

ABSTRACT
Flagellar biogenesis in Helicobacter pylori is regulated by a transcriptional hierarchy governed by three sigma factors, RpoD (σ80), RpoN (σ54), and FliA (σ28), that temporally coordinates gene expression with the assembly of the flagellum. Previous studies showed that loss of flagellar protein export apparatus components inhibits transcription of flagellar genes. The FlgS/FlgR two-component system activates transcription of RpoN-dependent genes though an unknown mechanism. To understand better the extent to which flagellar gene regulation is coupled to flagellar assembly, we disrupted flagellar biogenesis at various points and determined how these mutations affected transcription of RpoN-dependent (flaB and flgE) and FliA-dependent (flaA) genes. The MS ring (encoded by fliF) is one of the earliest flagellar structures assembled. Deletion of fliF resulted in the elimination of RpoN-dependent transcripts and an ∼4-fold decrease in flaA transcript levels. FliH is a cytoplasmic protein that functions with the C ring protein FliN to shuttle substrates to the export apparatus. Deletions of fliH and genes encoding C ring components (fliM and fliY) decreased transcript levels of flaB and flgE but had little or no effect on transcript levels of flaA. Transcript levels of flaB and flgE were elevated in mutants where genes encoding rod proteins (fliE and flgBC) were deleted, while transcript levels of flaA was reduced ∼2-fold in both mutants. We propose that FlgS responds to an assembly checkpoint associated with the export apparatus and that FliH and one or more C ring component assist FlgS in engaging this flagellar structure.
IMPORTANCE The mechanisms used by bacteria to couple transcription of flagellar genes with assembly of the flagellum are poorly understood. The results from this study identified components of the H. pylori flagellar basal body that either positively or negatively affect expression of RpoN-dependent flagellar genes. Some of these basal body proteins may interact directly with regulatory proteins that control transcription of the H. pylori RpoN regulon, a hypothesis that can be tested by examining protein-protein interactions in vitro.
INTRODUCTION
The bacterial flagellum is a complex nanomachine powered by an ion-driven rotary motor consisting of about 30 different types of proteins whose copy numbers range from a few to thousands (Fig. 1). The flagellum consists of three basic structures referred to as the basal body, hook, and filament (1). The basal body is an intricate complex that consists of the flagellar rod, rings, a motor, a switch complex, and a specialized type III secretion system (T3SS) that transports flagellar proteins across the cell membrane (1–3). The T3SS, also referred to as the flagellar protein export apparatus, consists of integral membrane proteins (FlhA, FlhB, FliO, FliP, FliQ, and FliR) which form an export pore located within the inner membrane, as well as cytoplasmic components (FliI, FliH, and FliJ) that deliver protein substrates to the export pore (4, 5). During flagellar assembly, the export apparatus initially transports rod- and hook-type substrates across the cell membrane into the lumen of the nascent flagellum (6, 7). Upon completion of the mature hook-basal body (HBB) structure, the export apparatus undergoes a conformational change that is accompanied by a switch in substrate specificity to filament-type substrates.
FIG 1.
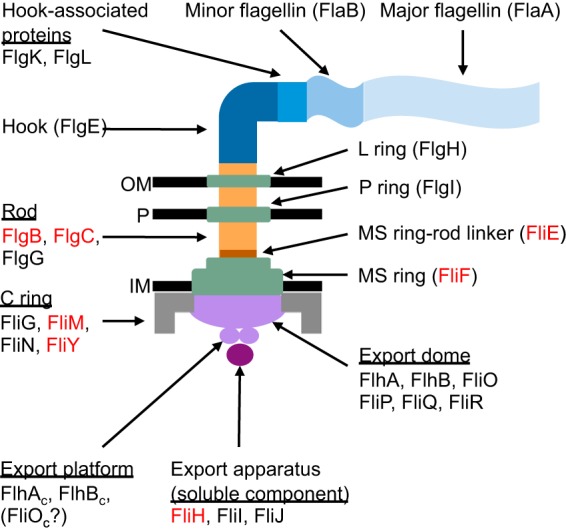
Structure of the H. pylori flagellum. The major components of the flagellum and the proteins that comprise these components in H. pylori are indicated. Proteins that were targeted for deletion are labeled in red. Relative positions of the outer membrane (OM), inner membrane (IM), and peptidoglycan (P) are indicated.
Biogenesis of the bacterial flagellum involves a transcriptional hierarchy that is responsive to checkpoints in assembly so that expression of specific flagellar genes occurs as their products are needed for formation of the nascent flagellum. In Salmonella enterica serovar Typhimurium (the model organism for flagellar biogenesis studies), flagellar genes needed early in assembly require the primary sigma factor RpoD (σ70) for their transcription, while the late flagellar genes (e.g., the flagellin gene) require the alternative σ factor FliA (σ28) for their transcription (reviewed in reference 8). Flagellar biogenesis in H. pylori and Campylobacter jejuni (both members of the subphylum Epsilonproteobacteria) similarly involves RpoD and FliA but also involves the alternative σ factor RpoN (σ54). Transcription of H. pylori and C. jejuni genes required early in flagellar assembly is dependent on RpoD (σ80), while transcription of genes needed midway through flagellar biogenesis is dependent on RpoN, and transcription of genes needed late in the assembly process is dependent on FliA (9–11). The organization of H. pylori flagellar genes into regulons based on the σ factor needed for transcription suggests a framework for a transcriptional hierarchy that operates in conjunction with assembly of the flagellum. The assembly checkpoints and mechanisms which regulate such a hierarchy, however, are poorly understood.
In H. pylori, the RpoN-dependent genes encode rod proteins (FlgBC), hook protein (FlgE), hook-associated proteins (FlgL and FlgK), hook-length control protein (FliK), a minor flagellin (FlaB), and enzymes required for flagellin glycosylation (9, 12, 13). Transcription of the RpoN regulon is regulated by a two-component system composed of a cytoplasmic sensor kinase, FlgS, and the response regulator FlgR (9, 12, 14, 15). In addition to FlgS and FlgR, several components of the flagellar export apparatus are required for transcription of the RpoN regulons in H. pylori and C. jejuni (16), although the export apparatus does not need to be competent for secretion to stimulate transcription of the RpoN-dependent genes (17, 18). FlgS is thought to respond to a signal within or near the flagellar T3SS to initiate signal transduction resulting in transcription of the RpoN regulon, though the exact signal that stimulates FlgS kinase activity is currently unknown. Boll and Hendrixson recently proposed a model in which the flagellar export apparatus is required for multimerization of FliF and FliG into the MS ring and rotor component of the C ring, respectively, and formation of this structure is sensed by FlgS to initiate signal transduction in C. jejuni (19). The signal sensed by FlgS may involve interactions of the sensor kinase with FliF, and FliG as FlgS can be cross-linked to these proteins in vivo (19).
Transcription of the FliA-dependent flagellar genes in Salmonella and Escherichia coli is also intimately linked with the flagellar T3SS, although in this case the flagellar T3SS must be competent for protein secretion for transcription of the FliA regulon. In the Salmonella/E. coli paradigm, the anti-σ28 factor, FlgM, inhibits transcription of the FliA-dependent genes by binding FliA and preventing it from engaging the promoter (20). Upon completion of the HBB complex, FlgM is exported from the cytoplasm by the flagellar T3SS, thereby allowing σ28-RNA polymerase holoenzyme to bind its target promoters to initiate transcription (21). H. pylori possesses a FlgM homolog (22), but the inhibitory effect of FlgM on FliA is thought to be alleviated through interactions between FlgM and the cytoplasmic domain of FlhA (FlhAC) rather than by secretion of FlgM (23).
