

ABSTRACT
Reactive oxygen species (ROS) promote the synthesis of the DNA lesion 8-oxo-G, whose mutagenic effects are counteracted in distinct organisms by the DNA glycosylase MutM. We report here that in Bacillus subtilis, mutM is expressed during the exponential and stationary phases of growth. In agreement with this expression pattern, results of a Western blot analysis confirmed the presence of MutM in both stages of growth. In comparison with cells of a wild-type strain, cells of B. subtilis lacking MutM increased their spontaneous mutation frequency to Rifr and were more sensitive to the ROS promoter agents hydrogen peroxide and 1,1′-dimethyl-4,4′-bipyridinium dichloride (Paraquat). However, despite MutM's proven participation in preventing ROS-induced-DNA damage, the expression of mutM was not induced by hydrogen peroxide, mitomycin C, or NaCl, suggesting that transcription of this gene is not under the control of the RecA, PerR, or σB regulons. Finally, the role of MutM in stationary-phase-associated mutagenesis (SPM) was investigated in the strain B. subtilis YB955 (hisC952 metB5 leuC427). Results revealed that under limiting growth conditions, a mutM knockout strain significantly increased the amount of stationary-phase-associated his, met, and leu revertants produced. In summary, our results support the notion that the absence of MutM promotes mutagenesis that allows nutritionally stressed B. subtilis cells to escape from growth-limiting conditions.
IMPORTANCE The present study describes the role played by a DNA repair protein (MutM) in protecting the soil bacterium Bacillus subtilis from the genotoxic effects induced by reactive oxygen species (ROS) promoter agents. Moreover, it reveals that the genetic inactivation of mutM allows nutritionally stressed bacteria to escape from growth-limiting conditions, putatively by a mechanism that involves the accumulation and error-prone processing of oxidized DNA bases.
INTRODUCTION
Reactive oxygen species (ROS), including hydrogen peroxide, superoxide, and hydroxyl radicals, are produced in all aerobic organisms as side products of oxidative metabolism or following exposure to environmental agents and are normally in balance with the cellular antioxidant defenses. Oxidative stress occurs when this critical balance is disrupted because of depletion of antioxidants or excess accumulation of ROS (1). Therefore, when antioxidant cellular defenses are deficient or overwhelmed, the damaging potential of ROS increases and they target different cellular biomolecules, including, lipids, proteins, carbohydrates, and DNA (2). One of the most common events resulting from attack of DNA by the hydroxyl radical is the formation of 7,8-dihydro-8-oxodeoxyguanosine (8-oxo-G), a DNA lesion extensively studied due to its strong mutagenic and genotoxic properties (3). However, the hydroxyl radicals can also impact the deoxyribonucleotide and ribonucleotide pools, generating the oxidized precursors 8-oxo-dGTP and 8-oxo-GTP, respectively (4, 5). The former is frequently incorporated opposite adenine during DNA synthesis, giving rise to G·C →T·A transversions, whereas 8-oxo-GTP has the potential of being used as a substrate by the RNA polymerase, generating oxidized mRNAs that may originate transcriptional errors (6, 7). In Escherichia coli, the mutagenic effects of 8-oxo-G are prevented by MutM, a DNA glycosylase that recognizes and hydrolyzes this oxidized base from DNA (3). Following this event, the repair of the apurinic/apyrimidinic (AP) site generated and the restitution of the undamaged guanine are carried out by downstream components of the base excision repair (BER) pathway (8, 9).
It has been shown that oxidative stress is a crucial factor that promotes mutagenesis in nutritionally stressed bacteria (10–13) and that the oxidized-guanine (GO) DNA repair system (composed of the DNA glycosylases MutM and MutY and the nucleotide diphosphohydrolase MutTA) is involved in this type of mutagenesis in B. subtilis (13). However, the individual contribution of MutM in preventing mutagenesis and its role in conferring protection against the toxic effects of oxidative stress in this microorganism are currently unknown. Here, we report that disruption of mutM sensitized B. subtilis to the noxious effects of the oxidizing agents hydrogen peroxide and Paraquat (1,1′-dimethyl-4,4′-bipyridinium dichloride [PQ]). Whereas in E. coli the superoxide radical induces the expression of mutM (14), our results showed that in B. subtilis the transcription of this gene is controlled in a temporal manner that keeps active the expression of mutM during the logarithmic and stationary phases of growth. Notably, the absence of this repair protein promoted the generation of mutations in nutritionally stressed cells of this bacterium.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. B. subtilis YB955 is a prophage-“cured” strain that contains the hisC952, metB5, and leuC427 auxotrophic mutations (15–17). B. subtilis strains were maintained on tryptic blood agar base (TBAB) (Acumedia Manufacturers, Inc., Lansing, MI). Liquid cultures of B. subtilis strains were grown in Penassay broth (PAB) (antibiotic A3 medium; Difco Laboratories, Sparks, MD). E. coli cultures were grown in Luria-Bertani (LB) medium. When required, neomycin (Neo; 10 μg ml−1), tetracycline (Tet; 10 μg ml−1), spectinomycin (Sp; 100 μg ml−1), kanamycin (Kan; 10 μg ml−1), ampicillin (Amp; 100 μg ml−1), chloramphenicol (Cm; 5 μg ml−1), erythromycin (Ery; 1 μg ml−1), rifampin (Rif; 10 μg ml−1), or isopropyl-β-d-thiogalactopyranoside (IPTG; 1 mM) was added to media. Hydrogen peroxide (H2O2) and 1,1′-dimethyl-4,4′-bipyridinium dichloride (Paraquat [PQ]) were obtained from Sigma-Aldrich (St. Louis, MO).
