

Abstract
In polycystic ovary syndrome (PCOS), oxidative stress is implicated in the development of β-cell dysfunction. However, the role of mononuclear cell (MNC)-derived inflammation in this process is unclear. We determined the relationship between β-cell function and MNC-derived nuclear factor-κB (NF-κB) activation and tumor necrosis factor-α (TNF-α) secretion in response to a 2-h 75-g oral glucose tolerance test (OGTT) in normoglycemic women with PCOS (15 lean, 15 obese) and controls (16 lean, 14 obese). First- and second-phase β-cell function was calculated as glucose-stimulated insulin secretion (insulin/glucose area under the curve for 0–30 and 60–120 min, respectively) × insulin sensitivity (Matsuda Index derived from the OGTT). Glucose-stimulated NF-κB activation and TNF-α secretion from MNC, and fasting plasma thiobarbituric acid-reactive substances (TBARS) and high-sensitivity C-reactive protein (hs-CRP) were also assessed. In obese women with PCOS, first- and second-phase β-cell function was lower compared with lean and obese controls. Compared with lean controls, women with PCOS had greater change from baseline in NF-κB activation and TNF-α secretion, and higher plasma TBARS. β-Cell function was inversely related to NF-κB activation (1st and 2nd) and TNF-α secretion (1st), and plasma TBARS and hs-CRP (1st and 2nd). First- and second-phase β-cell function also remained independently linked to NF-κB activation after adjustment for body fat percentage and TBARS. In conclusion, β-cell dysfunction in PCOS is linked to hyperglycemia-induced NF-κB activation from MNC and systemic inflammation. These data suggest that in PCOS, inflammation may play a role in impairing insulin secretion before the development of overt hyperglycemia.
Keywords: insulin secretion, glucose intolerance, androgens, insulin resistance, mononuclear cells, obesity, insulin sensitivity
up to 70% of women with polycystic ovary syndrome (PCOS) exhibit insulin resistance and are at risk for type 2 diabetes (T2D; see Refs. 7, 10, and 40). The conventional glucose-regulatory response to insulin resistance is a reciprocal rise in pancreatic β-cell insulin secretion that maintains normal blood glucose concentrations (29). While this compensatory hyperinsulinemia in PCOS has been reported and linked to elevated androgens (3, 38), defects in β-cell function are observed independent of body weight and the degree of insulin resistance (5, 6, 14, 27, 40). Consequently, it is not surprising that nearly 50% of women with PCOS develop prediabetes or T2D before the age of 40 (7, 10, 40). Further work is required to elucidate the mechanism involved in this attenuated β-cell function to better understand how interventions may prevent hyperglycemia in women with PCOS.
Hyperglycemia-induced reactive oxidative species (ROS) from mononuclear cells (MNC) is directly related to insulin resistance and circulating androgens in PCOS (20–23). The resultant oxidative stress leads to lipid peroxidation and DNA damage, and activation of nuclear factor-κB (NF-κB), the cardinal signal of inflammation. Activated NF-κB dissociates from inhibitory-κB (IκB) in the cytoplasm and undergoes nuclear translocation to promote the transcription of tumor necrosis factor-α (TNF-α), a proinflammatory cytokine known to impair insulin signaling and action (46). Although the link between insulin resistance and inflammation in the pathogenesis of T2D is well established, inflammation may also play an important role in disrupting pancreatic insulin secretion (13, 50). Within the β-cell, there is a readily available pool of insulin released upon initial glucose ingestion (1st phase) that is followed by synthesis of new insulin to manage postprandial glucose fluctuations (2nd phase) (30). Recent studies demonstrate that macrophages derived from circulating MNC infiltrate pancreatic islets in primates (39) and humans with T2D (15) and disrupt pancreatic insulin secretion. Indeed, TNF-α from MNC-derived macrophages activates NF-κB within the β-cell thereby inducing endoplasmic reticulum stress and subsequent β-cell apoptosis (50). We recently reported a link between hyperglycemia-induced MNC oxidative stress and low first-phase β-cell function in women with and without PCOS (34) and proposed that inflammation may trigger attenuated β-cell function in women with PCOS. However, to date, no study has specifically examined the interaction between hyperglycemia-induced MNC inflammation and decompensated first- or second-phase pancreatic β-cell function in humans. Therefore, we tested the hypothesis that MNC-derived NF-κB activation and TNF-α secretion in response to oral glucose ingestion would be associated with β-cell dysfunction independent of body fat and oxidative stress.
METHODS
Subjects.
Sixty women, 30 with PCOS (14 lean, 16 obese) and 30 ovulatory controls (16 lean, 14 obese), 18–40 yr of age volunteered for this cross-sectional study. Forty-seven of these subjects were involved in our previous work on PCOS and β-cell function and had complete data to assess inflammation (34). One-half of the cohort was also involved in our prior work with insulin resistance (22). Subjects were nonsmoking, weight stable (<2 kg weight loss in the previous 6 mo), free of T2D or cardiovascular disease, and not involved in habitual exercise for at least 6 mo before the study. Subjects were excluded if they were taking supplements or medications known to influence glucose metabolism or immune responses. Lean and obesity were defined as having a body mass index between 18–25 and or 30–40 kg/m2, respectively. Women with PCOS were selected using the National Institutes of Health criteria. As such, the presence of oligomenorrhea (i.e., intermenstrual intervals >35 days) and hyperandrogenemia [i.e., testosterone >60 ng/dl; androstenedione >3 ng/ml; or dehydroepiandrosterone-sulfate (DHEA-S) >300 μg/dl] was required after excluding nonclassic congenital adrenal hyperplasia, Cushing's Syndrome, hyperprolactinemia, and thyroid disease. All subjects with PCOS also exhibited polycystic ovaries on ultrasound. All control subjects had regular menses lasting 25–35 days and a luteal range serum progesterone level consistent with ovulation (>5 ng/ml). All control subjects exhibited normal circulating androgen levels and did not have any skin manifestations of androgen excess or polycystic ovaries on ultrasound. Subjects received both verbal and written information about the study before signing informed consent documents approved by our Institutional Review Board.
Body composition.
Body weight was recorded on a digital platform scale with subjects wearing a hospital gown to the nearest 0.1 kg. Height was measured without shoes using a wall-mounted stadiometer to the nearest 1.0 cm. All subjects underwent dual-energy X-ray absorptiometry to determine total body fat and truncal fat (Hologic, Waltham, MA). Truncal fat was defined as the area between the diaphragm and the top of the greater trochanter.
Pancreatic β-cell function.
