

Abstract
Regular exercise leads to systemic metabolic benefits, which require remodeling of energy resources in skeletal muscle. During acute exercise, the increase in energy demands initiate mitochondrial biogenesis, orchestrated by the transcriptional coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α). Much less is known about the degradation of mitochondria following exercise, although new evidence implicates a cellular recycling mechanism, autophagy/mitophagy, in exercise-induced adaptations. How mitophagy is activated and what role PGC-1α plays in this process during exercise have yet to be evaluated. Thus we investigated autophagy/mitophagy in muscle immediately following an acute bout of exercise or 90 min following exercise in wild-type (WT) and PGC-1α knockout (KO) animals. Deletion of PGC-1α resulted in a 40% decrease in mitochondrial content, as well as a 25% decline in running performance, which was accompanied by severe acidosis in KO animals, indicating metabolic distress. Exercise induced significant increases in gene transcripts of various mitochondrial (e.g., cytochrome oxidase subunit IV and mitochondrial transcription factor A) and autophagy-related (e.g., p62 and light chain 3) genes in WT, but not KO, animals. Exercise also resulted in enhanced targeting of mitochondria for mitophagy, as well as increased autophagy and mitophagy flux, in WT animals. This effect was attenuated in the absence of PGC-1α. We also identified Niemann-Pick C1, a transmembrane protein involved in lysosomal lipid trafficking, as a target of PGC-1α that is induced with exercise. These results suggest that mitochondrial turnover is increased following exercise and that this effect is at least in part coordinated by PGC-1α.
Anna Vainshtein received the AJP-Cell 2015 Paper of the Year award. Listen to a podcast with Anna Vainshtein and coauthor David A. Hood at http://ajpcell.podbean.com/e/ajp-cell-paper-of-the-year-2015-award-podcast/.
Keywords: biogenesis, endurance, mitochondria, Niemann-Pick C1, physical activity
the merits of physical activity are numerous and well documented. Regular exercise leads to improved cardiovascular health, cognition, metabolism, oxidative capacity, and fatigue resistance, among many other beneficial consequences. Moreover, increased contractile activity is also protective of muscle mass and function under a myriad of pathologies. Although alterations in skeletal muscle metabolism, as well as its secretome, have been documented to contribute to the pleiotropic benefits of regular exercise, the molecular mechanism underpinning these adaptations remains unclear.
Skeletal muscle possesses a remarkable capacity to adapt to alterations in contractile activity, a property referred to as muscle plasticity. This type of cellular remodeling often requires a shift in metabolic profile, with amendments to the structure of the mitochondrial network, as well as changes in mitochondrial content. Organelle density is determined by the balance between its synthesis and degradation. Mitochondrial biogenesis is regulated transcriptionally through the coordinated expression of nuclear and mitochondrial genes, orchestrated largely by the transcriptional coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) (18, 41). Conversely, organelle degradation is mediated by a selective form of macroautophagy known as mitophagy (13, 15). During mitophagy, dysfunctional mitochondria are first segregated from the healthy network by fission and are then tagged for elimination (14, 37). Mitochondrial targeting can occur via mitophagy-specific receptors such as Bcl-2/adenovirus E1B 19-kDa interacting protein 3-like (Nix/Bnip3) (14, 26, 30) or by ubiquitination of mitochondrial outer membrane proteins by E3 ligases such as Parkin and Mul1 (4, 21, 25). Tagged mitochondria are then recognized by, and engulfed into, double-membrane vesicles termed autophagosomes. Autophagosomes are subsequently delivered to the lysosome for proteolytic degradation.
Several recent studies have indicated that autophagy is activated following an acute bout of endurance exercise (5, 6, 8–10, 20, 22) and may contribute to chronic exercise-induced improvements in muscle health and oxidative capacity. Indeed, deficient autophagy results in lack of exercise-mediated metabolic benefits, as well as progressive degeneration of mitochondrial function and performance (6, 20, 22). Coincidentally, PGC-1α levels have also been demonstrated to increase following an acute bout of endurance exercise (2), and PGC-1α is localized to the nucleus during the postexercise recovery period. The significance of this is that the absence of PGC-1α also results in diminished exercise-induced metabolic benefits (2, 16). However, it is unknown whether PGC-1α plays a role in acute exercise-induced autophagy or in the regulation of mitophagy flux. Thus we set out to investigate the function of PGC-1α in mediating autophagy and mitophagy induction in skeletal muscle following an acute bout of exercise. Our results should shed light on how the mitochondrial biogenesis and degradation pathways may interact to ensure proper mitochondrial remodeling and, thus, contribute to muscle plasticity as a result of exercise.
MATERIALS AND METHODS
Animal generation, treatment, and exercise.
PGC-1α whole body knockout (KO) and C57BL/6 wild-type (WT) mice were housed in a 12:12-h light-dark cycle and given food and water ad libitum. Generation and characterization of PGC-1α KO mice have been previously described (1, 19). To evaluate autophagy flux, animals were injected with colchicine or an equal volume of vehicle (water) every 24 h (0.4 mg·kg−1·day−1) (12) for 2 days prior to the day of exercise/euthanization, with the final injection at 24 h prior to euthanization. After 2 days of habituation to the treadmill and colchicine or vehicle injections, the exercise (Ex) and exercise and recovery (Ex + R) groups were run to failure on a treadmill (Fig. 1C). The exercise bout was terminated when the animals could not continue running after three consecutive shocks. Animals were euthanized immediately after exercise (Ex) or 90 min postexercise (Ex + R). Muscle was then excised and immediately frozen in liquid nitrogen for later use in protein or gene expression analysis or used fresh for cellular fractionation. All procedures involving animals were approved and conducted in accordance with the regulations of the York University Animal Care Committee in compliance with the guidelines set forth by the Canadian Council on Animal Care.
Fig. 1.