TABLE 1.
Bacillus subtilis strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| YB955 | hisC952 metB5 leuC427 xin-1 | 23 |
| PS832 | Wild type; Trpr revertant of strain 168 | P. Setlow |
| PERM311 | Wild type; trpC2 | Laboratory stock |
| PERM571 | YB955 ΔmutM::tet Tetr | Laboratory stock |
| PERM572 | YB955 ΔmutM::tet ΔmutY::sp Tetr Spr | This study |
| PERM573 | YB955 ΔytkD::neo ΔmutM::tet ΔmutY::sp Neor Spr Tetr | 13 |
| PERM599 | PS832 ΔmutM::tet Tetr | P. Setlow |
| PERM659 | PERM311 containing pMUTIN4::mutM; Eryr | This study |
| PERM704 | YB955 ΔmutY::sp Spr | 18 |
| PERM794 | YB955 ΔytkD::neo ΔmutM::tet ΔmutY::sp with Phs-mutM inserted into the amyE locus; Neor Spr Tetr Cmr | This study |
| PERM796 | 168 pMUTIN-FLAG mutM Eryr | This study |
| PERM1199 | YB955 ΔmutM::tet with Phs-mutM; Tetr Cmr | This study |
| Plasmids | ||
| pdr-111-amyE-Phyperspank | bla- and sp-carrying Phs | 43 |
| pPERM617 | pCR-Blunt-II-TOPO with an EcoRI-BamHI promoter region in mutM of 405 bp; Kanr | This study |
| pPERM657 | pMUTIN4 carrying a 405-bp EcoRI-BamHI DNA fragment encompassing 264 bp upstream and 141 bp downstream of the mutM translational start codon; Ampr Eryr | This study |
| pPERM698 | pCR-Blunt II-TOPO with an 849-bp HindIII-KpnI PCR product containing mutM; Kanr | This study |
| pPERM735 | pCR-Blunt-II-TOPO containing the 936-bp SalI-SphI region of mutM; Kanr | This study |
| pPERM748 | pMUTIN-FLAG carrying an 849-bp HindIII-KpnI mutM fragment from pPERM707; Ampr Eryr | This study |
| pPERM792 | pdr-111-amyE-Phyperspank containing the 936-bp SalI-SphI fragment of mutM; Ampr Cmr | This study |
Construction of mutant strains.
To obtain a mutM mutant strain in the genetic background YB955, chromosomal DNA of strain B. subtilis PERM599 (PS832 ΔmutM::tet) was isolated and used to transform competent cells of strain B. subtilis YB955, generating the strain B. subtilis PERM571 (YB955 ΔmutM::tet).
For complementation of the mutM mutation, a copy of this gene was placed ectopically at the amyE locus under the control of the IPTG-inducible hyperspank promoter (Phs). To this end, the open reading frame of mutM was amplified by PCR using genomic DNA of B. subtilis YB955 as a template and Vent DNA polymerase (New England BioLabs, Beverly, MA). Oligonucleotide primers 5′-AAGTCGACGAGATAGGAAGTGATG-3′ (forward) and 5′-ATGCATGCGGGAAAGTGAAAAATC-3′ (reverse) containing SalI and SphI sites (underlined), respectively, were used in the PCR. The PCR product was ligated into the pCR-Blunt-II-TOPO vector (Invitrogen Life Technologies, Carlsbad, CA), generating plasmid pPERM735 (Table 1). The mutM gene was released from this plasmid and inserted into the integrative vector pdr-111-amyE-Phyperspank (a gift from David Rudner). The resulting construct, pPERM792, was introduced by transformation into the B. subtilis strains PERM571 and PERM573 to generate the strains B. subtilis PERM1199 and PERM794.
To obtain a mutM mutY double mutant, the strain PERM571 (YB955 ΔmutM::tet) was transformed with genomic DNA of PERM704 (YB955 mutY::sp) (18), generating the strain B. subtilis PERM572 (YB955 ΔmutM::tet mutY::sp) (Table 1). The double homologous-recombination event resulting in inactivation of the gene of interest was confirmed by PCR with specific oligonucleotide primers (data not shown).
Design of mutM-lacZ and mutM-FLAG constructs.
Construction of a transcriptional fusion between mutM and the lacZ gene was performed in the integrative plasmid pMUTIN4 (19). To this end, a 405-bp fragment, extending from 264 bp upstream to 141 bp downstream of the mutM open reading frame (ORF) start codon was amplified using Vent DNA polymerase (New England BioLabs, Beverly, MA) and oligonucleotide primers 5′-CGCGAATTCCGATTCAAGGAAGCGCCG-3′ (forward) and GCCGGATCCTCGCGCAAATTCCTCCGG-3′ (reverse) with EcoRI/BamHI restriction sites (underlined), respectively. The PCR product was ligated into pCR-Blunt-II-TOPO (Invitrogen Life Technologies, Carlsbad, CA), generating plasmid PERM617. The EcoRI/BamHI fragment was ligated into pMUTIN4, which had previously been digested with the same restriction enzymes. The resulting construct containing the mutM-lacZ fusion was designated pPERM657 and was introduced by transformation into competent cells of strain B. subtilis YB955 to generate strain B. subtilis PERM659 (Table 1).