To understand better the connection between flagellar gene expression and assembly of the flagellum in H. pylori, genes encoding various basal body components were disrupted, and motility, flagellar biogenesis, and transcript levels of selected flagellar genes were assessed in the resulting mutants. Genes targeted for mutagenesis included the MS ring protein (fliF), a soluble component of the flagellar T3SS (fliH), C ring elements (fliM and fliY), and axial components of the flagellum (fliE and flgBC, which encode the MS ring/rod linker and proximal rod proteins, respectively) (Fig. 1). In general, transcription of the FliA-dependent gene flaA was inhibited by mutations that blocked formation of the MS ring or rod but not by mutations that interfered with assembly of the C ring. Mutations that blocked formation of the MS or C rings inhibited transcription of RpoN-dependent genes, whereas mutations that blocked rod assembly stimulated transcription of RpoN-dependent genes. These findings contrasted with those reported for C. jejuni, where mutations in fliM and fliY either had no effect or stimulated expression of an RpoN-dependent reporter gene, and mutations in fliE, flgB, or flgC inhibited expression of the reporter gene (19). We postulate that FliH and the C ring facilitate interactions between FlgS and a target associated with the flagellar T3SS to initiate signal transduction in H. pylori but not in C. jejuni.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Escherichia coli DH5α was used for cloning and plasmid construction. E. coli strains were grown in Luria-Bertani broth or agar medium and were supplemented with ampicillin (100 μg/ml) and kanamycin (30 μg/ml) when appropriate. H. pylori strains were grown microaerobically at 37°C under an atmosphere consisting of 2% O2, 5% CO2, and 93% N2 on tryptic soy agar (TSA) supplemented with 10% horse serum or shaking under an atmosphere consisting of 8.3% O2, 4.6% CO2, 9.2% H2, and 77.9% N2 in brain heart infusion (BHI) broth supplemented with 0.4% β-cyclodextrin. Kanamycin (30 μg/ml) or chloramphenicol (30 μg/ml) was added to the medium used to culture H. pylori when appropriate.
Mutant construction.
Mutations in fliF, fliM, fliH, fliY, fliE, and flgBC were generated in H. pylori B128 by the following general procedure. HP locus numbers are included in Table 1. For each mutation, overlapping PCR was used to generate an amplicon that contained a chloramphenicol acetyltransferase (cat) gene flanked by ∼500-bp regions located upstream and downstream of the target gene. The cat cassette carries its own promoter and does not have a transcriptional terminator. The cat gene was inserted in the same orientation as the targeted gene. iProof high-fidelity DNA polymerase (Bio-Rad) was used for all PCR procedures. Primers used for PCR are listed in Table S1 in the supplemental material. The region upstream of the target gene was amplified using the upstream forward and the upstream reverse primers. The 5′ end of the upstream reverse primer contained sequence corresponding to one end of the cat cassette. The region downstream of the target gene was amplified using the downstream forward and the downstream reverse primers. The 5′ end of the downstream forward primer contained sequence corresponding to the other end of the cat cassette. Genomic DNA from H. pylori B128 was prepared using the Wizard genomic DNA purification kit (Promega) and was used as the template to amplify regions upstream and downstream of the target gene. The cat cassette was amplified from pUC20cat (24) using the cat forward and cat reverse primers. Overlapping PCR via the complementary regions of the amplified cat cassette and the amplicons of the regions flanking the target gene generated a PCR product with the cat cassette between the flanking regions. The resulting amplicons were introduced into H. pylori B128 by natural transformation using the following protocol. H. pylori cells were spotted onto a TSA plate and grown for 6 h. The amplicon was mixed with the cells and incubated for 18 h before cells were transferred onto TSA plates containing the appropriate antibiotics for selection. Plates were incubated for 5 days to allow colonies to appear. Replacement of the target gene with the cat cassette was confirmed by PCR using genomic DNA from chloramphenicol-resistant transformants as a template. The resulting amplicons were sequenced to verify that the mutant alleles were correct.
TABLE 1.
Strains constructed for this study
| Strain genotype | Descriptiona | Function of gene deleted |
|---|---|---|
| WT | H. pylori strain B128 | None |
| ΔfliF | B128 Δhp0351::cat | MS ring |
| ΔfliM | B128 Δhp1031::cat | C ring |
| ΔfliM/pfliM | B128 Δhp1031::cat/pfliM | |
| ΔfliM/pfliMY | B128 Δhp1031::cat/pfliMY | |
| ΔfliY | B128 Δhp1030::cat | C ring |
| ΔfliY/pfliMY | B128 Δhp1030::cat/pfliMY | |
| ΔfliH | B128 Δhp0353::cat | Soluble component of export apparatus |
| ΔfliE | B128 Δhp1557::cat | MS ring-rod linker protein |
| ΔfliE/pfliE | B128 Δhp1557::cat/pfliE | |
| ΔflgBC | B128 Δhp1559-hp1558::cat | Proximal rod |
| ΔflgBC/pflgBC | B128 Δhp1559-hp1558::cat/pflgBC |
HP locus numbers reference those in H. pylori strain 26695.
Mutations in flgR and rpoN were constructed by transformation and allelic exchange of plasmids containing the deletion alleles. The plasmid used to create the flgR insertion mutant was described by Brahmachary et al. (15) and contains the cat cassette inserted in a unique restriction site within flgR. This plasmid was transformed into H. pylori B128 for allelic exchange with the wild-type flgR allele. The rpoN mutant was created using a plasmid containing rpoN interrupted with a cat cassette. The rpoN gene was amplified from H. pylori 26695 and cloned into the NdeI and HindIII sites of pET28a. A unique EcoRI site was introduced into rpoN as described by Pereira et al. (25). The cat cassette was inserted into this site, and the resulting plasmid was transformed into H. pylori B128. Mutants were confirmed by PCR and sequencing of the resulting amplicons. Three different transformants were tested for all mutants. Mutants generated did not affect any experimentally verified sRNAs from Sharma and coworkers (26).
Complementation of mutants.
fliM, fliMY, fliE, and flgBC were amplified from genomic DNA isolated from H. pylori B128 using their respective forward and reverse primers indicated in Table S1 in the supplemental material. The fliE and flgBC regions amplified contained their native promoters. For the fliM and fliMY complementation plasmids, overlapping PCR was used to create a fusion which introduced the fliN promoter region upstream of these genes. The forward primer for each of the final PCR products contained a XhoI site at the 5′ end, and the reverse primer for each of the final PCR product contained a BamHI site at the 5′ end. All resulting PCR products were cloned into pGEM-T Easy, sequenced, and introduced into pHel3 by cloning the DNA into the unique XhoI and BamHI restriction sites in pHel3. The resulting plasmids were introduced into the deletion mutants by natural transformation. Plasmids pfliM, pfliMY, pfliE, and pflgBC contained the PCR products PfliN-fliM, PfliN-fliMY, PfliE-fliE, and PflgB-flgBC, respectively.
Motility assay.