Lack of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) results in diminished mitochondrial content and reduced exercise performance. A: representative images of cytochrome oxidase (COX) and succinate dehydrogenase (SDH) staining of extensor digitorum longus muscle from control [wild-type (WT)] and PGC-1α knockout (KO) animals. Scale bars = 100 μm. B: COX activity as a surrogate measure of mitochondrial content in WT and KO animals. C: after 2 days of habituation to the treadmill, animals were run to failure utilizing an incremental exercise protocol on a 0% incline. Animals began with a warm-up period of 5 min at 5 m/min and 10 min at 10 m/min followed by 45 min of endurance running at 15 m/min. Finally, running speed was increased by 1 m/min every 2 min until the animals refused to continue. D: running performance (i.e., total distance run) in WT and KO animals injected with water [vehicle (Veh)] or 0.4 mg/kg colchicine (Col). E: blood lactate levels in WT and KO animals prior to exercise (Con), immediately following exercise (Ex), and following 90 min of recovery (Ex + R). Values are means ± SE; n = 4–12 for all groups. *P < 0.05, significant effect of exercise. †P < 0.05, significant effect of genotype.
Blood lactate.
Blood lactate was measured prior to exercise, immediately postexercise, and following 90 min of recovery. A 0.2-μl blood sample was obtained by tail nick and immediately analyzed using the Lactate Scout+ analyzer (EKF Diagnostics, Magdeburg, Germany).
Histology.
Cytochrome oxidase (COX) and succinate dehydrogenase (SDH) staining was performed on 10-μm cross sections of extensor digitorum longus muscles as previously described (24). Briefly, frozen muscle sections adhered to glass slides were dried and subsequently incubated with COX or SDH reaction solutions for 30 min in darkness at 30°C. Each slide contained sections from all conditions to ensure equal staining across the groups. After they were washed with PBS, glass coverslips were mounted onto the slides with DPX Mountant for histology (catalog no. 44581, Fluka) and sealed. Images of stained muscle sections were captured with a Nikon 90i eclipse upright microscope using a ×20 objective.
COX activity.
COX enzyme activity was measured as previously described (38) as the rate of oxidation of fully reduced cytochrome c by isolated enzymatic extract, evaluated as a change in absorbance at 550 nm using a microplate reader (Synergy HT, BioTek).
Gene expression analysis.
Quantitative real-time PCR was performed to determine mRNA expression levels. Total RNA was isolated using TRIzol reagent (catalog no. 15596-026, Invitrogen). RNA was reverse-transcribed into cDNA using a SuperScript III First-Strand synthesis kit (catalog no. 18080-044, Invitrogen) according to the manufacturer's instructions. The primers used for gene expression analysis are listed in Table 1 and were designed on the basis of sequences available in GenBank (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi). Analyses were performed with SYBR Green chemistry (PerfeCTa SYBR Green Supermix, ROX, catalog no. 95055-500, Quanta BioSciences) in a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA). GAPDH (Gapdh) and β-actin (Actb) were used in combination as housekeeping genes.
Table 1.
Primer sequences based on gene transcripts available in GenBank
| Primer (5′ → 3′) |
||
|---|---|---|
| Gene | Forward | Reverse |
| Coxiv | CTCCAACGAATGGAAGACAG | TGACAACCTTCTTAGGGAAC |
| Tfam | GAAGGGAATGGGAAAGGTAGA | AACAGGACATGGAAAGCAGAT |
| Bnip3l | GGAAAGCGGCACAGAGAA | GAATGACGCCAGTGCTGAT |
| Park2 | GTCTGCAATTTGGTTTGGAGTA | GCATCATGGGATTGTCTCTTAAA |
| Sqstm1 | TGTGGTGGGAACTCGCTATAA | CAGCGGCTATGAGAGAAGCTAT |
| Maplc3b | GCTTGCAGCTCAATGCTAAC | CCTGCGAGGCATAAACCATGTA |
| Atg7 | TTTCTGTCACGGTTCGATAATG | TGAATCCTTCTCGCTCGTACT |
| Lamp2 | GCTGAACAACAGCCAAATTA | CTGAGCCATTAGCCAAATACAT |
| Catsd | TTTGCCAATGCTGTCGTACT | AGCGAGTGTGACTATGTGTGAG |
| TFEB | AGCTCCAACCCGAGAAAGAGTTTG | CGTTCAGGTGGCTGCTAGAC |
| Foxo3 | ATGGACGACCTGCTGGATAAC | GGAGCTCTTGGCGGTATATG |
| Beclin1 | AGGCTGAGGCGGAGAGATT | TCCACACTCTTGAGTTCGTCAT |
| NPC1 | AGCATCACCGCATCTTACAA | GCATGGCTGTTCTGGAAGTAA |
| Actb | TGTGACGTTGACATCCGTAA | GCTAGGAGCCAGAGCAGTAA |
| Gapdh | AACACTGAGCATCTCCCTCA | GTGGGTGCAGCGAACTTTAT |
mRNA array.
The gene expression of 84 key autophagic genes was profiled by RT2 Profiler autophagy PCR arrays (catalog no. PAMM-084, SABioscience) as recommended by the manufacturer. RT-PCRs were performed in 96-well-plate format using the StepOnePlus real-time PCR system. Fold changes in autophagic gene expression from KO samples relative to WT control samples were calculated using the comparative threshold (ΔΔCt) method with the integrated software package for PCR array systems provided by the manufacturer (RT2 Profiler PCR Array Data Analysis Template v3.3). ΔΔCt values from each sample were normalized by three housekeeping genes that did not change across the conditions.
Immunoblotting.
Protein extracts from frozen tibialis anterior cryosections (22), isolated mitochondria, or nuclear extracts were separated by SDS-PAGE and transferred to nitrocellulose membranes, which were blocked with 5% skim milk or 5% BSA solution. Membranes were incubated overnight at 4°C with the appropriate concentration of primary antibody. The following antibodies were used: phosphorylated (Thr172) AMP-activated kinase (AMPK), total AMPK, phosphorylated (Thr180/Tyr182) p38, total p38, long chain 3 (LC3), and Parkin (Cell Signaling Technology), GAPDH, voltage-dependent anion channel, and Niemann-Pick C1 (NPC1; Abcam), dynamin-related protein 1 (Drp-1; BD Transduction Laboratories), p62 (Sigma-Aldrich), PGC-1α (Millipore), and ubiquitin (Enzo Life Sciences). Membranes were subsequently washed and incubated with the suitable horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature and visualized with enhanced chemiluminescence. Quantification was performed with ImageJ software (National Institutes of Health, Bethesda, MD), and values were normalized to the appropriate loading control.