Subjects were provided weight-maintenance meals (resting metabolic rate ×1.2; ∼50% carbohydrate, 30% fat, and 20% protein) and were instructed to refrain from strenuous activity during the 3 days before testing. An oral glucose tolerance test (OGTT) was performed in all women 5–8 days following the onset of menstruation. After an 8- to 12-h overnight fast, a 75-g OGTT was performed, and blood samples were obtained from an antecubital vein at 0, 30, 60, 90, and 120 min to measure plasma glucose and insulin. Insulin sensitivity derived from the OGTT (ISOGTT) was estimated using the Matsuda Index (36), and total area under the curve (AUC) during the OGTT was calculated using the trapezoidal method. First- and second-phase glucose-stimulated insulin secretion (GSIS) was calculated by dividing plasma insulin by glucose AUC during the first 30 and last 60 min of the OGTT as previously described (35). Because the amount of insulin secreted to maintain normoglycemia is influenced by the ambient level of insulin sensitivity, the product of GSIS and ISOGTT (i.e., disposition index) was calculated to characterize pancreatic β-cell function.
MNC isolation and molecular assays.
MNCs were isolated by density gradient centrifugation in Polymorphprep separation media (Accurate Chemical and Scientific Corporation, Westbury, NY) from blood samples obtained during the OGTT at 0 and 120 min (2 h). As shown previously, these time points reflect baseline and peak or near-peak glucose-stimulated proinflammatory responses, respectively (2). Nuclear-bound NF-κB was quantified by electrophoretic mobility shift assay (EMSA) as described before (19, 22). EMSA band specificity was verified by incubating the samples with specific antibodies against the p65 (H-286) and p50 (H-119) subunits of the NF-κB complex (Santa Cruz Biotechnology, Santa Cruz, CA) to supershift the bands, and by competition with cold oligonucleotides. The protein content of IκB was quantified by Western blotting using a 1:1,000 dilution of a monoclonal antibody against IκB (Transduction Laboratories) and actin (Santa Cruz Biotechnology) (1). Densitometry after EMSA and Western blotting was performed on scanned films using Carestream Molecular Imaging software version 5.0.2.30 (Rochester, NY), and values for IκB were corrected for loading using those obtained from actin. A pooled control sample from all study subjects was also loaded on all Western blot gels to adjust for differences in exposure among gels.
MNC culture.
Isolated MNC were washed and resuspended in RPMI (Sigma) and seeded in coated cell culture plates as previously described (31). The cells were incubated for 24 h (humidified, 5% CO2, 37°C), and cell supernatants were collected (10,000 g for 2 min) and stored at −80°C for subsequent analysis.
Biochemical analysis.
Serum glucose was determined during the OGTT using a glucose oxidase assay (YSI 2300 STAT Plus, Yellow Springs, OH). All remaining blood was centrifuged at 1,000 revolutions/min for 10 min at 4°C, and stored at −80°C until analysis. Plasma insulin was measured by radioimmunoassay (Millipore, Billerica, MA). Plasma high-sensitivity C-reactive protein (hs-CRP) was determined by a high-sensitivity enzyme-linked immunosorbent assay (ELISA; Alpha Diagnostics International, San Antonio, TX). Serum luteinizing hormone (LH), testosterone, androstenedione, and DHEA-S concentrations were measured by radioimmunoassay (Siemens Healthcare Diagnostics, Deerfield, IL). Plasma thiobarbituric acid-reactive substances (TBARS), an index of oxidative stress related to lipid peroxidation, was measured by fluorescence (OXItex; ZeptoMetric, Buffalo, NY) (23). MNC-derived TNF-α was determined via high-sensitivity ELISA (R & D Systems, Minneapolis, MN). All samples from each subject were measured in duplicate in the same assay to minimize variance.
Statistical analysis.
Data were analyzed using the statistical program R (Leopard build 64-bit; The R Foundation, Vienna, Austria). Skewed data were log transformed for statistical analysis to meet normality requirements. Because prior work by our group suggests that obesity reduces insulin sensitivity in PCOS (2, 20–23), data from this study were compared across groups using ANOVA. In the event of statistical significance, pairwise comparisons with Bonferroni adjustments were used to identify the source of significance. The absolute change from baseline between pre- and 2-h post-glucose ingestion values was used to characterize TNF-α secretion from MNC. The percent change from baseline between alterations in NF-κB and IκB was determined for each subject to account for intersubject variability. Pearson's product moment correlation was used to determine associations. Linear regression analysis was used to adjust for body fat and oxidative stress to confirm the relationship between β-cell function and inflammation. Data are expressed as means ± SE, and significance was accepted as P ≤ 0.05.