Motility was assessed using a semisolid medium consisting of Mueller-Hinton broth and 0.4% Noble agar. After autoclaving, the medium was supplemented with sterile 10% heat-inactivated horse serum and 20 mM 2-(N-morpholino)ethanesulfonic acid (pH 6.0) to buffer the medium. The medium was also supplemented with 10 μM FeSO4, which we observed to enhance migration of H. pylori from the point of inoculation in the semisolid medium (our unpublished data). Kanamycin (30 μg/ml) or chloramphenicol (30 μg/ml) was added when appropriate. Sterile toothpicks were used to stab inoculating cells into the agar. Plates were incubated at 37°C under an atmosphere consisting of 2% O2, 5% CO2, and 93% N2. Diameters of the spreading H. pylori cells were measured after 7 days. Statistical significance was determined using the two-sample t test.
RNA extraction and cDNA synthesis.
H. pylori cells were grown on TSA plates supplemented with 10% horse serum for 18 h before harvesting and resuspended into 1 ml of nuclease-free water. Alternatively, H. pylori strains were grown in BHI liquid medium supplemented with 0.4% β-cyclodextrin to mid-log to late log phase, and 1 ml of cells were harvested. Cells were pelleted and resuspended in 100 μl of nuclease-free deionized water. The Aurum total RNA minikit (Bio-Rad) was used to isolate RNA, and the RNA solution was treated with the Turbo DNA-free kit (Ambion) to remove any contaminating DNA. RNA was quantified using a BioPhotometer (Eppendorf), and RNA quality was confirmed on a 1.2% agarose gel. Single-strand cDNA was synthesized from 200 ng of RNA using the iScript cDNA synthesis kit (Bio-Rad).
Quantitative reverse-transcription PCR.
Transcript levels of flaA, flaB, flgE, flgS, flgR, and fliA were monitored by quantitative reverse-transcription PCR (qRT-PCR) as described previously (27). Primers used are listed in Table S1 in the supplemental material. gyrA transcript levels were measured as a reference. Specificity and efficiency of each primer pair was confirmed by PCR using genomic DNA and by qRT-PCR on a serial dilution of wild-type RNA. Each qRT-PCR mixture, totaling 20 μl, consisted of 10 μl of iQ SYBR green Supermix (Bio-Rad), 5 μl of 100-fold diluted cDNA from the cDNA synthesis reaction, and a 200 nM concentration of each primer. A melt curve analysis was performed at the end of each experiment. Experiments were performed on the Bio-Rad iCycler iQ system in technical triplicate for three biological replicates of each strain. Gene expression levels were quantified by the 2−ΔΔCT method (28). Statistical significance was determined using the two-sample t test.
Electron microscopy.
Strains were grown to mid- to late-log phase in BHI supplemented with 0.4% β-cyclodextrin to an optical density at 600 nm (OD600) of 0.5 to 1.0. Kanamycin (30 μg/ml) and chloramphenicol (30 μg/ml) were included in the growth medium as necessary. Cells were spun down and resuspended in half-strength Karnovsky's fixative (2.5% glutaraldehyde, 2% paraformaldehyde, 0.1 M cacodylate buffer). Cells were fixed for 5 min and then incubated for 5 min on 300-mesh Formvar/carbon-coated copper grids. Grids were washed with 0.1 M cacodylate buffer, followed by a wash with deionized water. Excess liquid was wicked off with filter paper between washes. One drop of 1% uranyl acetate was applied to the grids for 30 s and then wicked off with filter paper. Grids were washed in deionized water and dried at room temperature overnight. Cells were visualized using the FEI Tecnai20 transmission electron microscope. For each strain, at least 115 cells were included for quantifying flagellated cells and determining the number of flagella per cell. Statistical analyses were performed using the Mann-Whitney U test to determine whether strains differed significantly from one another with regard to the distribution of the number of flagella per cell.
Detection of FlhA proteins.
H. pylori membrane fractions were collected as previously described (27). Cells were grown for 3 days on TSA plates supplemented with 10% horse serum. Cells were resuspended in a buffer containing 10% sucrose, 20 mM HEPES, and 1 mM EDTA, pH 7.4. Cells were passed three times through a French pressure cell at 10,000 kPa. Cellular debris and unlysed cells were removed by centrifugation two times for 15 min at 6,000 × g. Membrane fractions were separated from cytoplasmic proteins by two centrifugations for 60 min at 100,000 × g each. Protein concentrations were determined using the bicinchoninic acid protein assay (Thermo Scientific) following the manufacturer's instructions. Twenty micrograms of protein per sample was analyzed by Western blotting using affinity-purified antibodies directed against the N terminus of FlhA (18). Four micrograms of protein per sample was analyzed by Western blotting using antiserum directed against H. pylori KatA (29). Antigen-antibody complexes were detected by chemiluminescence using SuperSignal West Pico luminol/enhancer solution and SuperSignal West stable peroxide solution (Thermo Scientific). Blots were visualized on the FluoroChem E imager (ProteinSimple). Protein levels were quantified by densitometry using ImageJ (http://rsbweb.nih.gov/ij). Three biological replicates were analyzed, and statistical significance was determined using the two-sample t test.
Detection of RpoN proteins.
Antiserum generated against maltose-binding protein–RpoN (MBP-RpoN) was affinity purified prior to use using the AminoLink Plus immobilization kit (Thermo Scientific). MBP-RpoN was purified as previously described (25) and immobilized to the AminoLink Plus resin following the manufacturer's protocol. Antiserum generated against MBP-RpoN was buffer exchanged twice into phosphate-buffered saline (PBS) by diluting with 13 ml PBS and reducing the volume to 0.5 ml using an Amicon Ultra-15 centrifugal filter. The buffer-exchanged antiserum was incubated in the column to allow for binding to MBP-RpoN. The column was washed with 10 ml PBS before eluting the antibody with 8 ml of 0.1 M glycine, pH 2.5. The eluted antibody was dialyzed into citric acid-phosphate buffer (55 mM citric acid, 50 mM K2HPO4 [pH 5.5]), followed by Tris-buffered saline (50 mM Tris, 150 mM NaCl [pH 7.6]), and stored at −20°C.
H. pylori cytoplasmic fractions were prepared for immunoblotting. Cells were grown on agar medium for 24 h before resuspending into 3 ml PBS. Cells were lysed with three passages through a French press at 10,000 kPa. Unlysed cells were removed by centrifugation for 15 min at 6,000 × g. Membranes were separated from cytoplasmic proteins by two centrifugations for 60 min at 100,000 × g. The supernatant containing the cytoplasmic proteins was concentrated by trichloroacetic acid precipitation as described previously (17). Protein concentrations were determined using the bicinchoninic acid protein assay. Twenty micrograms of protein was analyzed by Western blotting using the affinity-purified RpoN antibody and goat anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (Bio-Rad). Western blots were visualized as described above.
RESULTS
FliF (MS ring) is required for transcription of the RpoN regulon.
The MS ring consists of a complex of FliF proteins (30) and acts as a mounting platform for the rotor, the switch complex (31), and the rod of the flagellum (32). Since the MS ring is one of the earliest flagellar structures assembled and is a scaffold for assembly of many of the basal body proteins, we wished to learn how the MS ring impacts flagellar gene expression in H. pylori.