An in-frame translational fusion between mutM and the FLAG epitope was constructed in the vector pMUTIN-FLAG (20). To this end, a DNA fragment from 15 bp upstream (including the Shine-Dalgarno sequence) of the translational start codon to the last codon of the mutM ORF was amplified by PCR by utilizing Vent DNA polymerase (New England BioLabs) and the oligonucleotide primers 5′-GGAAGCTTCAGAGATAGGAAGTCATGGAT-3′ (forward) and 5′-GGGGTACCGTTTTTTGTCTGGCACTTTCG-3′ (reverse), which inserted HindIII and KpnI sites (underlined) into the cloned DNA. The PCR-amplified DNA fragment (849 bp) was first ligated into pCR-Blunt-II-TOPO (Invitrogen, Carlsbad, CA) and then replicated in E. coli XL10-GOLD Kanr (Stratagene, Cedar Creek, TX). The resulting construct (PERM698) was treated with HindIII and KpnI, the 849-bp mutM insert was ligated into HindIII/KpnI-treated pMUTIN-FLAG, and the ligation products were introduced by transformation into competent cells of E. coli XL10-GOLD Kanr (Stratagene, Cedar Creek, TX). This strategy generated plasmid pPERM748, which was used to transform B. subtilis YB955, generating strain B. subtilis PERM796 (Table 1). The crossover events leading to insertion of the mutM-lacZ and mutM-FLAG fusions into the corresponding loci were confirmed by PCR with specific oligonucleotide primers (data not shown).
β-Galactosidase assays.
B. subtilis strain PERM659, containing a transcriptional mutM-lacZ fusion, was propagated in liquid A3 medium. Aliquots of 1 ml were collected from cultures at exponential growth phase, stationary phase, or sporulation. Cells were washed with 0.1 M Tris-HCl (pH 7.5), pelleted by centrifugation, and stored at 20°C until determination of β-galactosidase activity (21). Briefly, washed-cell samples were first disrupted with lysozyme and subjected to centrifugation; the β-galactosidase activity present in the supernatant was then determined as previously described, using ortho-nitrophenyl-β-d-galactopyranoside (ONPG) as a substrate.
RT-PCR experiments.
Total RNA from exponentially growing or stationary-phase B. subtilis YB955 cells grown in A3 medium was isolated by using TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH). Reverse transcription-PCRs (RT-PCRs) were performed with the RNA samples and a Master AMP RT-PCR kit (Epicentre Technologies, Madison, WI) according to the manufacturer's instructions. The primers used for RT-PCR were 5′-GGTCTTCATCAAGCCGAGGAG-3′ (forward) and 5′-ATTTCAGCGTGAAGGGTTTTG-3′ (reverse), which generated a 377-bp RT-PCR product extending from 249 bp downstream of the start codon of mutM to 626 bp downstream of this point. As a control, in each experiment, the absence of chromosomal DNA in the RNA samples was assessed by PCRs carried out with Vent DNA polymerase (New England BioLabs) and the set of primers described above. The size of the RT-PCR product was determined by utilizing the 1-kb-Plus DNA ladder (Life Technologies, Rockville, MD) during agarose gel electrophoresis.
Western blot assay.
B. subtilis strain YB955 was cultivated with shaking in liquid antibiotic A3 medium at 37°C. Aliquots of 1.5 ml were collected from cultures during the exponential, transition, or stationary phase of growth. Cells were collected by centrifugation (16,000 × g; 1 min), washed twice with 25 mM Tris-HCl (pH 7.5) buffer, and stored at 20°C. Bacterial pellets were resuspended in 0.3 ml of the same buffer supplemented with a protease inhibitor cocktail (Roche, Mannheim, Germany) and were disrupted by sonication with a VCX130-PB Vibra-Cell apparatus (Sonics and Materials Inc., Newton, CT). The cell lysate was subjected to centrifugation to eliminate undisrupted cells and cell debris. The supernatant was separated, and its protein concentration was determined with a Coomassie (Bradford) protein assay kit (Pierce, Rockford, IL). Protein aliquots (100 μg) were separated in SDS-12% polyacrylamide gels and then electrotransferred to polyvinylidene difluoride (PVDF) membranes. Western blot analyses were performed with a FLAG monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 10,000-fold and then processed with an enhanced-chemiluminescence (ECL) Western blotting system (Amersham Pharmacia, Little Chalfont, Buckinghamshire, United Kingdom).
Stationary-phase mutagenesis assays.
Essentially, cultures were grown in flasks containing antibiotic A3 medium with aeration (250 rpm) at 37°C until 90 min after the cessation of exponential growth (designated T90 [90 min after the time point in the culture when the slopes of the logarithmic and stationary phases of growth intercepted]). Growth was monitored with a spectrophotometer measuring the optical density at 600 nm (OD600). The stationary-phase mutagenesis assays were performed as previously described (22, 23) using solid Spizizen minimal medium (SMM; 1× Spizizen salts supplemented with 0.5% glucose and either 50 μg or 200 ng of the required amino acid per ml and 50 μg each of isoleucine and glutamic acid per ml). The concentration of the amino acid used depended on the reversion tested. For instance, in selecting His+ revertants, 50 μg (each) of methionine and leucine ml−1 was added to the medium and 200 ng of histidine ml−1 was added. Isoleucine and glutamic acid were added as described previously (16) in order to protect the viability of the cells. The number of revertants was scored daily. The initial number of bacteria plated for each experiment was estimated by serial dilution of the bacterial cultures and then plating of the cells on LB medium. The experiments were repeated at least three times.
Analysis of mutation frequencies.
