previously reported an increase in RPE mtDNA damage in donors with AMD (n = 46 donors) compared with age-matched controls (n = 26 donors; Karunadharma et al., 2010). The current study presents an evaluation of mtDNA damage in the macular RPE of 69 additional donors (27 controls, 42 donors with AMD). The results are consistent with our previous findings; mtDNA damage is significantly increased (p = 0.0005) in the RPE macula of donors with AMD (Fig. 1).
To determine whether there was a parallel increase in mtDNA damage with AMD progression in the neural retina, we measured the mtDNA damage in a 5 mm trephine punch centered over the macula of both the RPE and neural retina in a subset of donor samples (n = 7–10 per MGS group). Lesion frequency was calculated by dividing sample amplification (relative to mtDNA content) by the average relative amplification of MGS1 (control) RPE (n = 27). We observed that in the RPE of this subset of donors, the content of mtDNA lesion increased significantly with AMD progression (p = 0.03; Fig. 2A). In contrast, mtDNA lesions in the retina did not change with AMD progression (p = 0.27) and were ∼2-fold lower than RPE lesion content for donors with AMD. To test whether the extent of mtDNA damage measured in the RPE correlated with the damage in the retina, mtDNA lesions from the retina versus RPE for individual donors was plotted (Fig. 2B). As demonstrated from the nonsignificant fit of the data by linear regression (p = 0.90; R2 = 0.03), there is no relationship between the extent of mtDNA damage in the RPE and retina. Thus, increased mtDNA damage is limited to the RPE with AMD.
Similar RPE mtDNA damage in the macula and periphery
To determine whether retinal location can influence the extent of mtDNA damage, trephine punches of RPE cells were obtained from the macula, superior, and inferior sections of the retina for a subset of donors in each MGS group. Amplification of each mtDNA region (Regions I–IV) relative to the content of mtDNA provides an estimate of the extent of mtDNA damage, i.e., lower-amplification indicates more mtDNA damage. A paired t test was performed to determine whether there were differences between retinal punches for individual donors. Comparing amplification of the macula with the periphery (inferior or superior punches) for Regions I–IV (Fig. 3, left), there was no significant difference in relative amplification except for Region IV, where the inferior retina had higher-amplification indicative of less mtDNA damage compared with the macula (p = 0.02).
To help visualize the relationship between retinal sections, the ratio of amplification for the periphery (superior or inferior) and macula from individual donors was plotted (Fig. 3, right). As shown in the scatterplot and calculated median in Figure 3, most ratios were distributed ∼1 and fell within the twofold distribution (dashed lines) ∼1. These results indicate that for the majority of the donors, there was no difference in amplification. Together, these data support the idea that the extent of mtDNA damage is approximately equivalent for RPE sampled from different sections of the retina.
Localization of mt DNA damage within the mitochondrial genome
In our previous study (Karunadharma et al., 2010) and the results highlighted above, LX PCR was performed using four primer sets (referred to as Regions I– IV) that produced products in the range of 2.5–5.4 kb. With AMD, more mtDNA damage was observed in all regions of the RPE mt genome compared with age-matched controls. To more closely define and map regions of mtDNA damage, we used nine primer sets (A–I; Taylor et al., 2001) that produced smaller products (1.8–2 kb) and included the noncoding control region. For this study, donors from the intermediate stage (MGS3) of AMD were compared with age-matched controls. MGS 3 was selected because these donors were well into the disease but before secondary effects, such as choroidal neovascularization, which would have complicated interpretation of our findings.