A ΔfliF mutant was constructed in H. pylori B128 by replacement of fliF with the cat cassette, and the phenotype of the resulting mutant was analyzed. As expected, the resulting strain was nonmotile on soft-agar plates, and all cells analyzed by transmission electron microscopy were nonflagellated (Fig. 2A and B). Transcript levels of flaB and flgE (both RpoN dependent) and flaA (FliA dependent) were analyzed in the ΔfliF mutant and parental strain by qRT-PCR and normalized to the transcript levels of gyrA. Cells for the qRT-PCR assays were obtained from cells grown on agar medium and in broth. The trends in transcript levels were similar for cells grown under the two conditions, but there tended to be less variation between biological replicates for cells grown on agar medium (data not shown), and so for further qRT-PCR assays, we used cells grown on agar medium. Transcript levels of flaB and flgE in the ΔfliF mutant were similar to those in a ΔrpoN or ΔflgR mutant (Fig. 2C), indicating that the RpoN regulon is completely inactivated in the ΔfliF mutant. Boll and Hendrixson reported that deletion of fliF similarly results in the complete failure to express an RpoN-dependent reporter gene in C. jejuni (19).
FIG 2.

Effects of fliF deletion on flagellar gene transcription and motility. (A) Motility of ΔfliF mutant was assessed on 0.4% agar plates after 7 days of incubation under microaerobic conditions. Measurements indicate the diameter of the halo around the site of inoculation. The halo diameter for the wild type was 36 ± 2 mm, and that for the ΔfliF mutant was 7 ± 1 mm. (B) The number of flagella per cell was analyzed by electron microscopy after negative staining of cells. At least 115 cells were visualized per strain. (C) Transcript levels of flaB and flgE (left) and flaA (right) were detected by qRT-PCR. All results are significantly different from that for the wild type (P < 0.03). (D) Immunoblot of FlhA from membrane fractions. Each lane contains 20 μg protein. An immunoblot of KatA from the same membrane fractions was used as a loading control. Each lane contains 4 μg protein.
Levels of flaA transcript in the ΔfliF mutant were ∼4-fold lower than wild-type levels (Fig. 2C), in agreement with results from RNA slot blot hybridization assays for a fliF deletion mutant reported by Allan and coworkers (33). Transcript amounts of a second FliA-dependent gene (fliS) showed a trend similar to that of the levels of flaA transcripts in the ΔfliF mutant (see Fig. S1 in the supplemental material). Josenhans and coworkers showed that flaA transcripts were about 4-fold higher in a flgM mutant than in the wild type, indicating that FlgM is an antagonist of FliA activity (34). Colland and coworkers demonstrated that flaA transcript levels were increased ∼2.5-fold in a flgM deletion compared to the wild type and decreased only ∼2-fold in a flgM overexpression strain (22). On the other hand, deletion of fliA resulted in complete inhibition of flaA transcription (see Fig. S2 in the supplemental material), suggesting that FlgM modulates but does not completely inhibit expression of flaA.
The level of inhibition in the ΔfliF mutant was similar to that observed for H. pylori strains bearing deletions of the export apparatus genes flhA (18) or fliO (27), indicating that the phenotypes seen in the ΔfliF mutant may be due to a failure of the export apparatus to assemble properly. To test the hypothesis that FliF is required for the formation of the export apparatus, membrane fractions from the ΔfliF mutant and the wild-type strain were probed for FlhA by Western blotting. Levels of FlhA in the membrane fractions from the ΔfliF mutant were ∼3-fold lower than from those from the wild type (Fig. 2D), suggesting that FliF is not essential but may have a role in the assembly of the export apparatus.
The C ring proteins FliM and FliY are required for transcription of RpoN-dependent genes.
The C ring is a cup-like structure which extends into the cytoplasm and is localized near the MS ring and flagellar T3SS. In H. pylori, the C ring is comprised of four proteins, FliG, FliM, FliN, and FliY. The C ring has roles in rotor function, switching the rotational direction of the flagellum, and flagellar protein export (reviewed in reference 4).
Lowenthal and coworkers demonstrated that all four C ring proteins are required for wild-type flagellation in H. pylori (35) but did not report on whether the C ring proteins were required for transcription of the RpoN or FliA regulons. Allan and coworkers reported that FliG is required for flagellar biogenesis in H. pylori and showed that flaA transcript levels are reduced in the fliG mutant (33). To determine if other C ring proteins are required for flagellar gene expression, we individually replaced fliM and fliY with the cat cassette and analyzed the phenotypes of the resulting mutants. The ΔfliM and ΔfliY mutants were nonmotile (Fig. 3A). In the ΔfliM mutant, transmission electron microscopy revealed that most of the cells (77%) lacked flagella, while approximately 90% of the wild-type cells were flagellated (Fig. 3B). Of the cells that were flagellated, most possessed one or two flagella, while most of the flagellated wild-type cells possessed two to four flagella (Fig. 3B). The effect of the ΔfliY mutation on flagellar biogenesis was more severe than that of ΔfliM mutation. About 96% of the ΔfliY mutant cells lacked flagella, and cells that were flagellated possessed only a single flagellum (Fig. 3B). These results differed somewhat from those of Lowenthal and coworkers, who found that fliM mutants were almost completely nonflagellated (1% flagellated), whereas about 40% of the fliY mutant cells were flagellated (35). These differences between the results from the study by Lowenthal and coworkers and those of our study may be due to strain differences. The phenotypic differences between the ΔfliY and ΔfliM mutants in our experiments were unexpected, since fliM and fliY are in the same operon and the replacement of fliM with the cat cassette in the ΔfliM mutation was expected to have a polar effect on fliY. Complementation assays were consistent with the polarity of the ΔfliM mutation on fliY, as introduction of a plasmid-borne copy of fliMY into the ΔfliM mutant restored motility, but the introduction of a plasmid-borne fliM did not (Fig. 3A). Additionally, the same plasmid restored motility in the ΔfliY mutant (Fig. 3A).
FIG 3.

Effects of fliM, fliY, and fliH deletion on flagellar gene transcription and motility. (A) Motility of ΔfliM, ΔfliY, and ΔfliH mutants was assessed on 0.4% agar plates after 7 days of incubation under microaerobic conditions. The ΔfliM mutant was complemented with a plasmid containing either fliM or fliMY (ΔfliM/pfliM or ΔfliM/pfliMY, respectively), and the ΔfliY mutant was complemented with a plasmid containing fliMY (ΔfliY/pfliMY). Measurements indicate diameters of halos around sites of inoculation after 7 days of incubation under microaerobic conditions. Halo diameter are as follows: wild type (WT), 35 ± 2 mm; ΔfliM, 5 ± 1 mm; ΔfliY, 7 ± 1 mm; ΔfliM/pfliM, 5 ± 1 mm; ΔfliM/pfliMY, 24 ± 2 mm; ΔfliY/pfliMY, 43 ± 3 mm; and ΔfliH, 12 ± 1 mm. (B) The number of flagella per cell was analyzed by electron microscopy after negative staining of cells. At least 115 cells were visualized per strain. (C) Transcript levels of flaB and flgE (left) and flaA (right) were assessed by qRT-PCR. All transcript levels of flaB and flgE are significantly different from that of the wild type (P < 0.006). Transcript levels in the ΔfliY strain that are significantly different from those in the ΔfliM strain are marked with a circle (P < 0.05). The asterisk indicates significance relative to wild-type transcript levels of flaA (P < 0.05).
Transcript levels of flaB and flgE were about 3-fold lower in the ΔfliM mutant than the wild type (Fig. 3C), whereas flaA transcript levels in the ΔfliM mutant were comparable to those in the wild type (Fig. 3C). Levels of flaB and flgE transcripts in the ΔfliY mutant were ∼18-fold lower than wild-type levels (Fig. 3C), whereas flaA transcript levels in the ΔfliY mutant were ∼2-fold lower than in the wild type (Fig. 3C). Transcript levels of fliS showed a trend similar to that of the flaA transcripts in these mutants (see Fig. S1 in the supplemental material).