RESULTS
Age, body composition, and serum androgen levels.
Groups were similar in age, but body weight and total body fat were significantly greater (P < 0.05) in obese subjects compared with those who were lean (Table 1). Compared with weight-similar controls, women with PCOS exhibited significantly higher (P < 0.05) serum levels of LH, testosterone, androstenedione, and DHEA-S.
Table 1.
Body Composition, serum hormone levels, glucose homeostasis, and plasma hs-CRP and TBARS
| Lean Controls | Obese Controls | Lean PCOS | Obese PCOS | P Value | |
|---|---|---|---|---|---|
| n | 16 | 14 | 14 | 16 | |
| Age, yr | 29 ± 2 | 31 ± 2 | 27 ± 1 | 26 ± 1 | 0.10 |
| Weight, kg | 59.7 ± 1.6 | 90.4 ± 2.9† | 61.4 ± 1.7‡ | 97.0 ± 2.8†§ | <0.001 |
| BMI, kg/m2 | 22.3 ± 0.5 | 34.0 ± 0.9† | 23.2 ± 0.6‡ | 35.8 ± 0.7†§ | <0.001 |
| Fat mass, % | 32.2 ± 1.9 | 42.2 ± 1.1† | 30.9 ± 1.3‡ | 42.9 ± 1.0†§ | <0.001 |
| Trunk fat, % | 29.7 ± 2.3 | 41.8 ± 0.9† | 29.7 ± 1.8‡ | 44.2 ± 1.0†§ | <0.001 |
| Trunk-total fat ratio | 41.7 ± 1.4 | 46.7 ± 1.1 | 45.1 ± 2.1 | 50.7 ± 0.9†§ | <0.001 |
| LH, IU/ml# | 4.8 ± 0.4 | 3.6 ± 0.4 | 13.5 ± 1.1†‡ | 8.5 ± 1.0†‡§ | <0.001 |
| Testosterone, ng/dl | 41.7 ± 3.2 | 32.1 ± 2.8 | 72.9 ± 6.1†‡ | 76.3 ± 7.0†‡ | <0.001 |
| Androstenedione, ng/ml# | 1.3 ± 0.1 | 1.8 ± 0.1† | 3.4 ± 0.2†‡ | 3.3 ± 0.2†‡ | <0.001 |
| DHEA-S, μg/dl* | 161.7 ± 20.9 | 149.9 ± 17.6 | 321.0 ± 37.4†‡ | 332.0 ± 42.2†‡ | 0.001 |
| Glucose AUC0–30, mg·dl−1·min−1 | 3,411 ± 106 | 3,276 ± 132 | 3,439 ± 107 | 3,535 ± 142 | 0.59 |
| Insulin AUC0–30, μU·ml−1·min−1# | 743 ± 78 | 1,673 ± 331 | 1,334 ± 296 | 2,089 ± 365† | 0.01 |
| Glucose AUC60–120, mg·dl−1·min−1 | 6,904 ± 395 | 6,760 ± 401 | 7,292 ± 424 | 8,319 ± 342 | 0.84 |
| Insulin AUC60–120 (μU·ml−1·min−1# | 1,659 ± 170 | 4,015 ± 618† | 4,152 ± 526† | 8,478 ± 1,478†‡§ | <0.001 |
| ISOGTT | 9.8 ± 0.4 | 4.8 ± 0.6† | 4.9 ± 0.4† | 2.8 ± 0.4†‡§ | <0.001 |
| First-phase GSIS0–30# | 0.22 ± 0.02 | 0.51 ± 0.10 | 0.39 ± 0.09 | 0.60 ± 0.10† | 0.009 |
| Second-phase GSIS60–120# | 0.24 ± 0.02 | 0.62 ± 0.09† | 0.60 ± 0.08† | 1.01 ± 0.16†‡§ | 0.004 |
| hs-CRP, mg/l# | 1.1 ± 0.6 | 4.5 ± 0.8† | 1.3 ± 0.3 | 6.4 ± 1.0†§ | <0.001 |
| TBARS, nmol/ml | 0.6 ± 0.1 | 1.2 ± 0.1† | 1.2 ± 0.2† | 1.3 ± 0.2† | 0.002 |
Data are expressed as means ± SE; n, no. of subjects. BMI, body mass index; LH, luteinizing hormone; DHEA-S, dehydroepiandrosterone-sulfate. AUC, area under the curve, ISOGTT, insulin sensitivity derived from the Matsuda index; 1st and 2nd phase GSIS, glucose-stimulated insulin secretion was calculated as insulin/glucose area under the curve 0–30 min and 60–120 min of the oral glucose tolerance test, respectively; hs-CRP, high-sensitivity C-reactive protein; TBARS, thiobarbituric acid-acid reactive substances. P value represents ANOVA. #Data log-transformed for statistical analysis.
Significantly different compared with lean controls (P < 0.05).
Significantly different compared with obese controls (P < 0.05).
Significantly different compared with lean women with polycystic ovary syndrome (PCOS, P < 0.05).
Glucose regulation.
Glucose and insulin excursions during the OGTT are shown in Fig. 1, A and B, and clinical values are shown in Table 1. Fasting and 2-h glucose levels were comparable across groups, although glucose at 60 and 90 min was statistically higher in obese women with PCOS compared with lean and obese controls (P < 0.05). Compared with lean controls, women with PCOS and obese controls had higher fasting and 2-h insulin levels with a tendency towards higher second-phase insulin AUC (P = 0.11) and 2-h insulin levels (P = 0.07) in obese subjects regardless of PCOS status. First-phase GSIS was higher in obese women with PCOS (P < 0.05) and obese controls (P = 0.06) compared with lean controls, but was comparable in lean women with PCOS and lean controls. Second-phase GSIS was higher (P < 0.01) and ISOGTT was lower (P < 0.05) in women with PCOS compared with weight-similar controls (Table 1). First- and second-phase β-cell function (Fig. 2, A and B) was significantly lower (P < 0.05) in obese women with PCOS and modestly lower (P = 0.08, first phase only) in lean women with PCOS compared with weight-similar controls.
Fig. 1.
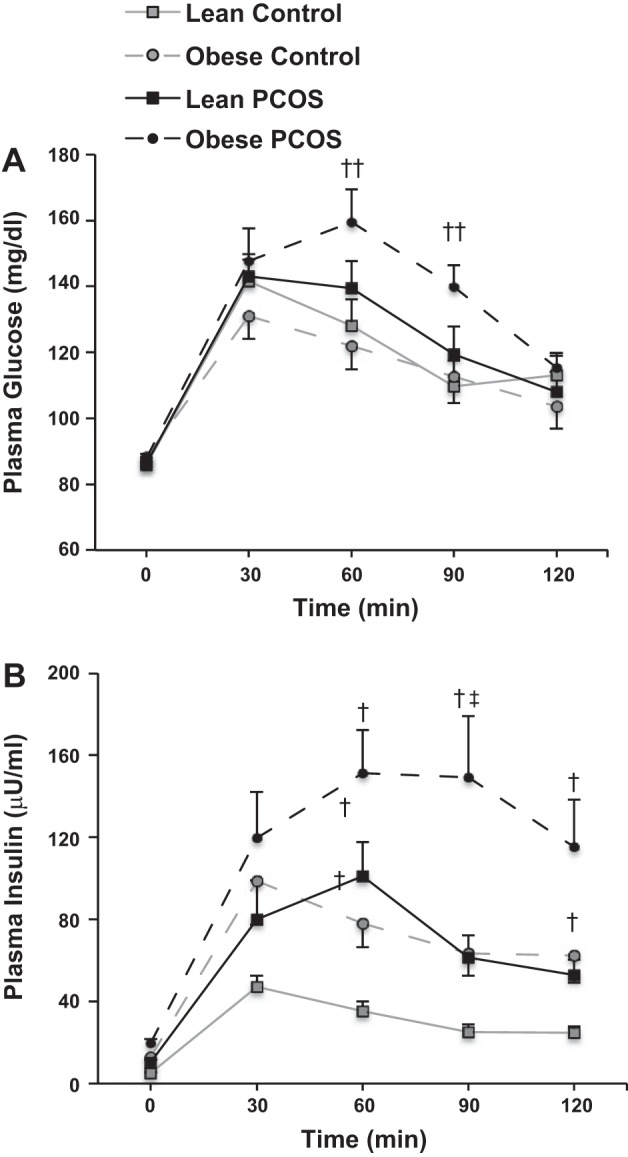
Plasma glucose (A) and insulin (B) excursions during the oral glucose tolerance test (OGTT). PCOS, polycystic ovary syndrome. Data are expressed as means ± SE. †Significantly different compared with lean controls (P < 0.05). ‡Significantly different compared with obese controls (P < 0.05). ††Significantly different compared with lean and obese controls (P < 0.05).
Fig. 2.