In the current analysis of nine regions of the mt genome, we identified four regions (amplified by primer sets B and G–I) that showed more mtDNA damage in MGS3 (disease) compared with MGS1 (age-matched control; Fig. 4). The regions within the mt genome covered by each primer set, as well as the genes located within each region, are summarized in Table 2. Also provided in Table 2 are the primers for Regions I–IV from our first mtDNA damage study (Karunadharma et al., 2010), noting where there is overlap with primers in the current study. Genes within the regions covered by primer sets B, G, and H contained multiple tRNAs and six proteins of the electron transport chain involved in oxidative phosphorylation. Of note, the only overlap for Region II occurred with a small portion of primer B, which included the gene for NADH dehydrogenase subunit I. Genes within primer I mainly included regulatory elements of the control region for mtDNA replication. These new results show that ∼54% of the mt genome is not significantly damaged in donors with AMD, thus refining our previous report of genome-wide damage to more discrete regions of the mt genome.
Table 2.
Genes associated with regions of mtDNA damage
| Primer | Genea | Starting | Ending | Function |
|---|---|---|---|---|
| Region I | 607b | 4256b | ||
| Region II | 4223b | 8188b | ||
| B | 2364 | 4262 | ||
| NADH dehydrogenase subunit 1 | 3307 | 4262 | ATP production | |
| 16S ribosomal RNA | 1671 | 3229 | Mitochondrial protein synthesis | |
| Humanin | 2634 | 2707 | Mitochondrially derived peptide | |
| tRNA leucine 1 (LUUA/G) | 3230 | 3304 | tRNA | |
| Transcription terminator | 3229 | 3256 | Transcription regulation | |
| Region III | 8148b | 13594b | ||
| Region IV | 13541b | 15996b | ||
| G | 11614 | 13638 | ||
| NADH dehydrogenase subunit 4 | 10760 | 12137 | ATP production | |
| NADH dehydrogenase subunit 5 | 12337 | 14148 | ATP production | |
| tRNA histidine | 12138 | 12206 | tRNA | |
| tRNA leucine 2 L(CUN) | 12266 | 12336 | tRNA | |
| tRNA serine 2 S(AGY) | 12207 | 12265 | tRNA | |
| Region IV | 13541b | 15996b | ||
| H | 13539 | 15431 | ||
| Cytochrome b | 14747 | 15887 | ATP production | |
| NADH dehydrogenase subunit 5 | 12337 | 14148 | ATP production | |
| NADH dehydrogenase subunit 6 | 14149 | 14673 | ATP production | |
| tRNA glutamic acid | 14676 | 14742 | tRNA | |
| Region IV | 13541b | 15996b | ||
| Region I | 607b | 4256b | ||
| I | 15331 | 815 | ||
| Mt3 H-strand control element | 384 | 391 | Control region | |
| Mt4 H-strand control element | 371 | 379 | Control region | |
| Control element | 16194 | 16208 | Control region | |
| 7S DNA | 16106 | 191 | Control region, D loop | |
| Conserved sequence block 1 | 213 | 235 | Transcription regulation | |
| Conserved sequence block 2 | 299 | 315 | Transcription regulation | |
| Conserved sequence block 3 | 346 | 363 | Transcription regulation | |
| Replication primer | 317 | 321 | Transcription regulation | |
| Major H-strand promoter | 545 | 567 | Control region | |
| Minor H-strand promoter | 645 | 645 | Control region | |
| Hypervariable segment 1 | 16024 | 16383 | Control region | |
| Hypervariable segment 2 | 57 | 372 | Control region | |
| Hypervariable segment 3 | 438 | 574 | Control region | |
| L-strand promoter | 392 | 445 | Control region | |
| L-strand control element | 16499 | 16506 | Control region | |
| mtTF1 binding site | 523 | 550 | Transcription regulation | |
| mtTF1 binding site | 418 | 445 | Transcription regulation | |
| mtTF1 binding site | 233 | 260 | Transcription regulation | |
| mtTF1 binding site | 276 | 303 | Transcription regulation | |
| tRNA phenylalanine | 577 | 647 | tRNA | |
| H-strand origin | 110 | 441 | Control region | |
| Control region, including D Loop | 16024 | 576 | Control region | |
| Termination associated sequence | 16157 | 16172 | Control region | |
| tRNA proline | 15956 | 16023 | tRNA | |
| tRNA threonine | 15888 | 15953 | tRNA | |
| Membrane attachment site | 15925 | 499 | Localization | |
| Cytochrome b | 14747 | 15887 | ATP production | |
| 12S ribosomal RNA | 648 | 1601 | Mitochondrial protein synthesis |
aMITOMAP: A Human Mitochondrial Genome Database; http://www.mitomap.org (2013).
bPrimer target regions on the mt genome (Karunadharma et al., 2010) that overlap with primers (B, G, H, I) used in the current study.