FliH is required for wild-type transcript levels of RpoN-dependent genes.
FliI is an ATPase that functions with FliH to facilitate the initial entry of protein substrates to the export gate of the flagellar T3SS (36). FliH prevents the ATPase activity of FliI when it is not engaged in secreting proteins (37, 38). FliH2FliI is proposed to deliver chaperone-substrate complexes from the cytoplasm to the export gate and is thought to be facilitated through interactions between FliH and FliN in Salmonella (5). Though interactions between the soluble components of the export apparatus are not as well characterized in H. pylori, homologs for these proteins exist and may function similarly to their Salmonella counterparts (39, 40). Given the interaction of FliH with the C ring protein FliN, we wished to determine if FliH influenced transcription of the RpoN-dependent genes.
A ΔfliH mutant was constructed, and the phenotype of the resulting mutant was analyzed. The ΔfliH mutant exhibited slight motility on the soft agar plate (Fig. 3A). Approximately 16% of the ΔfliH mutant cells were flagellated, most of which possessed either one or two flagella (Fig. 3B). Migration of H. pylori cells from the point of inoculation in the soft agar medium requires both formation of functional flagella and chemotaxis. The C ring is needed for chemotaxis (41), which likely explains why the ΔfliH mutant displayed some motility in the soft agar assay while the ΔfliM mutant did not, even though a higher proportion of the ΔfliM mutant cells were flagellated. Deletion of fliH resulted in an ∼6-fold decrease in flaB and flgE transcripts compared to the wild type but no change in the transcript levels of flaA or fliS (Fig. 3C; also, see Fig. S1 in the supplemental material). These findings suggest that FliH, similar to other basal body proteins, facilitates signal transduction by the FlgS/FlgR two-component system. We attempted to complement the ΔfliH mutation by introducing fliH carried on the shuttle vector pHel3 into the mutant but were unable to obtain transformants for unknown reasons. The gene downstream of fliH (hp0354; 1-deoxy-d-xylulose-5-phosphate synthase) is not related to flagellar biogenesis, so any polar effects on this gene are not expected to have influences on our flagellar gene regulation studies.
Early flagellar export substrates have different effects on transcript levels of RpoN- and FliA-dependent genes.
FliE, FlgB, and FlgC are the earliest substrates transported by the export apparatus (32), and we wished to determine how loss of these proteins might impact transcription of the RpoN and FliA regulons. FliE is a linker protein between the MS ring and rod (32), while FlgB and FlgC form the proximal rod (42). The fliE and flgBC genes were replaced with the cat cassette, and the phenotypes of the resulting mutants were analyzed. As expected, the ΔfliE and ΔflgBC mutant strains were nonmotile and nonflagellated (Fig. 4A and B). Motility was partially restored in the ΔfliE and ΔflgBC mutants by introducing fliE and flgBC, respectively, into the mutants via the shuttle vector pHel3 (Fig. 4A), verifying that deletion of the genes was responsible for loss of motility.
FIG 4.

Effects of fliE and flgBC deletion on flagellar gene transcription and motility. (A) Motility of ΔfliE and ΔflgBC mutants was assessed on 0.4% agar plates after 7 days of incubation under microaerobic conditions. The ΔfliE and ΔflgBC mutants were complemented with their respective genes on a plasmid. Measurements indicate the diameters of the halos around the sites of inoculation. Halo diameters are as follows: WT, 35 ± 2 mm; ΔfliE, 7 ± 1 mm; ΔfliE/pfliE, 15 ± 3 mm; ΔflgBC, 7 ± 1 mm; and ΔflgBC/pflgBC, 26 ± 1 mm. (B) The number of flagella per cell was analyzed by electron microscopy after negative staining of cells. At least 115 cells were visualized per strain. (C) Transcript levels of flaB and flgE (left) and flaA (right) were detected by qRT-PCR. All results are significantly different from that for the wild type (P < 0.05) with the exception of flaA levels in the ΔflgBC strain (P = 0.08).
The effects on flagellar gene transcript levels in the ΔfliE mutant were more pronounced than in the ΔflgBC mutant. The ΔfliE mutant showed an ∼9-fold increase in the levels of flaB and flgE transcripts, whereas the ΔflgBC mutant displayed an ∼4-fold increase in flaB and flgE transcripts compared to wild-type levels (Fig. 4C). Levels of flaA transcript in both the ΔfliE and the ΔflgBC mutants were ∼2-fold lower than that in the wild type (Fig. 4C), and levels of fliS transcript showed a trend similar to that of flaA transcript levels in these mutants (see Fig. S1 in the supplemental material). These results suggest that the axial structures of the flagellum affect transcription of the RpoN regulon in a manner different from that of the export apparatus and the C ring.
Mutations in basal body genes minimally affect expression of genes encoding regulatory proteins for the RpoN and FliA regulons.
To examine the possibility that the changes in flagellar gene expression observed for the various mutant strains were due to altered levels of regulatory proteins, transcript levels of flgS, flgR, and fliA and protein levels of RpoN were quantified in all mutants analyzed (Fig. 5). All strains used in this study had similar growth rates (see Fig. S3 in the supplemental material), so any differences in the levels of these flagellar regulatory proteins can be attributed solely to the mutations made in the flagellar biosynthesis pathway. With few exceptions, levels of flgS, flgR, and fliA transcripts in the mutants were similar to those in the wild-type strain. Transcript levels of flgS in the ΔfliE and ΔflgBC mutants were ∼2-fold higher than in the wild type (Fig. 5A), which may have, at least partially, accounted for the apparent upregulation of the RpoN-dependent genes in these mutants (Fig. 4C). Transcript levels of flgS were also slightly elevated (∼1.5-fold) in the ΔfliF mutant (Fig. 5A). H. pylori flgS is in an operon with two other class I flagellar genes (i.e., early flagellar genes transcribed by the σ80-RNA polymerase holoenzyme): flgI, which encodes P ring protein, and HP0245, whose product shares homology with the flagellar rod assembly protein/muramidase FlgJ. The apparent upregulation of flgS in the fliE and flgBC mutants may indicate that expression of at least some of the class I flagellar genes in H. pylori is coupled with rod assembly. This would be an interesting line of inquiry to pursue, as we are unaware of any reports of regulatory proteins that control expression of the class I flagellar genes in H. pylori. Transcript levels of flgR were slightly elevated (∼1.5-fold) in the ΔfliF mutant compared to the wild type (Fig. 5B) but not in any of the other mutants. Unlike flgS, flgR does not appear to be in an operon with other flagellar genes. Levels of fliA transcript were ∼2-fold lower in the ΔfliM mutant than in the wild type (Fig. 5C), but this decrease in the level of fliA transcript did not affect transcript levels of flaA (Fig. 3C). Western blot analysis of the H. pylori mutants indicated that levels of RpoN were unaffected by the mutations (Fig. 5D). Taken together, the results suggest the regulatory proteins that are known to control transcription of the RpoN-dependent and FliA-dependent genes are present at normal levels in the flagellar basal body mutants. These findings indicate that the downregulation of the RpoN-dependent genes in the fliF, fliM, fliY, and fliH mutants likely results from either the failure to create the flagellar structure which acts as the regulatory checkpoint for expression the RpoN regulon or the inability of the FlgS/FlgR two-component system to respond effectively to the regulatory checkpoint.
FIG 5.