Cellular fractionation.
Enriched mitochondrial and nuclear cellular subfractions were isolated by differential centrifugation, as previously described (38). Briefly, muscles were minced on ice and homogenized using a Teflon pestle and mortar and suspended in mitochondrial isolation buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, and 1 mM EGTA) supplemented with protease (cOmplete, catalog no. 1169749801, Roche) and phosphatase inhibitor cocktails 2 and 3 (catalog nos. P5726 and P0044, Sigma). The homogenates were centrifuged at 1,000 g for 10 min at 4°C to pellet the nuclei, while mitochondrial and cytosolic fractions were contained within the supernate. The supernate fraction was recentrifuged at 16,000 g for 20 min at 4°C to pellet the mitochondria. The mitochondrial pellet was washed twice and resuspended in a onefold dilution of mitochondrial isolation buffer. Mitochondria were subsequently sonicated three times for 3 s each to yield the enriched mitochondrial fraction. Pellets containing nuclei were resuspended in nuclear lysis buffer (1.5 mM MgCl2, 0.2 mM EDTA, 20 mM HEPES, 0.5 M NaCl, 20% glycerol, and 1% Triton X-100), incubated on ice for 30 min, sonicated three times for 10 s each, and then centrifuged for 15 min at 16,000 g. The supernate was collected to obtain the enriched nuclear fraction. Protein concentrations within the samples were determined using the Bradford method. Fraction purity was determined by Western blot analysis of each fraction for the loading controls of all fractions (unpublished data).
Statistics.
Comparisons between WT and KO and between control, Ex, and Ex + R animals were evaluated using two-way analyses of variance on each of the treatment conditions. Bonferroni's posttests were performed when applicable. Values are means ± SE. Data were considered statistically different if P < 0.05.
RESULTS
PGC-1α KO animals exhibit diminished mitochondrial content, reduced endurance capacity, and metabolic stress with exercise.
Deletion of PGC-1α resulted in significantly lower mitochondrial content in skeletal muscle, as demonstrated by reduced SDH, as well as COX, staining and a ∼40% decrease in COX activity (Fig. 1, A and B). To examine the involvement of PGC-1α in exercise-induced autophagy, 3-mo-old PGC-1α KO and WT animals were subjected to an acute bout of incremental treadmill running (Fig. 1, C–E). The lack of PGC-1α resulted in diminished endurance performance, as the KO animals ran significantly less than their WT counterparts (Fig. 1D). Moreover, KO animals exhibited a ∼40% higher blood lactate basally, which increased by 3.8-fold with exercise, compared with a 2.8-fold increase in WT animals (Fig. 1E). We also noted that the blood lactate of KO animals did not return to baseline as effectively as that of WT animals during the 90-min recovery period. This indicates increased metabolic stress and a greater reliance on glycolysis in the KO animals. Metabolic stress was evident from changes in the phosphorylation of the metabolically sensitive p38 MAPK and AMPK. Exercise resulted in a ∼2.5-fold increase in phosphorylation of AMPK in WT animals, while KO animals exhibited a ∼4-fold induction (Fig. 2, A and B). Phosphorylation of p38 MAPK was elevated by ∼50% with exercise in WT and KO animals. However, KO animals expressed higher levels of phosphorylated p38 overall, which did not return to baseline during recovery (Fig. 2, A and C).
Fig. 2.
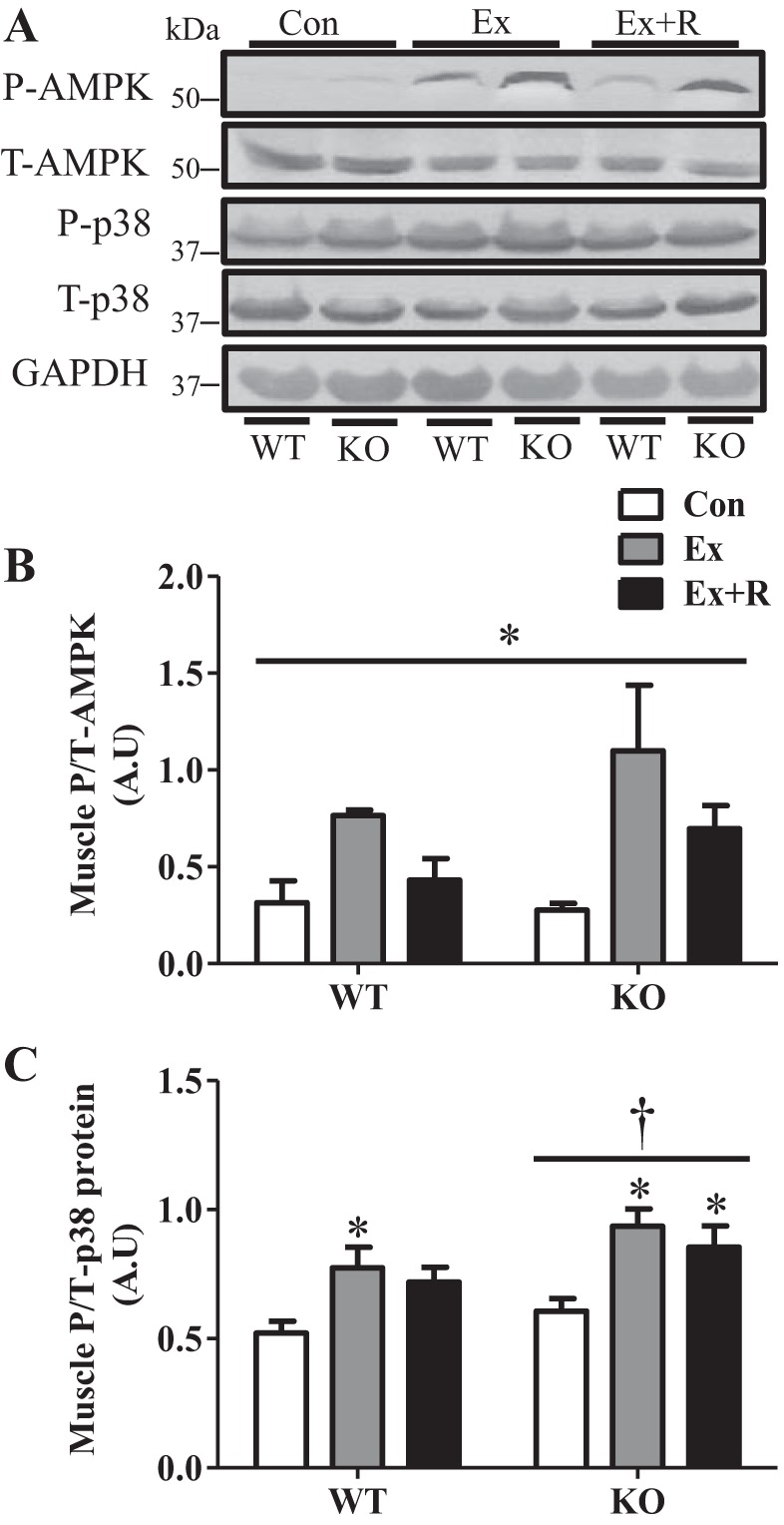
Signaling kinases are activated with exercise. A–C: blots and quantification of signaling kinases in WT and KO animals in Con, Ex, and Ex + R groups. A: representative blots of signaling kinases in WT and KO animals in Con, Ex, and Ex + R groups. Total protein and GAPDH were used as loading controls. P-AMPK and T-AMPK, phosphorylated and total AMP-activated kinase; P-p38 and T-p38, phosphorylated and total p38. B and C: quantification of signaling kinases in WT and KO animals in Con, Ex, and Ex + R groups. AU, arbitrary units. Values are means ± SE; n = 5–9. *P < 0.05, significant effect of exercise. †P < 0.05, significant effect of genotype.