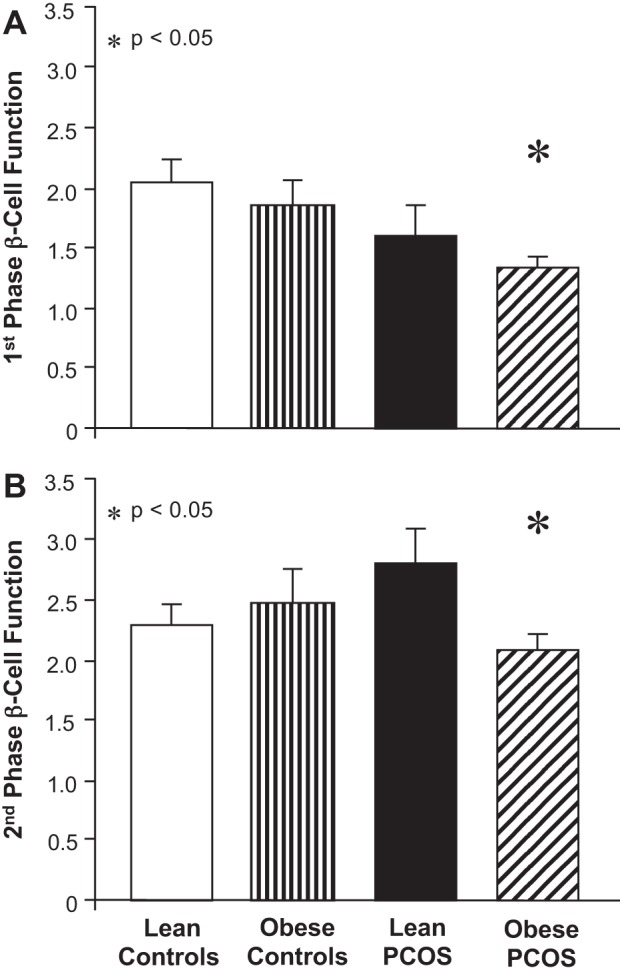
Effects of obesity and polycystic ovary syndrome on 1st-phase (A) and 2nd-phase (B) β-cell function. β-Cell function was calculated as 1st- or 2nd-phase glucose-stimulated insulin secretion (GSIS) × ISOGTT. GSIS was calculated as area under the curve for insulin divided by glucose during 0–30 and 60–120 min of the OGTT. ISOGTT, insulin sensitivity derived from the Matsuda index. Data are expressed as means ± SE. *Obese controls compared with obese women with PCOS (P < 0.05).
Inflammation and oxidative stress in MNC and plasma.
Basal and 2-h values of NF-κB, IκB, and TNF-α are shown in Table 2. Basal and 2-h values of NF-κB and IκB were similar across groups. TNF-α values were higher in lean controls compared with those of the other groups. In response to glucose ingestion, the change from baseline (%) in activated NF-κB was significantly greater (P < 0.01) in women with PCOS compared with weight-similar controls (Fig. 3A). The change from baseline (%) in IκB protein content was significantly lower (P < 0.05) and the absolute change in TNF-α secretion was significantly higher (P < 0.05) in both PCOS groups and obese controls compared with lean controls (Fig. 3, B and C). Fasting plasma TBARS was significantly higher (P < 0.05) in lean and obese women with PCOS and obese controls compared with lean controls (Table 1). In contrast, fasting plasma hs-CRP was higher (P < 0.05) in both obese groups compared with either lean group.
Table 2.
Baseline and 2-h post-glucose ingestion values of NF-κB, IκB protein content, and TNF-α secretion
| Lean Controls | Obese Controls | Lean PCOS | Obese PCOS | P Value | |
|---|---|---|---|---|---|
| NF-κB activation (densitometry) | |||||
| 0 Hour | 111 ± 17 | 107 ± 18 | 134 ± 18 | 110 ± 23 | 0.75 |
| 2 Hours | 104 ± 16 | 128 ± 26 | 158 ± 22 | 131 ± 24 | 0.41 |
| IκB protein content (densitometry) | |||||
| 0 Hour | 139 ± 20 | 176 ± 13 | 156 ± 22 | 161 ± 15 | 0.54 |
| 2 Hours | 138 ± 19 | 156 ± 12 | 120 ± 16 | 112 ± 11 | 0.17 |
| TNF-α, pg/ml | |||||
| 0 Hour | 23.6 ± 6.9* | 8.6 ± 2.6 | 2.6 ± 0.3 | 5.0 ± 1.1 | 0.001 |
| 2 Hours | 17.5 ± 6.0 | 11.9 ± 3.4 | 3.1 ± 0.7 | 9.9 ± 3.5 | 0.11 |
Data are expressed as means ± SE.
Significantly different compared with all other groups (P < 0.05).
Fig. 3.
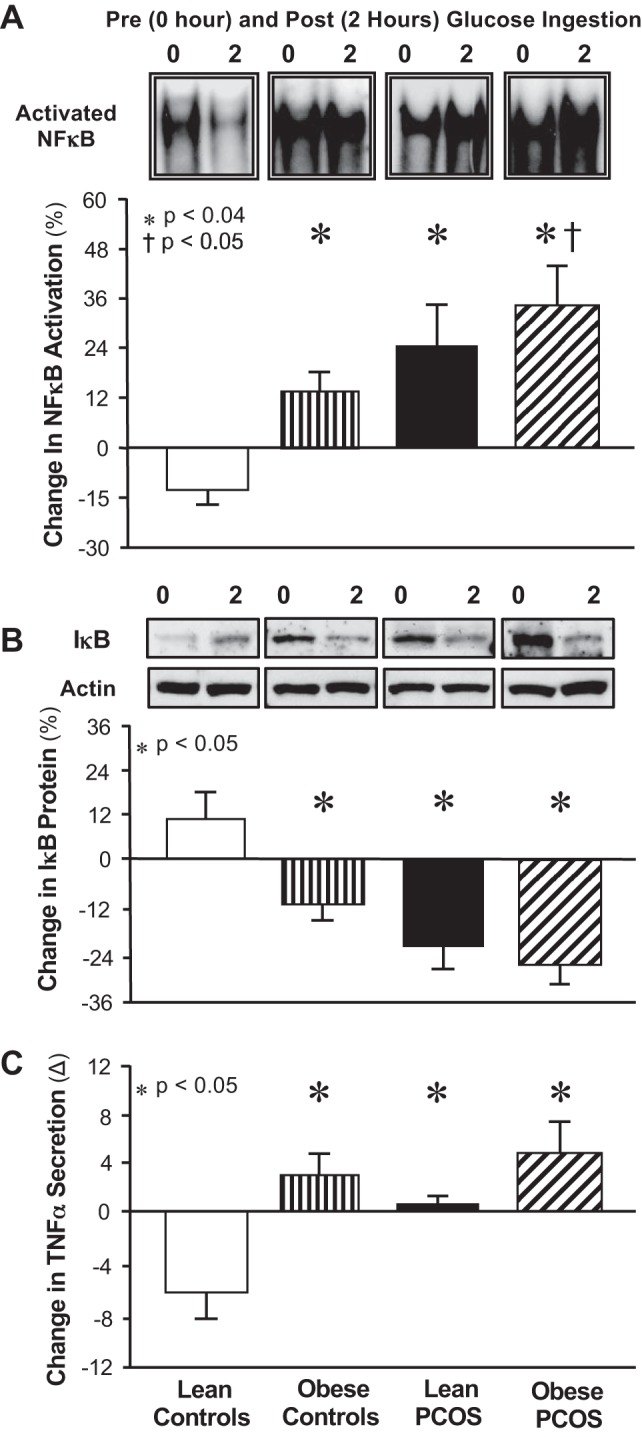
Mononuclear cell (MNC)-derived inflammation in response to glucose ingestion in women with PCOS and in weight-similar controls. Change from baseline (%) in NF-κB activation (A) and inhibitory-κB (IκB) protein content (B) based on a densitometric quantitative analysis between fasting and 2-h post-glucose ingestion samples. Representative electrophoretic mobility shift assay (EMSA) bands of intranuclear NF-κB and Western blots of IκB and actin protein are depicted above the histograms showing the change in content pre- and post-glucose ingestion. Samples from all four study groups were run on the same gel. Absolute change from baseline (Δ, pg/ml) in TNF-α secretion (C) measured in culture supernatants between fasting and 2-h post-glucose ingestion samples. Data are expressed as means ± SE. *Lean controls vs. obese controls, lean women with PCOS, and obese women with PCOS (P < 0.05). †Lean women with PCOS compared with obese women with PCOS (P < 0.05).