Frequencies of spontaneous mutation to rifampin resistance in growing cells were determined as previously described (13). Essentially, the appropriate strains were grown for 12 h at 37°C in antibiotic A3 medium with proper antibiotics. Mutation frequencies were determined by plating aliquots on six LB plates containing 10 μg ml−1 rifampin, and the rifampin-resistant (Rifr) colonies were counted after 1 day of incubation at 37°C. The number of cells used to calculate the frequency of mutation to Rifr was determined by plating aliquots of appropriate dilutions on LB plates without rifampin and incubating the plates for 24 to 48 h at 37°C. These experiments were repeated at least three times (24).
Assays of sensitivity to oxidative-stress inducers.
B. subtilis strains YB955, PERM571, and PERM1199 were grown in LB medium with aeration (250 rpm) at 37°C. Growth was monitored with a spectrophotometer measuring the optical density at 600 nm (OD600). Before cessation of exponential growth (OD600, ∼0.6), the cells were collected by centrifugation (6,500 × g, 5 min), washed twice with phosphate-buffered saline (PBS; pH 7.2), and resuspended in the same buffer. Cell aliquots of equal volumes were treated with different final concentrations of H2O2 or PQ and incubated for 30 min at 37°C with shaking. The total viable-cell numbers in each culture were determined by spotting serial dilutions of the cultures on LB agar plates. The number of colonies was counted after 24 h of incubation at 37°C.
Statistical analysis.
For determination of mutation frequencies and oxidative-stress sensitivity, differences were calculated by performing one-way analysis of variance (ANOVA) followed by a Tukey's post hoc analysis. Significance was set at a P of <0.05.
RESULTS AND DISCUSSION
MutM confers protection to B. subtilis from the toxicity promoted by oxidant agents.
ROS-promoted DNA lesions, including 8-oxo-G, may potentially generate mutagenesis and cell death (25, 26). The 8-oxo-G lesion is processed through the BER pathway with the specific participation of MutM, which eliminates this oxidized base from DNA (5, 27, 28). Thus, we analyzed whether MutM conferred protection to growing B. subtilis cells from the cytotoxic effects of oxidative stress. To this end, growing cells of a mutM knockout strain and its MutM-proficient parental strain were treated with increasing amounts of H2O2 or PQ. Results showed that disruption of mutM significantly sensitized exponentially growing cells of B. subtilis to these oxidizing agents (Fig. 1A and B). This result reveals a role for MutM in conferring to B. subtilis protection from the lethal effects of H2O2 and PQ. In support of this notion, the susceptibility to H2O2 and PQ of the mutM strain was reestablished to the level of the parental strain, YB955, following expression of the wild-type mutM gene from the IPTG-inducible Phs promoter (Fig. 1A and B). Taken together, these results strongly suggest that MutM plays a significant role in preventing the cytotoxic effects of 8-oxo-G and possibly of other related lesions, including the opened ring derivative formamidopyrimidine (FaPy) (29–32), thus contributing to B. subtilis survival. However, in addition to inducing the formation of oxidized bases, H2O2 and PQ may promote other types of DNA lesions, including 8-OxoG·A mispairs and apurinic/apyrimidinic (AP) sites, as well as single- and/or double-strand DNA breaks (33). Therefore, in addition to MutM, other repair proteins, including MutY, Nth, and the AP-endonucleases Nfo and ExoA, most probably contribute to protecting B. subtilis from the genotoxic effects of H2O2 (33).
FIG 1.

Contribution of MutM in the survival of B. subtilis to H2O2 and PQ treatment (A and B) and frequencies of spontaneous mutation to Rifr of different B. subtilis strains (C and D). (A and B) B. subtilis YB955 (parental; ●), PERM751 (ΔmutM; ○), and PERM1199 (ΔmutM amyE::Phs-mutM; ▲) strains were treated with different amounts of H2O2 (A) or PQ (B), and cell viability was determined as described in Materials and Methods. The values shown represent the means and standard deviations from three independent experiments done in triplicate. (C and D) B. subtilis YB955 (parental), PERM751 (ΔmutM), PERM1199 (ΔmutM amyE::Phs-mutM), PERM573 (GO system deletion [ΔGO]), and PERM794 (ΔGO amyE::Phs-mutM) were grown overnight in PAB medium, and frequencies of mutation to Rifr were determined as described in Materials and Methods. Each bar represents the mean of data collected from three independent experiments done in sextuplicate, and the error bars represent standard errors of the means (SEM). The statistical differences (a, b, c, and d) between the mutation frequencies of each strain and condition, as determined by ANOVA (P < 0.05), are shown above each bar.
Of note, the absence of MutM also decreased the H2O2 resistance of E. coli cells, and such effect was associated with an increased amount of 8-oxo-G lesions in the genome of this microorganism (34). However, the protective role conferred by MutM against H2O2-promoted DNA damage has also been described to occur in other bacteria, including Pseudomonas aeruginosa and Mycobacterium smegmatis. Thus, cells of these strains lacking MutM were significantly more susceptible to H2O2 treatment than their MutM-proficient counterparts (35, 36).
Spontaneous mutation frequencies in B. subtilis cells lacking MutM.