Discussion
Results from the current study, examining 27 age-matched control donors and 42 donors with AMD (Fig. 1), are consistent with our previous study (Karunadharma et al., 2010) reporting a significant increase in RPE mtDNA damage early in the disease. Importantly, the increase in mtDNA damage is not due to normal aging, but rather is associated with the pathology of AMD. Thus, results from these two studies, examining a total of 53 control and 88 donors with AMD, firmly establishes that RPE mtDNA damage occurs early in the disease. Additional findings include: (1) mtDNA damage is found in not only the macula but also peripheral RPE, and (2) damage is limited to discrete regions of the mt genome. The study's clinically relevant finding that mtDNA damage occurs in the RPE but not in the retina provides a scientific basis for targeting the RPE mitochondria as a strategy for treating AMD.
The increase in mtDNA damage could be due to ROS overproduction, inadequate protection, or diminished repair. mtDNA damage could also accumulate due to defects in processes required for maintaining mt homeostasis (mt biogenesis, fission/fusion, and autophagy). Although no comprehensive studies have been conducted in normal and diseased human donor tissue, evidence for defects in many of these key processes is beginning to emerge and provide a mechanistic explanation for the increased mtDNA damage associated with AMD.
Due to their extremely short half-life, direct measurement of ROS in vivo or in donor eyes is technically not feasible. However, a compensatory response to increased ROS is the activation of redox-sensitive transcription factors (i.e., Nrf-2, HSF-1) and subsequent upregulation of proteins that deter ROS-induced damage (Handa, 2012). In studies with human donor RPE, increased content of antioxidant enzymes (catalase, MnSOD), the electrophile-detoxifier glutathione-S-transferase, and multiple heat shock proteins that refold ROS-damaged proteins was observed in donors with AMD (Nordgaard et al., 2006, 2008; Decanini et al., 2007). No AMD-related change in content was observed for either catalase or CuZnSOD (Liles et al., 1991; Frank et al., 1999; Decanini et al., 2007) suggesting a protein-dependent difference in response to AMD.
Mt biogenesis is essential for replenishing damaged mitochondria. Data that support diminished mt biogenesis includes the decrease in number of mitochondria in RPE from AMD donors (Feher et al., 2006) and lower content of electron transport chain proteins (Nordgaard et al., 2006, 2008). Decreased content of heat shock proteins involved in the import of nuclear-encoded proteins that reside in the mitochondria has also been observed in RPE from AMD donors (Nordgaard et al., 2006, 2008). This process is integral to mt biogenesis because the vast majority of the ∼1500 proteins that reside in the mitochondria are produced in the cytosol.
Fission and fusion are required for remodeling and maintaining mt architecture, and for eliminating damaged mt segments. Evidence for defective fission/fusion includes the disorganized cristae (Feher et al., 2006) and increased content of mitofilin in AMD donor RPE (Nordgaard et al., 2008). Mitofilin is involved in stabilizing cristae, so its upregulation could be a compensatory response to altered mt remodeling and defects in fission/fusion.
Another potential site of defect is autophagy, which is a conserved catabolic process involving the degradation of damaged mitochondria. Evidence includes the decreased number of autophagic vesicles in AMD donor RPE compared with age-matched controls (Mitter et al., 2014) and accumulation of p62, a protein eliminated via autophagy, in AMD donor maculas (Viiri et al., 2013). Insulin signaling decreases autophagy (Dröge and Schipper, 2007), so the twofold increase in the insulin receptor β-subunit reported in AMD donor RPE implies diminished autophagy is possible (Decanini et al., 2007).