PGC-1α localization to the nucleus and gene expression are elevated in response to exercise, resulting in induction of mitochondrial biogenesis in WT animals.
Exercise induced localization of PGC-1α to the nucleus (Fig. 3A), which was accompanied by an 8.5-fold increase in PGC-1α (Ppargc1a) mRNA expression following recovery (Fig. 3B). The increase in PGC-1α with exercise resulted in ∼60% and ∼50% inductions in expression of the downstream targets COX subunit IV (Coxiv) and mitochondrial transcription factor A (Tfam), which were not evident in the KO animals (Fig. 3, C and D). Thus an acute bout of exercise was sufficient to induce the onset of mitochondrial biogenesis in a PGC-1α-dependent manner, an effect that was not observed in KO animals.
Fig. 3.

Exercise induces increased nuclear localization and expression of PGC-1α and mitochondrial biogenesis. A: blot and quantification of PGC-1α levels in the nuclear subfraction of WT animals in Con, Ex, and Ex + R groups. B: PGC-1α gene (Ppgargc1a) expression in WT animals in Ex and Ex + R groups compared with WT animals in Con group. C and D: COX subunit IV (Coxiv) and mitochondrial transcription factor A (Tfam) gene expression WT and KO animals in Ex and Ex + R groups compared with WT animals in Con group. GAPDH (Gapdh) and β-actin (Actb) were used as housekeeping genes. Values are means ± SE; n = 4–9. *P < 0.05, significant effect of exercise. †P < 0.05, significant effect of genotype.
Acute exercise results in induction of autophagy, which is differentially regulated in PGC-1α KO animals.
During autophagy, p62 and LC3 are consumed within the lysosome while accompanying their respective organelle targets. A continuous transcriptional contribution is, therefore, required to avoid exhaustion of these important autophagy factors. We observed ∼80% and ∼94% inductions of microtubule-associated protein 1 light chain 3 [Maplc3b (LC3)] and sequestosome 1 [Sqstm1 (p62)] transcript levels with exercise, respectively. However, this effect was abolished in KO animals (Fig. 4, A and B). In contrast, the transcript levels of additional autophagy [autophagy related 7 (Atg7) and beclin 1 (Becn1)] and lysosomal [cathepsin D (Catsd) and lysosome-associated membrane protein 2 (Lamp2)] markers were not different between WT and KO animals and did not change with exercise or recovery (data not shown). Our acute exercise protocol was sufficient to induce LC3 lipidation in the muscle of WT animals; however, we did not observe this increase in the KO mice until after the recovery period, indicating a delayed response to exercise in KO animals (Fig. 4, C and D). The levels of p62 did not change immediately following exercise in WT animals but increased with recovery (Fig. 4, C and E). Conversely, p62 levels were elevated immediately after exercise and following recovery in KO animals (Fig. 4, C and E). This is indicative of impaired autophagosome degradation of p62 in KO animals.
Fig. 4.

Expression of autophagy genes and induction of autophagy with exercise are differentially regulated in PGC-1α KO animals. A and B: gene expression of autophagy factors, microtubule-associated protein 1 light chain 3 [Maplc3b (LC3)] and sequestosome 1 [Sqstm1 (p62)], in WT and KO animals in Ex and Ex + R groups compared with WT animals in Con group. Gapdh and Actb were used as housekeeping genes. C–E: blots and quantification of autophagic protein in whole muscle extracts from WT and KO animals in Con, Ex, and Ex + R groups treated with vehicle or colchicine (0.4 mg·kg−1·day−1) for 2 days. F and G: autophagy flux as determined by percent change in protein content from colchicine- and vehicle-treated WT and KO animals in Con, Ex, and Ex + R groups. GAPDH was used as loading control. Values are means ± SE; n = 5–9. *P < 0.05, significant effect of exercise. †P < 0.05, significant effect of genotype.
One major difficulty in evaluating autophagic flux is the extremely dynamic and transient nature of autophagosomes, since the average half-life of this organelle is ∼10 min (20). Therefore, we evaluated autophagy flux by measuring the percent change in LC3II and p62 protein in vivo with interperitoneal administration of the microtubule-destabilizing drug colchicine, as previously described (12). Colchicine blocks autophagosome degradation by destabilizing the microtubule tracks on which they travel to the lysosome for degradation, resulting in accumulation of autophagosomes and, thus, a backlog of LC3II and p62. Interestingly, LC3II flux tended to increase immediately following exercise and after recovery in WT animals but was lower overall and did not follow a similar trend in KO animals (Fig. 4F). We also observed a trend for an increase in p62 flux with exercise in WT and KO animals (Fig. 4G); however, this tendency was attenuated in the KO animals. Taken together, these results indicate an induction of autophagy with exercise in WT animals that is impaired in mice lacking PGC-1α.