Due to an anticipated role of the MutM protein in preventing the mutagenic and cytotoxic effects of 8-oxo-G (13, 37), the mutation frequency to a Rifr phenotype was determined in growing cells of the MutM-deficient and parental YB955 strains. The results revealed that the loss of MutM increased the spontaneous mutation frequency to Rifr 5-fold in comparison with that of an isogenic strain that produced a functional MutM protein (Fig. 1C). From these results, we propose that MutM prevents the spontaneous mutagenic events promoted by oxidative stress in growing B. subtilis cells. Two observations support this contention: the levels of mutation to Rifr calculated in the MutM-deficient strain were restored to the levels of the parental strain following expression of mutM from the IPTG-inducible Phs promoter (Fig. 1C), and the overexpression of mutM induced a significant decrease in the frequency of mutation to Rifr of a hypermutagenic strain that was deficient for MutM, MutY, and MutT (Fig. 1D).
In agreement with a previous report (37), our results revealed that cells of the B. subtilis strain YB955 lacking MutM showed a slight but statistically significant increase in their frequency of spontaneous mutation to Rifr relative to that of the MutM-proficient parental strain. It must be pointed out that MutM-deficient strains of E. coli and Pseudomonas putida also presented a mutagenic Rifr phenotype; however, in these bacteria, as well as in B. subtilis (13), the single MutY deficiency conferred a stronger mutagenic effect than that observed in the strains lacking MutM (5, 36, 38, 39). These results suggest the existence of alternative repair pathways that compensate for the absence of MutM; in agreement with this notion, the genomes of the three microorganisms discussed above contain the gene for Nth, a DNA glycosylase capable of processing 8-oxo-G and AP sites (40–42). In the case of B. subtilis, it was recently shown that the genetic inactivation of Nth not only increases this bacterium's spontaneous Rifr mutation frequency but also sensitizes it to the ROS promoter agent H2O2 (33).
Stationary-phase mutagenesis in B. subtilis cells deficient for MutM.
We next investigated the role played by MutM in the stationary-phase-associated mutagenesis (SPM) of B. subtilis. These experiments were performed in strain B. subtilis YB955, which is auxotrophic for three amino acids due to the chromosomal mutations hisC952 (amber), metB5 (ochre), and leuC427 (missense). This strain has been validated and widely used as a model system to understand how mutations are generated in amino-acid-starved cells (18, 23, 43). Analysis of frequencies of reversion to his, met, and leu in cell cultures that were starved for each of these amino acids revealed that MutM contributes to mutagenesis in starved B. subtilis cells. As shown in Fig. 2, the MutM-deficient strain significantly increased the frequency of his, met, and leu reversions in reference to those generated by parental strain YB955. These results strongly suggest that unrepaired 8-oxo-G lesions that accumulate in the MutM-deficient strain promote stationary-phase-associated mutagenesis in B. subtilis. In a marked contrast with our results, the single absence of MutM did not promote mutagenesis in starved cells of E. coli and P. putida (11, 12). However, in E. coli and P. putida, the lack of MutY did induce a significant increase in the production of stationary-phase-associated mutations (11, 44), suggesting that accumulation of nonprocessed 8-oxo-G lesions contribute to stationary-phase-associated mutagenesis in these strains. In support of this notion, when the mutM mutation was combined with a deficiency in MutY, the mutation frequency was further enhanced in starved E. coli cells (11). Thus, despite the fact that the lack of MutY also contributes to SPM in B. subtilis (18), our results clearly indicate that the sole disruption of mutM also favored this type of mutagenesis in this microorganism. In support of this contention, an ectopic copy of mutM expressed from the IPTG-inducible Phs promoter diminished the numbers of His+, Met+, and Leu+ revertants relative to those produced by parental strain YB955 (Fig. 2). Moreover, we corroborated the finding that the genetic defect in mutM did not affect the survival of B. subtilis cells starved for his, met, and leu during the 10 days that the SPM experiments lasted (see Fig. S1 in the supplemental material).
FIG 2.
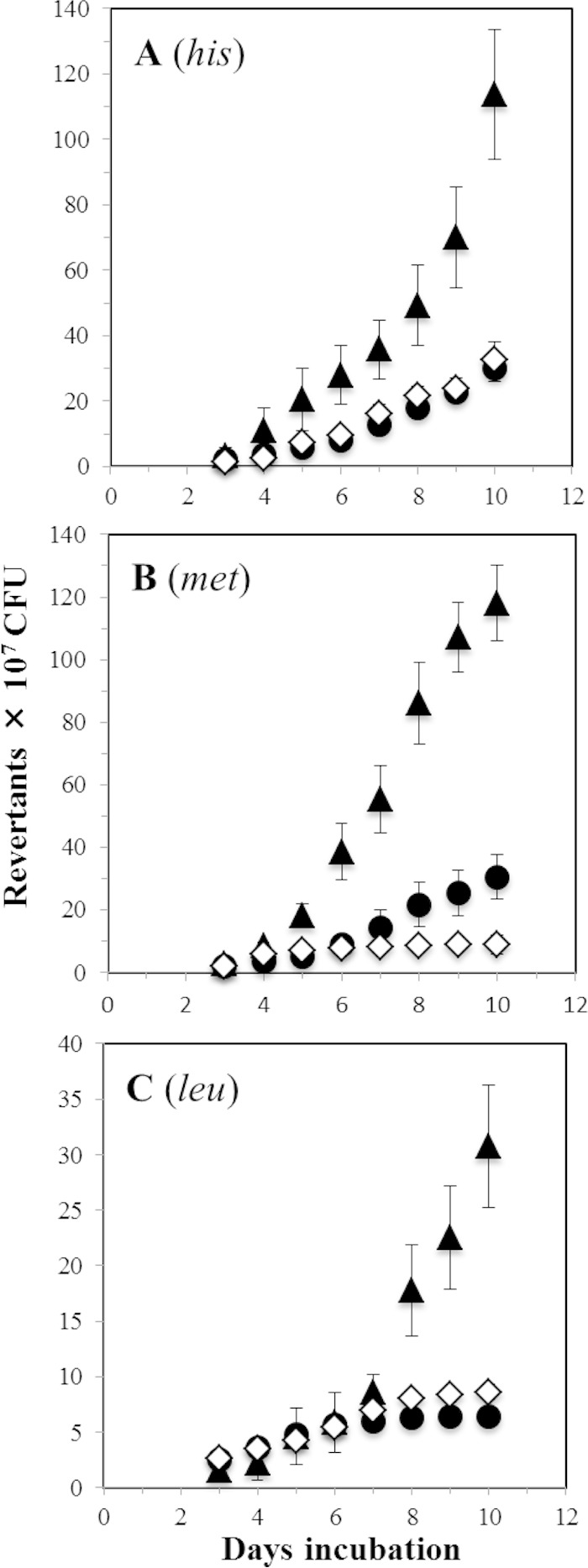
Stationary-phase-induced reversions to his (A), met (B), and leu (C) of the YB955 (◇), PERM571 (ΔmutM) (▲), and PERM1199 (ΔmutM amyE::Phs-mutM) (●) B. subtilis strains were determined as described in Materials and Methods. Data represent counts from six plates averaged from three separate tests normalized to initial cell titers ± standard deviations (SD).