Correlations.
First- and second-phase β-cell function was negatively correlated with glucose-stimulated NF-κB activation (Fig. 4, A and B), and first-phase β-cell function was positively correlated with IκB protein content (Table 2). First- and second-phase β-cell function remained independently linked to NF-κB activation after adjustment for body fat percentage (estimate: −0.001, t value: −1.95, P = 0.05) and TBARS (estimate: −0.001, t value: −2.7, P = 0.008). The relationship between NF-κB activation and first-phase β-cell function was also independent of the ratio of truncal fat to total body fat (estimate: −0.001, t value: −1.95, P = 0.05), and there was only a trend for impact on this relationship when trunk truncal fat percentage was used in the model (estimate: −0.001, t value: −1.90, P = 0.06). Furthermore, the relationship between NF-κB activation and second-phase β-cell function was independent of the ratio of truncal fat to total body fat and truncal fat percentage (estimate: −0.001, t value: −2.69, P = 0.009).
Fig. 4.
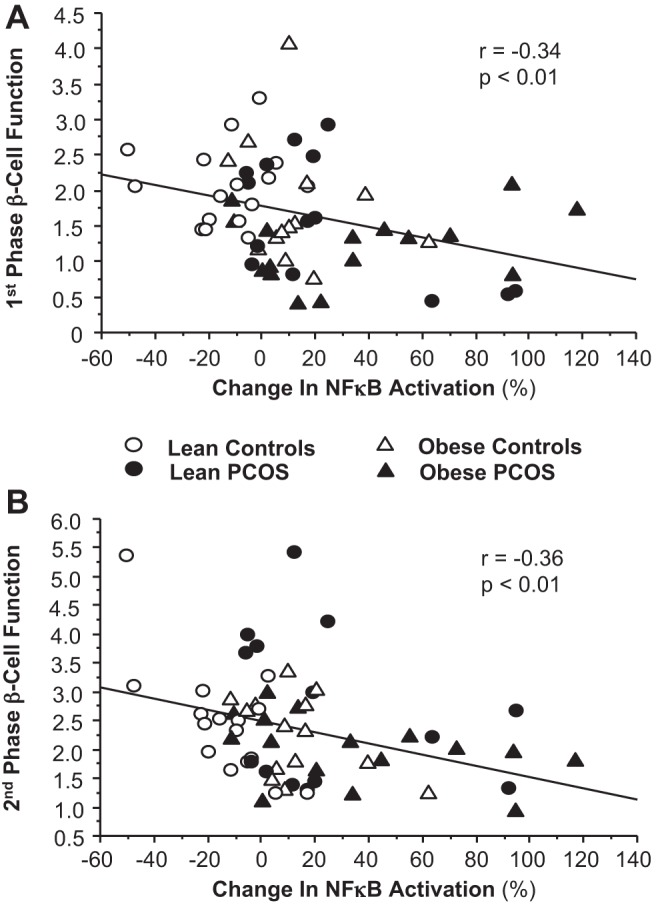
Correlations between the change from baseline (%) in MNC-derived NF-κB activation and 1st-phase (A) and 2nd-phase (B) β-cell function for the combined groups.
Glucose-stimulated NF-κB activation was negatively correlated with ISOGTT (r = −0.43, P = 0.006), and positively correlated with MNC-derived TNF-α secretion (r = 0.46, P < 0.001), plasma hs-CRP (r = 0.39, P < 0.001), and plasma TBARS (r = 0.35, P < 0.006). Serum levels of testosterone and androstenedione were positively correlated with NF-κB activation and TNF-α secretion, and negatively correlated with IκB protein content (Table 3). Serum androstenedione was also positively correlated with plasma TBARS and hs-CRP, and serum DHEA-S was positively correlated with plasma TBARS.
Table 3.
Pearson's correlations between insulin, body composition and serum androgen levels, and markers of inflammation and oxidative stress from MNC and plasma for the combined groups
| Correlation Coefficients | NF-κB Activation from MNC (Change from Baseline), % | IκB Protein from MNC (Change from Baseline), % | TNF-α Secretion from MNC (Change from Baseline) | Plasma TBARS | Plasma hs-CRP |
|---|---|---|---|---|---|
| Insulin metabolism | |||||
| Fasting insulin | 0.47‡ | −0.36† | 0.36† | 0.36† | 0.61‡ |
| 2-h Insulin | 0.21 | −0.31† | 0.19 | 0.12 | 0.46‡ |
| ISOGTT | −0.43† | 0.37† | −0.37† | −0.62‡ | 0.87‡ |
| First-phase GSIS | −0.08 | −0.08 | 0.09 | 0.36† | −0.06 |
| Second-phase GSIS | 0.12 | −0.27* | 0.23 | 0.17 | 0.39‡ |
| First-phase β-cell function | −0.37† | 0.31† | −0.27* | −0.41‡ | −0.26* |
| Second-phase β-cell function | −0.37† | 0.09 | −0.09 | −0.09 | −0.30* |
| Body composition | |||||
| BMI | 0.33† | 0.21 | 0.35† | 0.46† | 0.69‡ |
| Total body fat, % | 0.31* | 0.01 | −0.40† | 0.17 | 0.62‡ |
| Truncal fat, % | 0.35† | −0.02 | 0.21 | 0.21 | 0.68‡ |
| Androgens | |||||
| LH | 0.23 | −0.34† | 0.04 | 0.04 | 0.05 |
| Testosterone | 0.33† | −0.25* | 0.28* | 0.03 | −0.02 |
| Androstenedione | 0.38† | −0.44‡ | 0.27* | 0.28* | 0.28* |
| DHEA-S | 0.17 | 0.23 | 0.07 | 0.32† | 0.08 |
MNC, mononuclear cell; IκB, inhibitory-κB; β-cell function, disposition index (GSIS × ISOGTT). Fasting insulin, 2-h insulin, GSIS, hs-CRP, LH, DHEA-S, and androstenedione were log-transformed for statistical analysis. Statistically significant correlations are represented by
P < 0.05,
P < 0.01, and
P < 0.001.