Mitochondrial targeting for degradation is enhanced following an acute bout of exercise, but this signaling is attenuated in mice lacking PGC-1α.
To investigate mitophagy induction and the role of PGC-1α in this process, we measured the presence of autophagy markers in isolated mitochondrial subfractions. Exercise induced a ∼5.2-fold increase in localization of LC3II to the mitochondria in WT animals, which decreased with recovery (Fig. 5, A and B). Although mitochondrial localization of LC3II tended to increase with recovery in KO animals, this did not reach statistical significance. Moreover, LC3II flux was significantly elevated (∼4.4-fold) with exercise (Fig. 5C) in WT animals only, and this increase was completely abolished by the lack of PGC-1α. Localization of p62 to the mitochondria did not change with exercise or recovery in WT or KO animals (Fig. 5, A and D). However, p62 flux was increased following recovery by 65% and 56% in WT and KO animals, respectively (Fig. 5E).
Fig. 5.

Exercise-induced mitophagy signaling and flux are attenuated in PGC-1α KO animals. A–E: blots and quantification of autophagic proteins and flux in isolated mitochondrial fractions from WT and KO animals in Con, Ex, and Ex + R groups treated with vehicle or colchicine (0.4 mg·kg−1·day−1) for 2 days. Voltage-dependent anion channel (VDAC) was used as loading control. Values are means ± SE; n = 7–9. *P < 0.05, significant effect of exercise.
To elucidate the mechanisms behind mitochondrial targeting with exercise, we examined the mitochondrial E3 ubiquitin ligase Parkin, which has been well documented to be intimately involved in mitophagy (25). Interestingly, Parkin mRNA levels were elevated ∼3-fold, while mitochondrial localization of the protein was induced ∼3.5-fold, following exercise and remained elevated during the recovery period in WT animals (Fig. 6, A–C). Parkin mRNA tended to increase with exercise in KO animals, but this increase was not statistically significant (Fig. 6A). Importantly, Parkin localization to the mitochondria was both delayed and attenuated in the KO animals, increasing by 2.6-fold only after recovery (Fig. 6, B and C). Elevated Parkin in WT animals translated to a ∼60% increase in ubiquitination of mitochondrial proteins following exercise, and this effect was absent in KO animals (Fig. 6, B and D). Since an important prerequisite for mitochondrial degradation by autophagy is organelle fragmentation (37), we also examined mitochondrially localized Drp-1, a fission protein that translocates to mitochondria and facilitates their fragmentation. We found that localization of Drp-1 to the mitochondrial subfraction increased ∼2.2-fold following exercise and returned to baseline during recovery in WT animals (Fig. 6. B and E). No change in mitochondrial Drp-1 levels was observed in KO animals. Thus these results indicate an increased targeting of mitochondria for mitophagy postexercise, which is, at least in part, dependent on PGC-1α.
Fig. 6.

Lack of PGC-1α results in attenuated exercise-mediated mitophagy signaling. A: Parkin (Park2) gene expression in WT and KO animals in Con, Ex, and Ex + R groups compared with WT animals in Con group. Gapdh and Actb were used as housekeeping genes. B–E: blots and quantification of proteins in isolated mitochondrial subfractions. Drp-1, dynamin-related protein 1; Ub, ubiquitin. VDAC was used as loading control. Values are means ± SE; n = 4–9. *P < 0.05, significant effect of exercise.
Exercise-mediated alterations in transcriptional regulators of autophagy are not different between WT and KO animals.
Transcriptional regulation of autophagy with exercise has not been thoroughly examined. Therefore, we investigated Forkhead box O3 (FoxO3) and transcription factor EB (TFEB), two well-known transcriptional regulators of the autophagy-lysosome system. Foxo3 expression was induced 2.8-fold with exercise in WT animals and was consistently higher in KO animals across all conditions (Fig. 7A). TFEB mRNA expression was not altered during exercise in WT or KO animals but tended to increase after recovery in WT animals only (Fig. 7B). Thus, both TFEB and FoxO3 may play a role in transcriptional regulation of autophagy, but neither appears to be regulated by PGC-1α in this context.
Fig. 7.
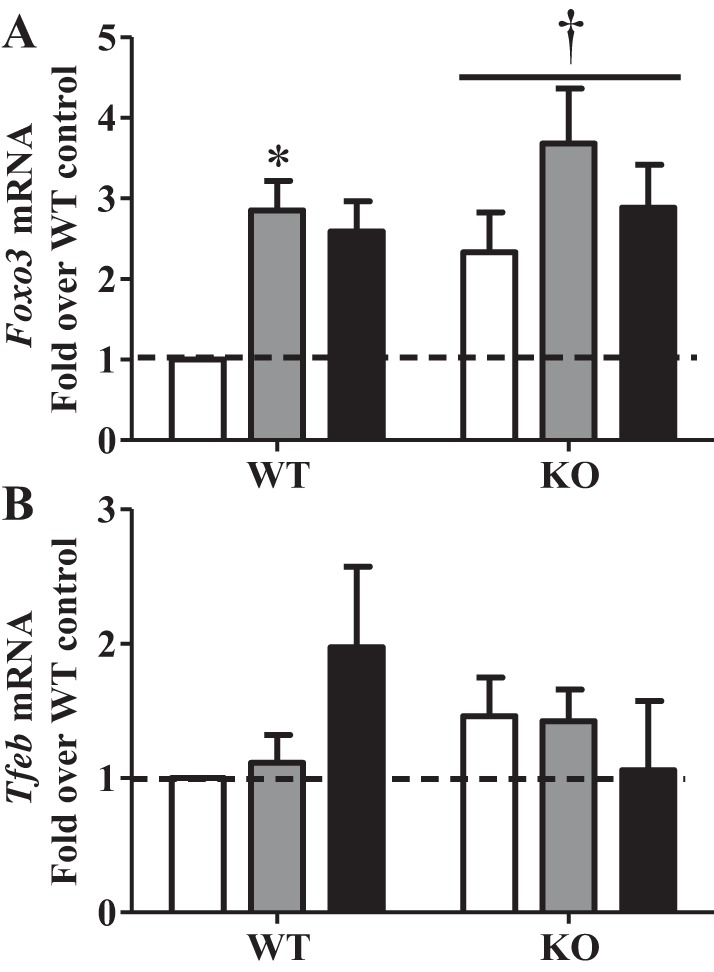
Transcriptional regulators of autophagy with exercise. A and B: gene expression of transcriptional regulators of autophagy [Forkhead box O3 (Foxo3) and transcription factor EB (TFEB)] in WT and KO animals in Con, Ex, and Ex + R groups compared with WT animals in Con group. Gapdh and Actb were used as housekeeping genes. Values are means ± SE; n = 4–9. *P < 0.05, significant effect of exercise. †P < 0.05, significant effect of genotype.