Analysis of mutM expression during the life cycle of B. subtilis.
As described above, MutM confers protection to growing B. subtilis cells from the toxic effects of H2O2, and its deficiency promotes adaptive mutagenesis in nutritionally stressed cells. These results suggest that mutM may be expressed in the exponential and stationary phases of growth of this microorganism. To explore this notion, we analyzed the temporal pattern of expression of mutM and determined the levels of its encoded product during the life cycle of B. subtilis. The levels of transcription were determined by employing B. subtilis strain PERM659, which harbors a genomic copy of a transcriptional mutM-lacZ fusion (Table 1). The results showed that this strain expressed barely similar levels of β-galactosidase during the exponential transition (from exponential to stationary phase) and the first hours of stationary phases of growth (Fig. 3A). However, the expression levels of the reporter lacZ gene commenced to diminish during the late stationary phase of growth. Results from an RT-PCR experiment performed with RNA samples collected during exponential growth as well as during the transition and stationary phases of growth confirmed the presence of mutM mRNAs during the three developmental phases analyzed (Fig. 3B). In agreement with this result, we also detected a MutM-FLAG protein in actively growing cells of a B. subtilis strain and in cells in the stationary phase of growth harboring an in-frame translational mutM-FLAG fusion (Fig. 3C). Based on these and previous results (45, 46), it is feasible to propose that B. subtilis expresses mutM during its entire life cycle to contend with the genotoxic and cytotoxic effects of ROS. However, despite the role displayed by mutM in protecting B. subtilis from oxidatively induced DNA damage, we did not find evidence that this gene is part of the gene circuitries that respond to distinct types of stressful conditions, including DNA damage and oxidative or osmotic stress (47–53). This conclusion was deduced from experiments showing that H2O2 (0.1%), mitomycin C (0.5 μg ml−1), and NaCl (4%) did not turn on the transcription of a mutM-lacZ fusion inserted into the genome of strain B. subtilis YB955 (Fig. S2). Therefore, in conjunction with previous reports (47, 54–56), it is feasible to conclude that expression of mutM is not under the control of the master regulator RecA/DinR, PerR, or σB.
FIG 3.
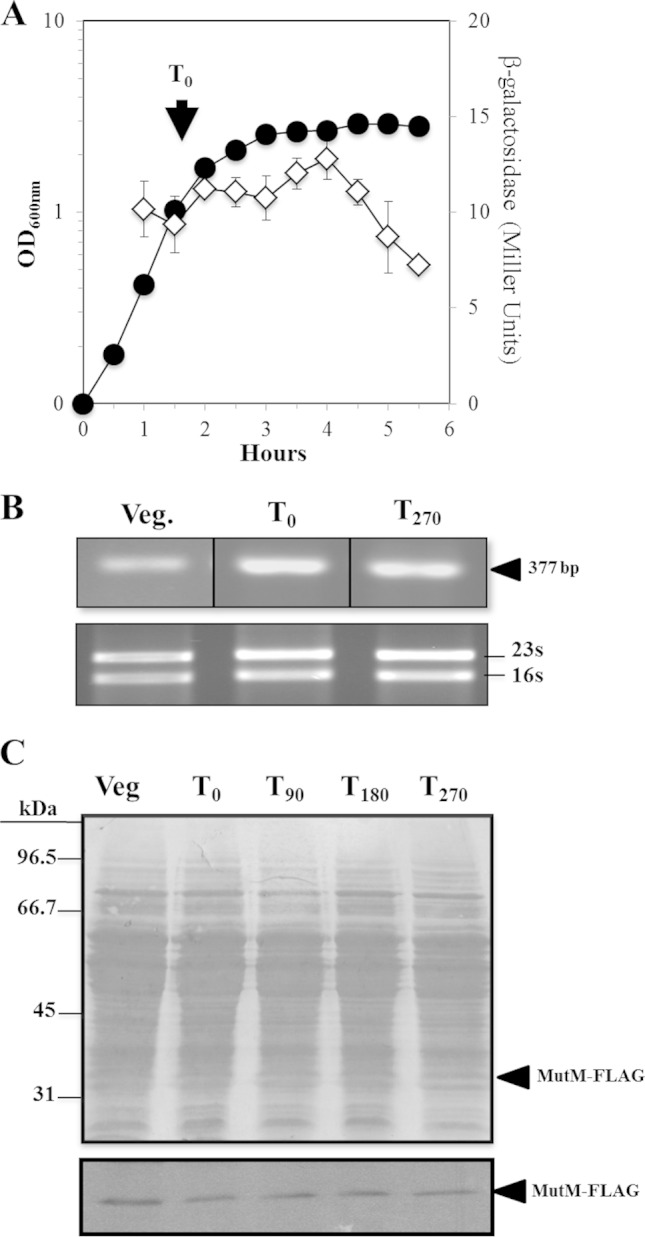
(A). Levels of β-galactosidase in a mutM-lacZ transcriptional fusion during the vegetative and stationary phases of growth. B. subtilis strain PERM659 was grown in liquid antibiotic (A3) medium. Cell samples were collected at the indicated times and treated with lysozyme, and the extracts were assayed for β-galactosidase as described in Materials and Methods. Data shown are average values from triplicate independent experiments ± SD for β-galactosidase specific activity (◇) and for A600 values (●). (B) RT-PCR analysis of mutM transcription during the vegetative and stationary phases of growth. RNA samples (1 μg) isolated from a B. subtilis YB955 A3 culture, at the steps indicated, were processed for RT-PCR analysis as described in Materials and Methods. The arrowhead shows the size of the expected RT-PCR product. 