Transcript and protein levels of flgS, flgR, fliA, and RpoN among mutants. (A to C) Transcript levels of flgS (A), flgR (B), and fliA (C) were quantified by qRT-PCR. Asterisks indicate significant differences from the value for the wild type (P < 0.05). (D) Immunoblot of RpoN from H. pylori wild-type and mutant cytoplasmic protein fractions. Each lane contains 20 μg protein. The immunoblot was probed with affinity-purified antibody directed against the MBP-tagged RpoN.
DISCUSSION
We show here that in addition to the export apparatus, other flagellar basal body substructures have potential roles in modulating transcription of the RpoN and FliA regulons, possibly by affecting the structure and/or activity of the export apparatus. Given the conformational changes that occur within the basal body during flagellar biogenesis, it is reasonable to speculate that such conformational changes communicate the status of flagellar assembly to the transcription machinery that controls expression of the RpoN and FliA regulons.
Effects of basal body proteins on the FliA flagellar regulon.
One of the earliest events in flagellar biogenesis is the coordinated assembly of the MS ring and the flagellar protein export apparatus (43). The export apparatus protein FlhA is proposed to promote oligomerization of FliF monomers into the MS ring in E. coli (44). We show here that in the absence of FliF, the amount of FlhA in the membrane is significantly reduced (Fig. 2D), suggesting that FliF is needed to support assembly or stability of the export apparatus. Deletion of fliF in H. pylori resulted in reduced amounts of transcripts of two FliA-dependent genes, flaA and fliS (Fig. 2C; also, see Fig. S1 in the supplemental material). The inhibitory effect of FlgM on the H. pylori FlhA regulon is proposed to be alleviated by interactions between FlgM and FlhA (23). Since FliF is needed for normal expression or localization of FlhA to the membrane (Fig. 2D), we postulate that the reduced expression of the FliA regulon in the ΔfliF mutant results from the failure of FlhA to assemble properly.
In contrast to the loss of FliF, loss of FliM or FliH had no effect on expression of the FliA-dependent genes flaA and fliS (Fig. 3C; also, see Fig. S1 in the supplemental material). Loss of FliY did appear to inhibit expression of the FliA-dependent genes, but this inhibition was modest compared to the inhibition observed for the RpoN-dependent genes in the ΔfliY mutant (Fig. 3C; also, see Fig. S1). Taken together, these findings suggest that these C ring components and soluble components of the export apparatus do not play a significant role in alleviating FlgM inhibition of FliA in H. pylori.
Loss of the exported flagellar structures FliE and FlgBC resulted in an ∼2-fold reduction of flaA transcript levels (Fig. 4C). We postulate that in the absence of FliE or FlgBC, FlhA does not interact appropriately with FlgM to alleviate its inhibitory effect on FliA.
Effects of basal body proteins on the RpoN flagellar regulon.
Transcripts of RpoN-dependent genes were abolished in the ΔfliF mutant (Fig. 2C), suggesting that the MS ring is essential for transcription of the RpoN regulon. FliF could have a direct role in controlling transcription of the RpoN regulon through interactions with FlgS, a hypothesis consistent with the observation that C. jejuni FlgS can be cross-linked to FliF in vivo (19). Alternatively, FliF could have an indirect effect on expression of the RpoN regulon, as the assembly of the MS ring and the export apparatus may occur simultaneously.
The H. pylori C ring is comprised of FliG, FliM, FliN, and FliY subunits (35). The organization of the C ring proteins has not been examined in H. pylori. In Salmonella, however, FliG is located closest to the cell membrane and interacts with FliF (45), while FliN is most peripheral protein in the C ring, and FliM is situated between FliG and FliN, where it interacts with both proteins (46). Unlike H. pylori, Salmonella does not possess a FliY homolog. Given that FliY shares homology with FliN and that some bacteria that have FliY homologs lack FliN (35, 47), we postulate that FliY and FliN both interact with FliM at the periphery of the C ring. Based on the arrangement of C ring proteins in Salmonella, we believe that FliY and FliN are unable to assemble onto the C ring in the H. pylori ΔfliM mutant, leaving FliG as the only protein assembled into the C ring. If this is the case, it is somewhat puzzling why the loss of fliY had a more profound effect on flagellar biogenesis and expression of RpoN-dependent genes than did disruption of fliM. One possible explanation is that FliN and FliY compete for binding to FliM and in the absence of FliY, extra copies of FliN are incorporated into the C ring, which interferes with signal transduction.
In contrast to our results, where deletions of fliM and fliY inhibited expression of the RpoN regulon (Fig. 3C), Boll and Hendrixson observed that deletion of fliM or fliY either had no effect or stimulated expression of an RpoN-dependent flaB::astA transcriptional reporter in C. jejuni (19). The phenotypic differences between H. pylori and C. jejuni C ring protein mutants were unexpected, since mechanisms for flagellar biogenesis in these two bacteria appear to be quite similar. One possible explanation for this distinction is that there is a greater demand for synthesis of flagellar proteins in H. pylori, since it produces multiple flagella per cell while C. jejuni produces only a single flagellum per cell. H. pylori may require a more robust signal transduction response by the FlgS/FlgR two-component system to ensure that enough flagellar subunits are made for the simultaneous assembly of multiple flagella. To elicit this robust signaling response, the C ring in H. pylori but not C. jejuni may facilitate protein-protein interactions needed for activation of the FlgS/FlgR two-component system.
FliN interacts with the soluble export apparatus component FliH to help shuttle protein substrates to the export apparatus (5). Studies in E. coli and Salmonella have shown that FliI forms a heterotrimer with FliH (FliH2-FliI) which forms a hexameric ring (48) around FliJ (FliH12-FliI6-FliJ complex) (49). FliH is also thought to interact with FlhA to anchor the FliH12-FliI6-FliJ complex to the export platform of the flagellar T3SS (50). Bai and coworkers and proposed a model in which the FliH12-FliI6-FliJ complex in Salmonella remains associated with the export gate and FliH2-FliI complexes carrying flagellar substrates are interchangeable to deliver new substrates to the export gate (51). Since loss of fliH resulted in reduced amounts of transcripts from RpoN-dependent genes (Fig. 3C), we hypothesize that the activity of the FliH12-FliI6-FliJ complex may enhance expression of the RpoN regulon by facilitating interactions between components of the basal body and FlgS or FlgR.
Among the basal body mutants we have analyzed, mutants in fliE and flgBC were the only ones that resulted in elevated transcript levels of RpoN-dependent genes. In contrast to our results, Boll and Hendrixson reported that disruption of fliE, flgB, or flgC in C. jejuni inhibits expression of the RpoN-dependent flaB::astA reporter gene (19). It is not clear why disrupting fliE or flgBC has such radically different effects on expression of the RpoN-dependent genes in H. pylori and C. jejuni. Given that FliE and the rod proteins are external to the cell membrane, they likely exert their effects on expression of the RpoN regulon through interactions with the MS ring or membrane components of the export apparatus. The observation that disruption of fliE had the most profound effect on expression of the RpoN regulon in both H. pylori and C. jejuni is consistent with this hypothesis since FliE is located more proximal to the MS ring and export apparatus than the rod proteins.
Proposed mode for activation of the RpoN regulon.