NPC1 is a novel autophagy factor regulated by PGC-1α.
To identify additional autophagy factors that may be under the control of PGC-1α, we performed an unbiased PCR-based mRNA array and compared the expression of 84 autophagy-related genes (for a complete list of genes see Supplemental Table S1 in Supplemental Material for this article, available online at the Journal website) in WT and PGC-1α KO animals. Several genes, including mammalian target of rapamycin (mTOR), NF-κB (Nfkb1), phosphatidylinositol 4,5-bisphosphate 3-kinase (Pik3cg), and NPC1, were dramatically downregulated in KO compared with WT animals (Fig. 8A). We chose to further investigate NPC1, a novel transmembrane protein responsible for cholesterol trafficking in late endosomes and lysosomes. Mutations in NPC1 result in Niemann-Pick disease type C, an autosomal recessive neurovisceral lipid storage disorder, which is accompanied by impaired autophagy (17, 33). We first confirmed our array findings with real-time PCR and found that NPC1 expression was indeed diminished by the lack of PGC-1α (Fig. 8B). Interestingly, NPC1 expression was induced by ∼80% with exercise and returned to basal levels during recovery in WT animals. The exercise-mediated increase in NPC1 was abolished in KO animals. We also found that NPC1 protein levels were significantly lower in KO animals than in their WT counterparts (Fig. 8C). Thus we have identified NPC1 as a novel autophagy factor that is induced with exercise, and this induction appears to be PGC-1α-dependent.
Fig. 8.
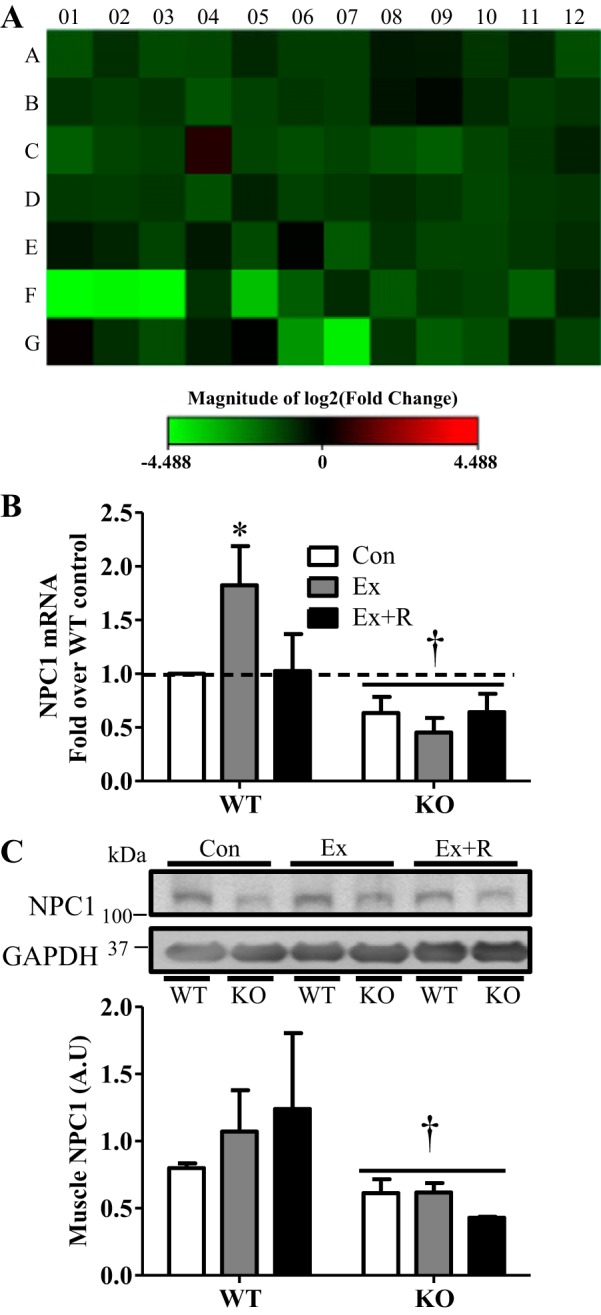
Identification of Niemann-Pick C1 (NPC1) as a PGC-1α-regulated autophagy factor through PCR-array analysis. A: heat map showing expression of 84 autophagy-related genes in WT and KO animals. Green indicates reduction, while red indicates increase, in gene expression; the brighter the color, the greater the change in gene expression. (For the full list of fold changes and statistical significance see Supplemental Table S1.) B: gene expression of NPC1 in WT and KO animals in Con, Ex, and Ex + R groups compared with WT animals in Con group. Gapdh and Actb were used as housekeeping genes. C: representative blot and quantification of NPC1 in tibialis anterior muscle extracts. GAPDH was used as loading control. Values are means ± SE; n = 3–4. *P < 0.05, significant effect of exercise. †P < 0.05, significant effect of genotype.