16S and 23S rRNA bands are shown in the lower panel. (C) Western blot analysis of MutM-FLAG synthesis during the vegetative and stationary phases of growth. B. subtilis strain YB955 was grown in liquid A3 medium. Cell extract samples (∼100 μg of protein; see Materials and Methods), harvested at the steps indicated, were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad, Hercules, CA). The blots were stained with Ponceau red (top), probed with a FLAG monoclonal antibody diluted 10,000-fold, and then processed with an ECL Western blot system (bottom). The positions of molecular mass markers are indicated to the left of the stained membrane. T0 is the time point in the culture when the slopes of the logarithmic and stationary phases of growth intercepted. T90, T180, and T270 indicate times in minutes after T0. Veg., vegetative growth.
In contrast, in E. coli, mutM is under the negative transcriptional control of the Fur, Fnr, and ArcA regulators; thus, the mRNA levels of mutM are enhanced in this bacterium by ROS-producing chemicals, including Paraquat. It is noteworthy that the levels of expression of mutY are repressed under the stressful conditions that activate mutM (14, 57, 58).
Thus, the ability of B. subtilis to keep active the synthesis of MutM during the logarithmic and stationary phases is in agreement with our results that demonstrated antimutagenic roles of this repair protein in both stages of growth (Fig. 1 and 2). Moreover, the presence of MutM in the stationary phase of B. subtilis but its apparent absence in E. coli (57) may explain why the single disruption of mutM did not promote mutagenesis in starved cells of E. coli unless combined with a mutation in MutY (11). Alternatively, the existence in E. coli of repair proteins that process 8-oxo-G lesions, including Nth and Nei (11, 59), may suppress mutagenesis in starved E. coli cells deficient for MutM.
Our analysis of his, met, and leu reversions in nutritionally stressed B. subtilis cells showed that deficiencies in mutM significantly increased the mutagenesis levels in the three alleles tested. However, the mechanisms involved in generating such reversions may be different; thus, for the his and met alleles, ROS-promoted synthesis of 8-oxo-G may be responsible for these reversions. In support of this contention, genetic inactivation of mutY in the MutM-deficient strain dramatically increased the production of His and Met revertants in the resulting mutM mutY mutant (Fig. 4). In contrast, the levels of reversion of the leu allele in the mutM mutY strain were reduced compared to those observed in the mutM and parental YB955 strains (Fig. 4C). This result suggest that MutY promotes reversions in the leuC allele; in support of this notion, the levels of Leu+ revertants were almost completely ablated in the MutY-deficient strain (Fig. 4C). Furthermore, a previous study demonstrated that processing of accumulated G·A mismatches in starved B. subtilis by MutY is involved in generating stationary-phase-associated Leu+ revertants (18).
FIG 4.