DISCUSSION
These data show for the first time that MNC-derived NF-κB activation in response to glucose ingestion is inversely related to in vivo measures of first- and second-phase β-cell function. Suppression of postprandial inflammation appears to be a normal physiological response to glucose ingestion, since this is associated with adequate matching of GSIS to the level of insulin sensitivity. This is reflected by decreases in NF-κB activation and TNF-α secretion and increases in IκB protein content from MNC in normoglycemic lean ovulatory controls. This is in contrast to lean women with PCOS where glucose ingestion increases NF-κB activation and TNF-α secretion and decreases IκB protein content from MNC compared with lean controls. These findings are consistent with our previous reports in women with PCOS (19, 23) and older adults (32), and in impaired glucose tolerance (16) and type 1 diabetes (26). This observation is important because it illustrates that, in PCOS, feeding alone is capable of triggering inflammation in the absence of obesity (17, 48).
Elevated β-cell mass is a typical consequence of insulin resistance that allows for increased insulin secretion to maintain normoglycemia (42). In fact, insulin-resistant obese controls in our study cohort exhibit greater first- and second-phase GSIS compared with lean controls. In contrast, lean women with PCOS with a comparable degree of insulin resistance to obese controls do not exhibit greater first-phase GSIS. This latter finding suggests that, in PCOS, early β-cell decompensation can occur before the development of obesity and that MNC-derived inflammation may reduce the readily available pool of insulin to respond to glucose ingestion (41, 44). Interestingly, second-phase GSIS reflecting the synthesis of new insulin to manage fluctuations in postprandial glucose is increased in lean PCOS women compared with lean controls and comparable to that of obese controls. Despite differences between first- and second-phase GSIS, these observations should be viewed with caution given that the amount of insulin that is secreted to maintain normoglycemia is influenced by the prevailing insulin sensitivity. Consequently, β-cell function is best characterized by the product of GSIS and ISOGTT. In this context, only first-phase β-cell dysfunction is evident after correcting for insulin resistance in lean women with PCOS. Thus, our data suggest the possibility that the stark differences in first- vs. second-phase β-cell function in lean women with PCOS are linked to inflammation-induced deficits in β-cell mass and/or function (37, 49).
Obese women with PCOS exhibit the greatest degree of insulin resistance and compromised β-cell function, which may confer the greatest risk for developing T2D compared with obese controls or lean women with PCOS. Because obesity is associated with elevated β-cell mass (42), it is not surprising that obese women with PCOS have exaggerated first- and second-phase GSIS compared with lean women with PCOS. It is evident after correction of GSIS for insulin resistance that first- and second-phase β-cell function is significantly lower compared with both lean groups and obese controls. This decrease in β-cell function may very well reflect the initial sign of disruption in the balance between inflammation-induced β-cell injury and compensatory β-cell function (9, 28). TBARS, a common index of lipid peroxidation, was similar in both obese groups and lean women with PCOS, suggesting that differences in insulin resistance among groups are independent of oxidative stress alone. Instead, the combination of excess total body fat and/or trunk truncal fat and PCOS may promote greater inflammation to account for the profound decrease in insulin sensitivity and β-cell function in obese women with PCOS. Glucose-stimulated NF-κB activation from MNC is greater in obese women with PCOS compared with lean women with PCOS. Furthermore, measures of adiposity, including abdominal adiposity, in our study cohort are negatively associated with insulin sensitivity and positively associated with MNC-derived NF-κB activation and TNF-α secretion as well as plasma TBARS and hs-CRP. Abdominal adiposity has been linked to elevations in circulating mediators of inflammation such as hs-CRP that have been shown to impair β-cell function, although our study cannot rule out a role of adipokines (e.g., adiponectin or leptin) or elevated free fatty acids in this process (9, 28). Nevertheless, our data indicate that the regulation of early and late-phase blood glucose control in response to glucose ingestion appears to be synergistically hampered by inflammation unique to PCOS in conjunction with the inflammatory load of excess body fat. Our findings are of clinical relevance because they highlight the unique pancreatic endocrine phenotypes in lean and obese women with PCOS that may differentially elevate risk of developing fasting vs. postprandial glucose intolerance (30).
Hyperandrogenism in PCOS may contribute to β-cell dysfunction. It remains controversial whether androgens are capable of imparting a direct effect on the pancreatic β-cell. In rodents, androgen receptors have been identified in β-cells, and prenatal androgen exposure decreases β-cell sensitivity to GSIS (2, 3, 18, 25, 27, 43, 45). In humans, however, induction of hyperandrogenism in healthy individuals has no effect on β-cell function or viability (11). The ability of hyperandrogenism to contribute to pancreatic dysfunction in PCOS is most likely through the induction of oxidative stress and inflammation from MNC that may ultimately promote β-cell failure in susceptible individuals (33). Circulating androgens are positively associated with glucose-stimulated NF-κB activation and TNF-α secretion from MNC along with plasma TBARS and hs-CRP, and negatively associated with glucose-stimulated IκB protein content. This corroborates similar results from our other studies (2, 20–23). Furthermore, we have recently reported that hyperandrogenism is capable of activating MNC to increase MNC sensitivity to glucose ingestion in a receptor-dependent fashion (19, 20, 24). Thus, it appears that in PCOS, hyperandrogenism serves as an additional indirect mechanism to accentuate the deleterious effects of oxidative stress and inflammation on β-cell function.