DISCUSSION
Skeletal muscle is a malleable tissue that rapidly adapts to its metabolic environment. Much research has focused on establishing the mechanism responsible for this remarkable plasticity, as it has great therapeutic potential for a vast array of muscle and metabolic pathologies. The energy demands stemming from muscle contraction are known to initiate signaling cascades, which lead to increased mitochondrial biogenesis to enhance the energetic potential of muscle. This exercise signaling largely converges on PGC-1α, a transcriptional coactivator responsible for orchestrating the mitochondrial biosynthesis program. However, it is not known how mitochondrial degradation is regulated during exercise, and the role of PGC-1α in this process has not been conclusively determined. Thus the purpose of this study was to examine the activation of autophagy and mitophagy during an acute bout of exercise and to evaluate the involvement of PGC-1α in this process.
PGC-1α has been documented to drive mitochondrial biogenesis and expression of oxidative genes (2, 18, 28, 29). It is no surprise, then, that KO animals were found to be profoundly deficient in mitochondria, as observed biochemically by less intense SDH and COX staining, as well as diminished COX activity. This reduction in mitochondrial content translated to a functional deterioration of endurance performance. PGC-1α KO mice exhibited metabolic distress during exercise, evident by elevated lactic acid levels that did not effectively resolve with a 90-min recovery period. This was accompanied by reduced endurance performance compared with WT animals. KO animals also exhibited greater increases in activation of the metabolic sensor AMPK and the cellular stress sensor p38, further supporting metabolic distress in these animals. WT animals displayed normal elevations in activation of these kinases with exercise, which returned to basal levels during the recovery period. Our results confirm that mitochondrial biogenesis is initiated with the first bout of exercise and that PGC-1α is instrumental in this process (2, 28). PGC-1α has been documented to localize to the nucleus shortly after commencement of exercise (7, 31). We noted an increase in nuclear PGC-1α in WT animals immediately after exercise that was followed by a strong induction of the transcript levels of the coactivator, particularly following the recovery period. These increases in PGC-1α transcript and nuclear localization were accompanied by induction of mitochondrial genes encoded by the nuclear genome. This increase was abolished in animals lacking PGC-1α. These findings add further substantive support for the importance of PGC-1α in mediating adaptations in oxidative capacity within muscle as a result of exercise.
Several studies have documented induction of autophagy following an acute bout of exercise (5, 6, 8, 9). Here we demonstrate, for the first time, that the transcript levels of the autophagy factors LC3B and p62 are induced immediately following an acute bout of exercise and that this increase is mediated by PGC-1α. We did not detect alterations in the transcript levels of these factors in KO animals 90 min postexercise. We also noted an increase in LC3 lipidation with exercise in WT animals. This response was delayed in KO animals and did not occur until after the recovery period, when LC3II levels returned to baseline in WT mice. Interestingly, we did not note a decrease in p62 levels in WT animals with exercise, as has been previously described (6, 22). This could be a result of differences in the exercise protocol between the studies and, perhaps, increases in p62 mRNA that we noted with exercise. In KO animals we observed the opposite effect: p62 levels were elevated following exercise. Because of the lack of increase in p62 mRNA levels in KO animals, we conclude that the increase in p62 protein with exercise is likely due to impaired degradation by autophagy in these animals. We did not note basal differences in LC3 or p62 levels between the genotypes, indicating that this is an exercise-mediated effect. Moreover, we also observed a trend for increases in LC3II and p62 flux in WT animals as determined with colchicine treatment experiments, but this trend was not found in KO mice. Indeed, LC3II flux was lower in KO animals. These results indicate that autophagy induction, gene expression, and flux are induced by exercise and that this induction is compromised in mice lacking PGC-1α. Thus PGC-1α appears to play a role in the regulation of exercise-induced autophagy.
Activation of mitophagy with exercise has only recently been documented and was deemed to be required for removal of dysfunctional mitochondria following damaging downhill running exercise (22). However, regulation of mitophagy in exercising muscle has not been thoroughly examined. Our findings demonstrate that mitophagy signaling, as well as flux, is induced with an acute bout of exercise and that this effect is diminished in animals deficient in PGC-1α. In our hands, exercise-induced mitochondrial localization and flux of LC3II were abolished in KO animals. Interestingly, no alterations in p62 localization to the mitochondria were observed with exercise in either genotype, and p62 flux was similarly elevated in WT and KO animals following recovery only. We also noted that Parkin plays a role in exercise-induced mitophagy, as there was an increase in localization of this E3 ligase to the mitochondria. Enhanced abundance of ubiquitinated proteins within the mitochondrial subfraction with exercise was also observed in the WT animals. In contrast, Parkin localization to mitochondria was delayed in PGC-1α KO animals, occurring following recovery, and no increase in mitochondrial protein ubiquitination was observed. It is interesting to note that the Parkin-PGC-1α axis presents a potential point of communication between mitochondrial biogenesis and degradation following exercise. Parkin has been documented to target the transcriptional repressor Paris (ZNF746) for degradation, thus releasing the Paris-mediated repression of PGC-1α transcription (36). This would act to enhance mitochondrial biogenesis to compensate for increased mitochondrial degradation.
The impairment in mitophagy that we observed in KO animals could also be due, in part, to altered organelle dynamics. Fission is a prerequisite for mitophagy (37), and in our model the localization of the fission protein Drp-1 to the mitochondria was increased after exercise in WT, but not KO, animals. This further supports our findings that mitophagy is activated with exercise and that PGC-1α is involved in this process.
Although several factors have been identified to participate in the transcriptional regulation of autophagy (3, 23, 35, 42), little is known about this process in skeletal muscle, and the regulation of this process with exercise has not been investigated. Therefore, we examined the expression of FoxO3 and TFEB, two transcriptional regulators that have been well documented to induce autophagic gene expression in different cells and tissues. FoxO3 transcript levels were induced with exercise, suggesting a role for this protein in mediating the increased expression of autophagy genes with exercise. However, we also noted an overall higher expression of FoxO3 in KO animals, indicating that FoxO3 is not likely to mediate PGC-1α-induced autophagy. This is in line with previous evidence implicating PGC-1α in the suppression of FoxO3 under atrophic conditions (32). We also investigated TFEB, a master transcriptional regulator of the autophagy-lysosome system (27, 35), previously documented to play a role in the induction of PGC-1α expression in the liver during nutrient deprivation (34). We noted a trend for increased TFEB transcript levels during recovery only in WT animals. The lack of this trend in KO animals suggests a potential role for TFEB in PGC-1α-mediated autophagic induction. Indeed, we previously observed reduced TFEB protein levels in KO animals compared with their WT controls (unpublished observations) under basal conditions. It is possible that PGC-1α may bind and coactivate TFEB on the promoter of autophagy-related genes; however, this warrants further investigation.