Based on our results, we propose a model for how H. pylori couples the expression of the RpoN regulon with assembly of the flagellum. We postulate that FlgS is in an inactive state prior to assembly of the MS ring, C ring, and flagellar T3SS. Formation of these structures creates a regulatory checkpoint sensed by FlgS and results in a stimulation of FlgS autokinase activity, culminating in transcriptional activation of the RpoN regulon. FlhA, FliF, and FliG are potential components for such a regulatory checkpoint, as FlgS can be cross-linked to FliF and FliG in C. jejuni in vivo (19), and FlgS binds a peptide corresponding to the N terminus of FlhA with high affinity (52). Since FlhA, FliF, and FliG are located at the base of the cavity formed by the C ring, we postulate that FliH and one or more of the C ring proteins facilitate interactions between FlgS and the regulatory checkpoint. Alternatively, FlgS may remain bound to the regulatory checkpoint, where FliH and C ring proteins assist FlgR in interacting with FlgS. Once the flagellar subunits encoded by RpoN-dependent genes are no longer needed for flagellar assembly, the signal sensed by FlgS is presumably switched off. Formation of the HBB may be a cellular cue that turns off expression of the RpoN regulon when the proteins encoded by RpoN-dependent genes either are incorporated into the nascent flagellum or are awaiting transport at this point in flagellar biogenesis. We plan to test the validity of the proposed model by examining interactions of FlgS and FlgR with flagellar proteins.
Supplementary Material
ACKNOWLEDGMENTS
We thank Rob Maier and Zach Lewis for use of lab equipment, Jonathan McMurry for comments on the manuscript, and Sierra Jennings for her work in construction of the flgBC, fliH, and fliF mutants.
This work was supported by grant number MCB-1244242 from the National Science Foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02533-14.
REFERENCES
- 1.Macnab RM. 1996. Flagella and motility, p 123–145. In Neidhardt FC, Curtiss R III, Ingraham JL, Linn ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella Cellular and Molecular Biology, 2nd ed ASM Press, Washington, D. C. [Google Scholar]
- 2.Minamino T, Imada K, Namba K. 2008. Mechanisms of type III protein export for bacterial flagellar assembly. Mol Biosyst 4:1105–1115. doi: 10.1039/b808065h. [DOI] [PubMed] [Google Scholar]
- 3.Minamino T, Imada K, Namba K. 2008. Molecular motors of the bacterial flagella. Curr Opin Struct Biol 18:693–701. doi: 10.1016/j.sbi.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Macnab RM. 2003. How bacteria assemble flagella. Annu Rev Microbiol 57:77–100. doi: 10.1146/annurev.micro.57.030502.090832. [DOI] [PubMed] [Google Scholar]
- 5.McMurry JL, Murphy JW, Gonzalez-Pedrajo B. 2006. The FliN-FliH interaction mediates localization of flagellar export ATPase FliI to the C ring complex. Biochemistry 45:11790–11798. doi: 10.1021/bi0605890. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki T, Iino T, Horiguchi T, Yamaguchi S. 1978. Incomplete flagellar structures in nonflagellate mutants of Salmonella typhimurium. J Bacteriol 133:904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones CJ, Macnab RM. 1990. Flagellar assembly in Salmonella typhimurium: analysis with temperature-sensitive mutants. J Bacteriol 172:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chilcott GS, Hughes KT. 2000. Coupling of flagellar gene expression to flagellar assembly in Salmonella enterica serovar Typhimurium and Escherichia coli. Microbiol Mol Biol Rev 64:694–708. doi: 10.1128/MMBR.64.4.694-708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niehus E, Gressmann H, Ye F, Schlapbach R, Dehio M, Dehio C, Stack A, Meyer TF, Suerbaum S, Josenhans C. 2004. Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol Microbiol 52:947–961. doi: 10.1111/j.1365-2958.2004.04006.x. [DOI] [PubMed] [Google Scholar]
- 10.Hendrixson DR, Akerley BJ, DiRita VJ. 2001. Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol Microbiol 40:214–224. doi: 10.1046/j.1365-2958.2001.02376.x. [DOI] [PubMed] [Google Scholar]
- 11.Hendrixson DR, DiRita VJ. 2003. Transcription of sigma54-dependent but not sigma28-dependent flagellar genes in Campylobacter jejuni is associated with formation of the flagellar secretory apparatus. Mol Microbiol 50:687–702. doi: 10.1046/j.1365-2958.2003.03731.x. [DOI] [PubMed] [Google Scholar]
- 12.Spohn G, Scarlato V. 1999. Motility of Helicobacter pylori is coordinately regulated by the transcriptional activator FlgR, an NtrC homolog. J Bacteriol 181:593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schirm M, Soo EC, Aubry AJ, Austin J, Thibault P, Logan SM. 2003. Structural, genetic and functional characterization of the flagellin glycosylation process in Helicobacter pylori. Mol Microbiol 48:1579–1592. doi: 10.1046/j.1365-2958.2003.03527.x. [DOI] [PubMed] [Google Scholar]
- 14.Beier D, Frank R. 2000. Molecular characterization of two-component systems of Helicobacter pylori. J Bacteriol 182:2068–2076. doi: 10.1128/JB.182.8.2068-2076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brahmachary P, Dashti MG, Olson JW, Hoover TR. 2004. Helicobacter pylori FlgR is an enhancer-independent activator of sigma54-RNA polymerase holoenzyme. J Bacteriol 186:4535–4542. doi: 10.1128/JB.186.14.4535-4542.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joslin SN, Hendrixson DR. 2009. Activation of the Campylobacter jejuni FlgSR two-component system is linked to the flagellar export apparatus. J Bacteriol 191:2656–2667. doi: 10.1128/JB.01689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith TG, Pereira L, Hoover TR. 2009. Helicobacter pylori FlhB processing-deficient variants affect flagellar assembly but not flagellar gene expression. Microbiology 155:1170–1180. doi: 10.1099/mic.0.022806-0. [DOI] [PubMed] [Google Scholar]
- 18.Tsang J, Smith TG, Pereira LE, Hoover TR. 2013. Insertion mutations in Helicobacter pylori flhA reveal strain differences in RpoN-dependent gene expression. Microbiology 159:58–67. doi: 10.1099/mic.0.059063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boll JM, Hendrixson DR. 2013. A regulatory checkpoint during flagellar biogenesis in Campylobacter jejuni initiates signal transduction to activate transcription of flagellar genes. mBio 4:e00432-13. doi: 10.1128/mBio.00432-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillen KL, Hughes KT. 1991. Negative regulatory loci coupling flagellin synthesis to flagellar assembly in Salmonella typhimurium. J Bacteriol 173:2301–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes KT, Gillen KL, Semon MJ, Karlinsey JE. 1993. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science 262:1277–1280. doi: 10.1126/science.8235660. [DOI] [PubMed] [Google Scholar]
- 22.Colland F, Rain J-C, Gounon P, Labigne A, Legrain P, De Reuse H. 2001. Identification of the Helicobacter pylori anti-σ28 factor. Mol Microbiol 41:477–487. doi: 10.1046/j.1365-2958.2001.02537.x. [DOI] [PubMed] [Google Scholar]