Our finding that the retina did not accumulate mtDNA damage with AMD suggests differences in how the disease is manifested in specific ocular tissues. Tissue-related differences in repair capacity, antioxidant protection, or processes associated with mt homeostasis could contribute to the observed results. In support of this idea, proteomic analyses of donor RPE showed significant changes in mt proteins with AMD (Nordgaard et al., 2006), whereas in the retina, AMD-related changes involved the microtubule complex, chaperones, and stress-related proteins (Ethen et al., 2006). Our data are also consistent with observations by Udar et al. (2009) following amplification of mtDNA using LX PCR. They reported no significant difference in rearrangements, degradation, or deletion in retinal mtDNA in AMD donors compared with age-matched controls. However, they did observe increased staining for 8-hydroxydeoxyguanidine (8-OH-dG), a product of DNA damage, in retinal sections from AMD donors. The 8-OH-dG staining colocalized with mitochondria in photoreceptor inner segments, suggesting damaged mtDNA. Importantly, 8-OH-dG modification does not interfere with polymerase amplification (Kovalenko and Santos, 2009), so this type of damage would not be evident using the LX PCR assay. Another limitation is that our assay includes an average contribution from all cells in the retina. This prevents identification of specific cells whose level of damage may correlate with AMD, especially if the cells are a minor contributor to the overall total mtDNA.
The current investigation also tested a long-held belief that the macula is selectively damaged with AMD. This idea was likely promoted due to the anatomical differences in the macula versus peripheral retina. For example, the greater choroidal bloodflow, high ambient oxygen, and intensely focused light in the macula create the potential for production of excessive ROS (Curcio et al., 2009).
Although two studies support the idea that the macula accumulates more mtDNA damage, results from other investigations, including the current study, differ in their conclusions. Lin et al. (2011) reported more mtDNA lesion in primary RPE cultures from the macula compared with the periphery in aged human donors. In an assay that detects a 3895 bp deletion in the mt genome, Gendron et al. (2013) observed higher macular levels in both the retina and RPE from aged donors. This is in contrast to the study by Barreau et al. (1996) who found the 4977 bp deletion was actually greatest in the peripheral RPE compared with the macula. Results from the current study revealed no obvious differences between the extent of mtDNA damage in the macula and surrounding periphery. This suggests other factors unique to the macular environment promote the localization of the degenerative phenotype to the macula with AMD. However, this does not rule out the possibility that the combined effect of mtDNA damage within the microenvironment of the macula contributes to the disease.
One of the limitations of using donor tissue is that direct measures of mt function are not feasible. However, possible consequences of damage can be inferred when the sites of damage are defined. Using nine primer sets that cover the entire mt genome, we identified four regions that exhibited increased mtDNA damage, including the control region. This region encompasses the noncoding region and includes many of the cis-elements required for mt transcription and replication. Thus, damage could lead to abnormalities in expression of mt genes and/or production of new mtDNA.
Other damaged regions of the mt genome included genes for the 16S and 12S ribosomal RNAs, and 8 of 22 tRNAs. Because both RNAs are required for translation, defects in these genes could adversely affect synthesis of the 13 mt-encoded proteins. The 16S rRNA region also produces mt-derived peptides; the best characterized is humanin. This peptide has demonstrated neuroprotective and cytoprotective properties in both cultured cells and mouse models of disease (Yen et al., 2013). Damage to the 16S rRNA gene could diminish production of this cytoprotective peptide.