In an attempt to further characterize the endogenous role of PGC-1α in autophagy regulation, we performed an unbiased mRNA array to examine 84 genes involved in various aspects of autophagy. Our array data reveal multiple genes that were downregulated in KO animals. We further focused on NPC1, a novel transmembrane protein involved in regulation of cholesterol trafficking from late endosomes and lysosomes. NPC1 protein and mRNA were strongly downregulated in PGC-1α KO animals but were induced with exercise in WT animals, suggesting a role for this protein in exercise-induced adaptations. NPC1 has not been previously studied in this context, but mutations in NPC1 result in Niemann-Pick disease, a devastating lysosomal storage disease characterized by defective autophagy and increased cholesterol load (39). Moreover, polymorphisms or haploinsufficiency in the NPC1 gene have been correlated with obesity and type 2 diabetes (11, 40). Very little is known about NPC1 and its role in skeletal muscle.
Taken together, our findings indicate that exercise-induced metabolic adaptations involve augmented mitochondrial turnover that engages concomitant increases in degradation and biogenesis. Our results also demonstrate that the transcriptional coactivator PGC-1α coordinates mitochondrial biogenesis and mitophagy immediately following exercise and that both of these processes are compromised in the absence of the coactivator. This study sheds light on the mechanisms underpinning mitochondrial turnover induced by exercise, implicating PGC-1α in orchestrating this process.
GRANTS
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) to D. A. Hood. D. A. Hood holds a Canada Research Chair in Cell Physiology. A. Vainshtein was a recipient of a graduate scholarship from NSERC.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.V., L.D.T., and M.P. performed the experiments; A.V. analyzed the data; A.V. and D.A.H. interpreted the results of the experiments; A.V. prepared the figures; A.V. and D.A.H. drafted the manuscript; A.V. and D.A.H. edited and revised the manuscript; A.V., L.D.T., M.P., and D.A.H. approved the final version of the manuscript; D.A.H. developed the concept and designed the research.
Supplementary Material
REFERENCES
- 1.Adhihetty PJ, Uguccioni G, Leick L, Hidalgo J, Pilegaard H, Hood DA. The role of PGC-1α on mitochondrial function and apoptotic susceptibility in muscle. Am J Physiol Cell Physiol 297: C217–C225, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16: 1879–1886, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Chua JP, Reddy SL, Merry DE, Adachi H, Katsuno M, Sobue G, Robins DM, Lieberman AP. Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum Mol Genet 23: 1376–1386, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, Bonaldo P. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 7: 1415–1423, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hood DA. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl Physiol Nutr Metab 34: 465–472, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Jamart C, Benoit N, Raymackers JM, Kim HJ, Kim CK, Francaux M. Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur J Appl Physiol 112: 3173–3177, 2012. [DOI] [PubMed] [Google Scholar]
- 9.Jamart C, Francaux M, Millet GY, Deldicque L, Frère D, Féasson L. Modulation of autophagy and ubiquitin-proteasome pathways during ultra-endurance running. J Appl Physiol 112: 1529–1537, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Jamart C, Naslain D, Gilson H, Francaux M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab 305: E964–E974, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Jelinek D, Millward V, Birdi A, Trouard TP, Heidenreich RA, Garver WS. Npc1 haploinsufficiency promotes weight gain and metabolic features associated with insulin resistance. Hum Mol Genet 20: 312–321, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 6: 929–935, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462: 245–253, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee Y, Lee HY, Hanna RA, Gustafsson ÅB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301: H1924–H1931, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8: 3–5, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, Chen Z, Holloszy JO, Medeiros DM, Schmidt RE, Saffitz JE, Abel ED, Semenkovich CF, Kelly DP. PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3: e101, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao G, Yao Y, Liu J, Yu Z, Cheung S, Xie A, Liang X, Bi X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1−/− mouse brain. Am J Pathol 171: 962–975, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 418: 797–801, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119: 121–135, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, Yan Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27: 4184–4193, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M, Kambadur R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab 16: 613–624, 2012. [DOI] [PubMed] [Google Scholar]
- 22.LoVerso F, Carnio S, Vainshtein A, Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 10: 11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6: 458–471, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Menzies KJ, Singh K, Saleem A, Hood DA. Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288: 6968–6979, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dötsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 20: 3852–3866, 2011. [DOI] [PubMed] [Google Scholar]
- 28.Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol 546: 851–858, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O'Malley B, Spiegelman BM. Activation of PPARγ coactivator-1 through transcription factor docking. Science 286: 1368–1371, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson ÅB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 18: 721–731, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA. Exercise increases mitochondrial PGC-1α content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem 286: 10605–10617, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA 103: 16260–16265, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, Cohen MA, Chakraborty S, Wang H, Spooner E, Ploegh H, Gsponer J, Korolchuk VI, Jaenisch R. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep 5: 1302–1315, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15: 647–658, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell 144: 689–702, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27: 433–446, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vainshtein A, Kazak L, Hood DA. Effects of endurance training on apoptotic susceptibility in striated muscle. J Appl Physiol 110: 1638–1645, 2011. [DOI] [PubMed] [Google Scholar]
- 39.Watari H, Blanchette-Mackie EJ, Dwyer NK, Glick JM, Patel S, Neufeld EB, Brady RO, Pentchev PG, Strauss JF. Niemann-Pick C1 protein: obligatory roles for N-terminal domains and lysosomal targeting in cholesterol mobilization. Proc Natl Acad Sci USA 96: 805–810, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu L, Xi B, Zhang M, Shen Y, Zhao X, Cheng H, Hou D, Sun D, Ott J, Wang X, Mi J. Associations of six single nucleotide polymorphisms in obesity-related genes with BMI and risk of obesity in Chinese children. Diabetes 59: 3085–3089, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6: 472–483, 2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.