- 23.Rust M, Borchert S, Niehus E, Kuehne SA, Gripp E, Bajceta A, McMurry JL, Suerbaum S, Hughes KT, Josenhans C. 2009. The Helicobacter pylori anti-sigma factor FlgM is predominantly cytoplasmic and cooperates with the flagellar basal body protein FlhA. J Bacteriol 191:4824–4834. doi: 10.1128/JB.00018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Taylor DE. 1990. Choramphenicol resistance in Campylobacter coli: nucleotide sequence, expression, and cloning vector. Gene 94:23–28. doi: 10.1016/0378-1119(90)90463-2. [DOI] [PubMed] [Google Scholar]
- 25.Pereira L, Hoover TR. 2005. Stable accumulation of sigma54 in Helicobacter pylori requires the novel protein HP0958. J Bacteriol 187:4463–4469. doi: 10.1128/JB.187.13.4463-4469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 27.Tsang J, Hoover TR. 2014. Requirement of the flagellar protein export apparatus component FliO for optimal expression of flagellar genes in Helicobacter pylori. J Bacteriol 196:2709–2717. doi: 10.1128/JB.01332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 29.Alamuri P, Maier RJ. 2004. Methionine sulphoxide reductase is an important antioxidant enzyme in the gastric pathogen Helicobacter pylori. Mol Microbiol 53:1397–1406. doi: 10.1111/j.1365-2958.2004.04190.x. [DOI] [PubMed] [Google Scholar]
- 30.Ueno R, Oosawa K, Aizawa S-I. 1992. M ring, S ring and proximal rod of the flagellar basal body of Salmonella typhimurium are composed of subunits of a single protein. J Mol Biol 227:672–677. doi: 10.1016/0022-2836(92)90216-7. [DOI] [PubMed] [Google Scholar]
- 31.Oosawa K, Ueno T, Aizawa S. 1994. Overproduction of the bacterial flagellar switch proteins and their interactions with the MS ring complex in vitro. J Bacteriol 176:3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minamino T, Yamaguchi S, Macnab RM. 2000. Interaction between FliE and FlgB, a proximal rod component of the flagellar basal body of Salmonella. J Bacteriol 182:3029–3036. doi: 10.1128/JB.182.11.3029-3036.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allan E, Dorrell N, Foynes S, Anyim M, Wren BW. 2000. Mutational analysis of genes encoding the early flagellar components of Helicobacter pylori: evidence for transcriptional regulation of flagellin A biosynthesis. J Bacteriol 182:5274–5277. doi: 10.1128/JB.182.18.5274-5277.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Josenhans C, Niehus E, Amersbach S, Horster A, Betz C, Drescher B, Hughes KT, Suerbaum S. 2002. Functional characterization of the antagonistic flagellar late regulators FliA and FlgM of Helicobacter pylori and their effects on the H. pylori transcriptome. Mol Microbiol 43:307–322. doi: 10.1046/j.1365-2958.2002.02765.x. [DOI] [PubMed] [Google Scholar]
- 35.Lowenthal AC, Hill M, Sycuro LK, Mehmood K, Salama NR, Ottemann KM. 2009. Functional analysis of the Helicobacter pylori flagellar switch proteins. J Bacteriol 191:7147–7156. doi: 10.1128/JB.00749-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan F, Macnab RM. 1996. Enzymatic characterization of FliI. An ATPase involved in flagellar assembly in Salmonella typhimurium. J Biol Chem 271:31981–31988. [DOI] [PubMed] [Google Scholar]
- 37.Minamino T, MacNab RM. 2000. FliH, a soluble component of the type III flagellar export apparatus of Salmonella, forms a complex with FliI and inhibits its ATPase activity. Mol Microbiol 37:1494–1503. doi: 10.1046/j.1365-2958.2000.02106.x. [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez-Pedrajo B, Fraser GM, Minamino T, Macnab RM. 2002. Molecular dissection of Salmonella FliH, a regulator of the ATPase FliI and the type III flagellar protein export pathway. Mol Microbiol 45:967–982. doi: 10.1046/j.1365-2958.2002.03047.x. [DOI] [PubMed] [Google Scholar]
- 39.Lane MC, O'Toole PW, Moore SA. 2006. Molecular basis of the interaction between the flagellar export proteins FliI and FliH from Helicobacter pylori. J Biol Chem 281:508–517. doi: 10.1074/jbc.M507238200. [DOI] [PubMed] [Google Scholar]
- 40.Jenks PJ, Foynes S, Ward SJ, Constantinidou C, Penn CW, Wren BW. 1997. A flagellar-specific ATPase (FliI) is necessary for flagellar export in Helicobacter pylori. FEMS Microbiol Lett 152:205–211. doi: 10.1111/j.1574-6968.1997.tb10429.x. [DOI] [PubMed] [Google Scholar]
- 41.Magariyama Y, Yamaguchi S, Aizawa S. 1990. Genetic and behavioral analysis of flagellar switch mutants of Salmonella typhimurium. J Bacteriol 172:4359–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Homma M, Kutsukake K, Hasebe M, Iino T, Macnab RM. 1990. FlgB, FlgC, FlgF and FlgG. A family of structurally related proteins in the flagellar basal body of Salmonella typhimurium. J Mol Biol 211:465–477. [DOI] [PubMed] [Google Scholar]
- 43.Morimoto YV, Ito M, Hiraoka KD, Che YS, Bai F, Kami-Ike N, Namba K, Minamino T. 2014. Assembly and stoichiometry of FliF and FlhA in Salmonella flagellar basal body. Mol Microbiol 91:1214–1226. doi: 10.1111/mmi.12529. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Sourjik V. 2011. Assembly and stability of flagellar motor in Escherichia coli. Mol Microbiol 80:886–899. doi: 10.1111/j.1365-2958.2011.07557.x. [DOI] [PubMed] [Google Scholar]
- 45.Francis NR, Irikura VM, Yamaguchi S, DeRosier DJ, Macnab RM. 1992. Localization of the Salmonella typhimurium flagellar switch protein FliG to the cytoplasmic M-ring face of the basal body. Proc Natl Acad Sci U S A 89:6304–6308. doi: 10.1073/pnas.89.14.6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marykwas DL, Schmidt SA, Berg HC. 1996. Interacting components of the flagellar motor of Escherichia coli revealed by the two-hybrid system in yeast. J Mol Biol 256:564–576. doi: 10.1006/jmbi.1996.0109. [DOI] [PubMed] [Google Scholar]
- 47.Bischoff DS, Ordal GW. 1992. Identification and characterization of FliY, a novel component of the Bacillus subtilis flagellar switch complex. Mol Microbiol 6:2715–2723. doi: 10.1111/j.1365-2958.1992.tb01448.x. [DOI] [PubMed] [Google Scholar]
- 48.Claret L, Calder SR, Higgins M, Hughes C. 2003. Oligomerization and activation of the FliI ATPase central to bacterial flagellum assembly. Mol Microbiol 48:1349–1355. doi: 10.1046/j.1365-2958.2003.03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ibuki T, Imada K, Minamino T, Kato T, Miyata T, Namba K. 2011. Common architecture of the flagellar type III protein export apparatus and F- and V-type ATPases. Nat Struct Mol Biol 18:277–282. doi: 10.1038/nsmb.1977. [DOI] [PubMed] [Google Scholar]
- 50.Hara N, Morimoto YV, Kawamoto A, Namba K, Minamino T. 2012. Interaction of the extreme N-terminal region of FliH with FlhA is required for efficient bacterial flagellar protein export. J Bacteriol 194:5353–5360. doi: 10.1128/JB.01028-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai F, Morimoto YV, Yoshimura SD, Hara N, Kami-Ike N, Namba K, Minamino T. 2014. Assembly dynamics and the roles of FliI ATPase of the bacterial flagellar export apparatus. Sci Rep 4:6528. doi: 10.1038/srep06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsang J, Hirano T, Hoover TR, McMurry JL. 2015. Helicobacter pylori FlhA binds the sensor kinase and flagellar gene regulatory protein FlgS with high affinity. J Bacteriol 197:1886–1892. doi: 10.1128/JB.02610-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.