Other damaged regions included genes for multiple subunits of Complex I (NADH dehydrogenase S1, S4, S5, S6) and complex III (cytochrome b). These mt-encoded proteins were not detected in our previous analysis of the mt proteome, where we reported a decrease in ATP synthase subunits α, β, and δ of Complex V, and cytochrome c oxidase subunit 6B in Complex IV (Nordgaard et al., 2008). These proteins are all nuclear-encoded and reside outside the membrane. In contrast, all 13 mt-encoded proteins are embedded in the mt membrane and are often not detected in proteomic studies due to the limited resolution of membrane proteins on 2D gels. Damage to genes in Complexes I/III could result in decreased subunit content, somatic mutations, and reduced ATP production. Although the consequences of somatic mutations to these genes have not been experimentally tested, genetic evidence from SNPs in the mt genome infers sensitivity to changes in the sequence. For example, SNPs in the genome encoding the NADH dehydrogenase subunits (A11812G, MT-ND4 and A14233G, MT-ND6) have been associated with a 2.5-fold increased risk for developing advanced AMD (SanGiovanni et al., 2009). Together, the consequences of the combined defects in protein synthesis and somatic mutation of electron transport chain proteins could reduce ATP production and create an energy crisis in the cell.
In summary, results from our laboratory support the idea that the RPE mitochondria suffer early insults in AMD and provide a scientific basis for targeting therapies to the RPE. Because mt damage occurs before vision loss, early intervention would likely protect or rescue RPE mt function and could therefore attenuate disease progression.
Footnotes
This work was supported by funding from the Arnold and Mabel Beckman Initiative for Macular Research, Foundation Fighting Blindness, a University of Minnesota Academic Health Center Grant for Faculty Development, an anonymous benefactor for AMD Research, Minnesota Lions Vision Foundation, National Institutes of Health (MRT, T32-AG029796), University of Minnesota Diversity of Views and Experiences Fellowship (C.G.), and an unrestricted grant to the Department of Ophthalmology and Visual Neurosciences from the Research to Prevent Blindness. We thank the Minnesota Lions Eye Bank personnel for their assistance in procuring eyes, Kathy Goode and Sung Lee for photographing and processing eye tissue, and Dr Timothy Olsen for grading donor eyes before 2011.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Alliance for Eye and Vision Research. 20th Anniversary Special Report. 2013. October, 2013. http://www.eyeresearch.org/pdf/ValueBrochure/Value_of_Vision_Research_Brochure_Page_1.pdf.
- Ayala-Torres S, Chen Y, Svoboda T, Rosenblatt J, Van Houten B. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods. 2000;22:135–147. doi: 10.1006/meth.2000.1054. [DOI] [PubMed] [Google Scholar]
- Barreau E, Brossas JY, Courtois Y, Tréton JA. Accumulation of mitochondrial DNA deletions in human retina during aging. Invest Ophthalmol Vis Sci. 1996;37:384–391. [PubMed] [Google Scholar]
- Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–812. doi: 10.1016/S0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- Colby SL, Ortman JM. Current Population Reports. Washington, DC: US Census Bureau; 2014. The baby boom cohort in the United States: 2012 to 2060. P25-1141. [Google Scholar]
- Curcio CA, Johnson M, Huang JD, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009;28:393–422. doi: 10.1016/j.preteyeres.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- Droge W, Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007;6:361–370. doi: 10.1111/j.1474-9726.2007.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethen CM, Reilly C, Feng X, Olsen TW, Ferrington DA. The proteome of central and peripheral retina with progression of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:2280–2290. doi: 10.1167/iovs.05-1395. [DOI] [PubMed] [Google Scholar]
- Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006;27:983–993. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Frank RN, Amin RH, Puklin JE. Antioxidant enzymes in the macular retinal pigment epithelium of eyes with neovascular age-related macular degeneration. Am J Ophthalmol. 1999;127:694–709. doi: 10.1016/S0002-9394(99)00032-X. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O'Colmain BJ, Muñoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Eye diseases prevalence research group: prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Gendron SP, Bastien N, Mallet JD, Rochette PJ. The 3895-bp mitochondrial DNA deletion in the human eye: a potential involvement in corneal ageing and macular degeneration. Mutagenesis. 2013;28:197–204. doi: 10.1093/mutage/ges071. [DOI] [PubMed] [Google Scholar]
- Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med. 2012;33:418–435. doi: 10.1016/j.mam.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51:5470–5479. doi: 10.1167/iovs.10-5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko OA, Santos JH. Analysis of oxidative damage by gene-specific quantitative PCR. Curr Protoc Hum Genet. 2009;19:1. doi: 10.1002/0471142905.hg1901s62. [DOI] [PubMed] [Google Scholar]
- Leibowitz HM, Krueger DE, Maunder LR, Milton RC, Kini MM, Kahn HA, Nickerson RJ, Pool J, Colton TL, Ganley JP, Loewenstein JI, Dawber TR. The Framingham eye study monograph: an ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, and macular degeneration, and visual acuity in the general population of 2631 adults, 1973–1975. Surv Ophthalmol. 1980;24:335–610. doi: 10.1016/0039-6257(80)90015-6. [DOI] [PubMed] [Google Scholar]
- Liles MR, Newsome DA, Oliver PD. Antioxidant enzymes in the aging human retinal pigment epithelium. Arch Ophthalmol. 1991;109:1285–1288. doi: 10.1001/archopht.1991.01080090111033. [DOI] [PubMed] [Google Scholar]
- Lin H, Xu H, Liang FQ, Liang H, Gupta P, Havey AN, Boulton ME, Godley BF. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:3521–3529. doi: 10.1167/iovs.10-6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, Lewin A, Grant M, Dunn W, Jr, Ding J, Bowes Rickman C, Boulton M. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10:1989–2005. doi: 10.4161/auto.36184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgaard CL, Berg KM, Kapphahn RJ, Reilly C, Feng X, Olsen TW, Ferrington DA. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:815–822. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2848–2855. doi: 10.1167/iovs.07-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen TW, Feng X. The Minnesota grading system of eye bank eyes for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:4484–4490. doi: 10.1167/iovs.04-0342. [DOI] [PubMed] [Google Scholar]
- Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanGiovanni JP, Arking DE, Iyengar SK, Elashoff M, Clemons TE, Reed GF, Henning AK, Sivakumaran TA, Xu X, DeWan A, Agrón E, Rochtchina E, Sue CM, Wang JJ, Mitchell P, Hoh J, Francis PJ, Klein ML, Chew EY, Chakravarti A. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS One. 2009;4:e5508. doi: 10.1371/journal.pone.0005508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol Biol. 2002;197:159–176. doi: 10.1385/1-59259-284-8:159. [DOI] [PubMed] [Google Scholar]
- Santos JH, Meyer JN, Skorvaga M, Annab LA, Van Houten B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell. 2004;3:399–411. doi: 10.1111/j.1474-9728.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Taylor GA, Durham SE, Turnbull DM. The determination of complete human mitochondrial DNA sequences in single cells: implications for the study of somatic mitochondrial DNA point mutations. Nucleic Acids Res. 2001;29:E74–4. doi: 10.1093/nar/29.15.e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udar N, Atilano SR, Memarzadeh M, Boyer DS, Chwa M, Lu S, Maguen B, Langberg J, Coskun P, Wallace DC, Nesburn AB, Khatibi N, Hertzog D, Le K, Hwang D, Kenney MC. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:2966–2974. doi: 10.1167/iovs.08-2646. [DOI] [PubMed] [Google Scholar]
- Viiri J, Amadio M, Marchesi N, Hyttinen JM, Kivinen N, Sironen R, Rilla K, Akhtar S, Provenzani A, D'Agostino VG, Govoni S, Pascale A, Agostini H, Petrovski G, Salminen A, Kaarniranta K. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One. 2013;8:e69563. doi: 10.1371/journal.pone.0069563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Lee C, Mehta H, Cohen P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol. 2013;50:R11–R9. doi: 10.1530/JME-12-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]