Our study has certain limitations that may affect our interpretation. We recognize that associations do not equate to causality and that further studies are needed to elucidate the role of MNC-derived inflammation on β-cell function in PCOS. In addition, we acknowledge that our study cannot completely rule out the specific role of visceral fat as opposed to subcutaneous fat on pancreatic function, since we did not use magnetic resonance imaging or computed tomography scans to determine abdominal fat. We also recognize that subtle differences in plasma glucose during 60 and 90 min of the OGTT could cause some degree of impairment in pancreatic insulin secretion between PCOS and control subjects. It is likely that the higher blood glucose in obese women with PCOS is an effect of excess body fat that promotes insulin resistance and β-cell dysfunction. In contrast, it is unlikely that hyperglycemia per se is the primary factor explaining our results since lean women with PCOS exhibit β-cell dysfunction in the face of glucose levels comparable to lean and obese controls. Moreover, the distinct differences in metabolic and inflammation parameters we report in women with PCOS compared with weight-similar controls lend support to the contention that, in PCOS, a distinct β-cell dysfunction is present before the development of overt impaired glucose intolerance or T2D. The reason for increased basal TNF-α secretion from MNC of lean healthy controls is unclear. However, data should be interpreted with caution as opposed to inferring that healthy controls have greater inflammation than PCOS or obese counterparts. We have not observed increased basal TNF-α secretion from MNC in lean controls compared with other groups in our previous work (19), and there is no clinical correlate with TNF-α secretion from MNC. In fact, hs-CRP, a well-established biomarker of inflammation, was low in healthy controls, showing that these individuals are not characterized by clinical inflammation. Most importantly, TNF-α secretion from MNC decreased in response to glucose ingestion, suggesting that suppression of MNC-derived inflammation goes hand-in-hand with adequate pancreatic β-cell function. We also acknowledge that use of plasma C-peptide as a measure of prehepatic insulin secretion may provide more direct measures of β-cell function. Nevertheless, we have previously shown that hepatic insulin extraction does not affect insulin-derived calculations of β-cell function from the OGTT before or after lifestyle modification (35). Use of the euglycemic and hyperglycemic clamp or frequently sampled intravenous glucose tolerance test may provide a more accurate assessment of insulin resistance and GSIS, respectively, compared with OGTT-derived measures (8). However, these intravenous techniques exclude the gastrointestinal tract, which limits the physiological understanding of in vivo β-cell function. Nevertheless, the ISOGTT has been shown to correlate strongly with euglycemic clamp-derived measures of insulin sensitivity (36), and our ISOGTT results for lean and obese women with PCOS compared with weight-similar controls mimic those from seminal articles that used the euglycemic clamp (12). Thus, use of the OGTT may provide more “real-world” advantage to discerning links between MNC and systemic markers of inflammation and in vivo β-cell function.
In conclusion, lean women with PCOS exhibit MNC-derived inflammation and lower first- but not second-phase β-cell function, suggesting that inflammation may affect distant pathways regulating insulin secretion physiology before the development of obesity. In contrast, obese women with PCOS exhibit greater insulin resistance than lean subjects along with first- and second-phase β-cell dysfunction, indicating that the combination of PCOS and obesity induces greater decompensation of pancreatic responses to insulin resistance. Most importantly, β-cell function is directly related to MNC-derived NF-κB activation in response to glucose ingestion along with plasma TBARS and hs-CRP, and inversely related to IκB protein content, which implicates inflammation in the impairment of β-cell function in PCOS (4, 44, 47). Our findings highlight the need for further investigation to determine the mechanism by which inflammation interacts with the pancreatic β-cells to increase diabetes risk in PCOS.
GRANTS
This research was supported by National Institutes of Health Grants HD-048535 (F. González), RO1-AG-12834 (J. P. Kirwan), and T32-DK-007319 (S. K. Malin).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.K.M., J.P.K., and F.G. conception and design of research; C.L.S., and F.G. performed experiments; S.K.M., and F.G. analyzed data; S.K.M., and F.G. interpreted results of experiments; S.K.M. and F.G. prepared figures; S.K.M. and F.G. drafted manuscript; S.K.M., J.P.K., and F.G. edited and revised manuscript; S.K.M., J.P.K., C.L.S., and F.G. approved final version of manuscript.
ACKNOWLEDGMENTS
This paper was presented at the 74th Annual Meeting of the American Diabetes Association, San Francisco, CA, June 13–17, 2014.
Current Address for S. K. Malin: Department of Kinesiology & Division of Endocrinology and Metabolism, University of Virginia, Charlottesville, VA 22902.
REFERENCES
- 1.Aljada A, Ghanim H, Dandona P. Translocation of p47phox and activation of NADPH oxidase in mononuclear cells. Methods Mol Biol 196: 99–103, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Aljada A, Friedman J, Ghanim H, Mohanty P, Hofmeyer D, Chaudhuri A, Dandona P. Glucose ingestion induces an increase in intranuclear nuclear factor kappaB, a fall in cellular inhibitor kappaB, and an increase in tumor necrosis factor alpha messenger RNA by mononuclear cells in healthy human subjects. Metabolism 55: 1177–1185, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab 50: 1: 113–116, 1980. [DOI] [PubMed] [Google Scholar]
- 4.Ceriello A, Testa R. Antioxidant anti-inflammatory treatment in type 2 diabetes. Diabetes Care 32, Suppl 2: S232–S236, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciampelli M, Fulghesu AM, Cucinelli F, Pavone V, Caruso A, Mancuso S, Lanzone A. Heterogeneity in beta cell activity, hepatic insulin clearance and peripheral insulin sensitivity in women with polycystic ovary syndrome. Hum Reprod 12: 1897–1901, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Ciampelli M, Fulghesu AM, Cucinelli F, Pavone V, Ronsisvalle E, Guido M, Caruso A, Lanzone A. Impact of insulin and body mass index on metabolic and endocrine variables in polycystic ovary syndrome. Metabolism 48: 167–172, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Ciampelli M, Leoni F, Cucinelli F, Mancuso S, Panunzi S, De Gaetano A, Lanzone A. Assessment of insulin sensitivity from measurements in the fasting state and during an oral glucose tolerance test in polycystic ovary syndrome and menopausal patients. J Clin Endocrinol Metab 90: 1398–1406, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Cobelli C, Toffolo G, Dalla Man C, Campioni M, Denti P, Caumo A, Butler P, Rizza R. Assessment of beta-cell function in humans, simultaneously with insulin sensitivity and hepatic extraction, from intravenous and oral glucose tests. Am J Physiol Endocrinol Metab 293: E1–E15, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Despres JP, Lemieux I, Prud'homme D. Treatment of obesity: need to focus on high risk abdominally obese patients. Br Med J 322: 716–720, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeUgarte C, Bartolucci A, Azziz R. Prevalence of insulin resistance in the polycystic ovary syndrome using the homeostasis model assessment. Fertil Steril 83: 1454–1460, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Diamond MP, Grainger D, Sherwin RS, DeFronzo RA. Effects of methyltestosterone on insulin secretion and sensitivity in women. J Clin Endocrinol Metab 83: 4420–4425, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 38: 1165–1174, 1989. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi KM, Manabe I. Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes Metab 15, Suppl 3: 152–158, 2013. [DOI] [PubMed] [Google Scholar]
- 14.Ehrmann DA, Sturis J, Byrne MM, Karrison T, Rosenfield RL, Polonsky KS. Insulin secretory defects in polycystic ovary syndrome. Relationship to insulin sensitivity and family history of non-insulin-dependent diabetes mellitus. J Clin Invest 96: 1: 520–527, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehses J, Perren A, Eppler E, Ribaux P, Pospisilik J, Maor Cahn R, Gueripel X, Ellingsgaard H, Schneider MKJ, Biollaz G, Fontana A, Reinecke M, Homo Delarche F, Donath M. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56: 2356–2370, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106: 2067–2072, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23: 599–622, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Franks S, Gilling Smith C, Watson H, Willis D. Insulin action in the normal and polycystic ovary. Endocrinol Metab Clin North Am 28: 361–378, 1999. [DOI] [PubMed] [Google Scholar]
- 19.González F, Minium J, Rote N, Kirwan JP. Hyperglycemia alters tumor necrosis factor-alpha release from mononuclear cells in women with polycystic ovary syndrome. J Clin Endocrinol Metab 90: 5336–5342, 2005. [DOI] [PubMed] [Google Scholar]
- 20.González F, Nair KS, Daniels J, Basal E, Schimke J. Hyperandrogenism sensitizes mononuclear cells to promote glucose-induced inflammation in lean reproductive-age women. Am J Physiol Endocrinol Metab 302: E297–E306, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.González F, Nair KS, Daniels J, Basal E, Schimke J, Blair H. Hyperandrogenism sensitizes leukocytes to hyperglycemia to promote oxidative stress in lean reproductive-age women. J Clin Endocrinol Metab 97: 2836–2843, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.González F, Rote N, Minium J, Kirwan JP. Reactive oxygen species-induced oxidative stress in the development of insulin resistance and hyperandrogenism in polycystic ovary syndrome. J Clin Endocrinol Metab 91: 336–340, 2006. [DOI] [PubMed] [Google Scholar]
- 23.González F, Sia C, Shepard M, Rote N, Minium J. Hyperglycemia-induced oxidative stress is independent of excess abdominal adiposity in normal-weight women with polycystic ovary syndrome. Hum Reprod 27: 3560–3568, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.González F, Sia C, Shepard M, Rote N, Minium J. Inflammation in response to glucose ingestion is independent of excess abdominal adiposity in normal-weight women with polycystic ovary syndrome. J Clin Endocrinol Metab 97: 4071–4079, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodarzi MO, Erickson S, Port SC, Jennrich RI, Korenman SG. beta-Cell function: a key pathological determinant in polycystic ovary syndrome. J Clin Endocrinol Metab 90: 310–315, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann MA. Insufficient glycemic control increases nuclear factor-kappa B binding activity in peripheral blood mononuclear cells isolated from patients with type 1 diabetes. Diabetes Care 21: 1310–1316, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Holte J, Bergh T, Berne C, Berglund L, Lithell H. Enhanced early insulin response to glucose in relation to insulin resistance in women with polycystic ovary syndrome and normal glucose tolerance. J Clin Endocrinol Metab 78: 1052–1058, 1994. [DOI] [PubMed] [Google Scholar]
- 28.Jourdan T, Godlewski G, Cinar R, Bertola A, Szanda G, Liu J, Tam J, Han T, Mukhopadhyay B, Skarulis MC, Ju C, Aouadi M, Czech MP, Kunos G. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat Med 19: 1132–1140, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC, Palmer JP. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42: 1663–1672, 1993. [DOI] [PubMed] [Google Scholar]
- 30.Kanat M, Norton L, Winnier D, Jenkinson C, DeFronzo RA, Abdul-Ghani MA. Impaired early- but not late-phase insulin secretion in subjects with impaired fasting glucose. Acta Diabetol 48: 209–217, 2011. [DOI] [PubMed] [Google Scholar]
- 31.Kelly KR, Haus JM, Solomon TPJ, Patrick Melin A, Cook M, Rocco M, Barkoukis H, Kirwan JP. A low-glycemic index diet and exercise intervention reduces TNF(alpha) in isolated mononuclear cells of older, obese adults. J Nutr 141: 1089–1094, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirwan JP, Krishnan RK, Weaver J, Del Aguila LF A, Evans WJ. Human aging is associated with altered TNF-alpha production during hyperglycemia and hyperinsulinemia. Am J Physiol Endocrinol Metab 281: E1137–E1143, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Liu S, Navarro G, Mauvais-Jarvis F. Androgen excess produces systemic oxidative stress and predisposes to beta-cell failure in female mice PLoS One 5: e11302–e11302, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malin SK, Kirwan JP, Sia C, González F. Glucose-stimulated oxidative stress in mononuclear cells is related to pancreatic B-cell dysfunction in polycystic ovary syndrome. J Clin Endocrinol Metab 99: 322–329, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malin SK, Solomon TPJ, Blaszczak A, Finnegan S, Filion J, Kirwan JP. Pancreatic beta cell function increases in a linear dose-response manner following exercise training in adults with prediabetes. Am J Physiol Endocrinol Metab 305: E1248–E1254, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22: 1462–1470, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Meier JB, Riccardo RC. Role of reduced B-cell mass versus impaired B-cell function in the pathogenesis of type 2 diabetes. Diabetes Care 36, Suppl 2: S113–S119, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab 83: 2001–2005, 1998. [DOI] [PubMed] [Google Scholar]
- 39.Nicol LE, Grant WR, Comstock SM, Nguyen ML, Smith MS, Grove KL, Marks DL. Pancreatic inflammation and increased islet macrophages in insulin-resistant juvenile primates. J Endocrinol 217: 207–213, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ovalle F, Azziz R. Insulin resistance, polycystic ovary syndrome, and type 2 diabetes mellitus. Fertil Steril 77: 1095–1105, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta 1801: 289–298, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prentki M, Nolan C. Islet beta cell failure in type 2 diabetes. J Clin Invest 116: 1802–1812, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rae M, Grace C, Hogg K, Wilson L, McHaffie S, Ramaswamy S, MacCallum J, Connolly F, McNeilly A, Duncan C. The pancreas is altered by in utero androgen exposure: implications for clinical conditions such as polycystic ovary syndrome (PCOS). PLoS One 8: e56263–e56263, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson RP. Beta-cell deterioration during diabetes: what's in the gun? Trends Endocrinol Metab 20: 388–393, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roland AV, Nunemaker CS, Keller SR, Moenter SM. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol 207: 213–223, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 107: 181–189, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang C, Han P, Oprescu A, Lee S, Gyulkhandanyan A, Chan GNY, Wheeler M, Giacca A. Evidence for a role of superoxide generation in glucose-induced beta-cell dysfunction in vivo. Diabetes 56: 2722–2731, 2007. [DOI] [PubMed] [Google Scholar]
- 48.Tirosh A, Potashnik R, Bashan N, Rudich A. Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3–L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J Biol Chem 274: 10595–10602, 1999. [DOI] [PubMed] [Google Scholar]
- 49.Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53, Suppl 3: S16–S21, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Westwell-Roper C, Nackiewicz D, Dan M, Ehses JA. Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol Cell Biol 92: 314–323, 2014. [DOI] [PubMed] [Google Scholar]