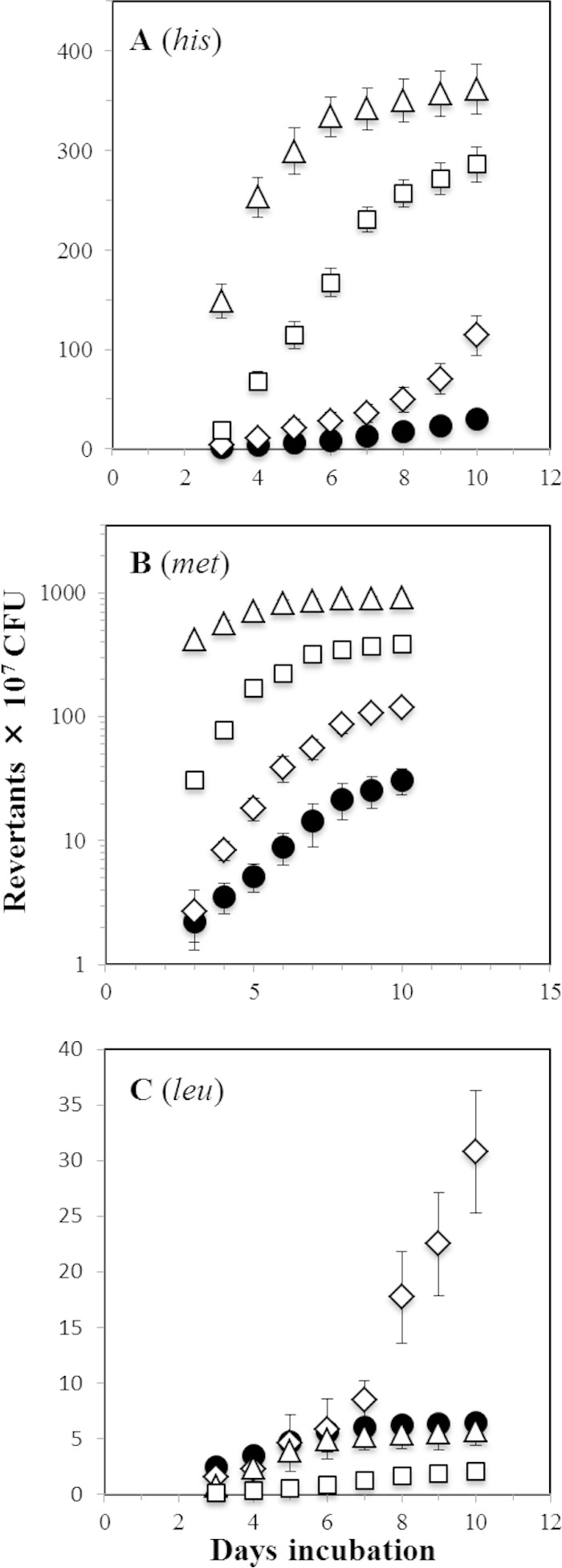
Stationary-phase-induced reversions to his (A), met (B), and leu (C) of the YB955 (●), PERM571 (ΔmutM) (◇), PERM704 (ΔmutY) (□), and PERM573 (ΔmutM ΔmutY) (△) B. subtilis strains were determined as described in Materials and Methods. Data represent counts from six plates averaged from three separate tests normalized to initial cell titers ± SD.
As shown in this and previous reports (13, 18), DNA repair proteins that process ROS-induced DNA damage play prominent roles in modulating mutagenesis in starved bacterial cells. Nevertheless, current reports have shown that in B. subtilis, this type of mutation is also dependent on Mfd, a protein that couples transcription with the DNA repair machinery (43, 60). It was recently found that production of Leu+ prototrophs in MutY-deficient B. subtilis cells of strain YB955 are fully dependent on a functional Mfd protein (M. Gómez-Marroquín, E. A. Robleto, and M. Pedraza-Reyes, unpublished results). Therefore, we are currently investigating how Mfd coordinates the activities of repair proteins of the GO system to generate mutations that occur in nutritionally stressed B. subtilis cells.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the University of Guanajuato (grants DAIP-324-2013 and 602-2015) and by the Consejo Nacional de Ciencia y Tecnología (CONACYT) (grants 205744 and 221231) of Mexico. Work at the laboratory of E.A.R is supported by grant GM110624 from the NIH. M.G.-M., F.S.-E., B.N.D., and L.E.V. were supported by scholarships from CONACYT.
We acknowledge the excellent technical assistance of Fernando H. Ramírez-Guadiana.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00147-15.
REFERENCES
- 1.Scandalios JG. 2002. Oxidative stress responses—what have genome-scale studies taught us? Genome Biol 3:REVIEWS1019. doi: 10.1186/gb-2002-3-7-reviews1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saran M, Bors W. 1990. Radical reactions in vivo—an overview. Radiat Environ Biophys 29:249–262. doi: 10.1007/BF01210406. [DOI] [PubMed] [Google Scholar]
- 3.Michaels ML, Miller JH. 1992. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J Bacteriol 174:6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castellanos-Juarez FX, Alvarez-Alvarez C, Yasbin RE, Setlow B, Pedraza-Reyes M. 2006. YtkD and MutT protect vegetative cells but not spores of Bacillus subtilis from oxidative stress. J Bacteriol 188:2285–2289. doi: 10.1128/JB.188.6.2285-2289.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michaels ML, Cruz C, Grollman AP, Miller JH. 1992. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci U S A 89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maki H, Sekiguchi M. 1992. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 355:273–275. doi: 10.1038/355273a0. [DOI] [PubMed] [Google Scholar]
- 7.Taddei F, Hayakawa H, Bouton M, Cirinesi A, Matic I, Sekiguchi M, Radman M. 1997. Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 278:128–130. doi: 10.1126/science.278.5335.128. [DOI] [PubMed] [Google Scholar]
- 8.Bohr VA. 2002. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med 32:804–812. doi: 10.1016/S0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 9.Seeberg E, Eide L, Bjørås M. 1995. The base excision repair pathway. Trends Biochem Sci 20:391–397. doi: 10.1016/S0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- 10.Bridges BA. 1996. Elevated mutation rate in mutT bacteria during starvation: evidence for DNA turnover? J Bacteriol 178:2709–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridges BA, Sekiguchi M, Tajiri T. 1996. Effect of mutY and mutM/fpg-1 mutations on starvation-associated mutation in Escherichia coli: implications for the role of 7,8-dihydro-8-oxoguanine. Mol Gen Genet 251:352–357. doi: 10.1007/s004380050176. [DOI] [PubMed] [Google Scholar]
- 12.Saumaa S, Tover A, Tark M, Tegova R, Kivisaar M. 2007. Oxidative DNA damage defense systems in avoidance of stationary-phase mutagenesis in Pseudomonas putida. J Bacteriol 189:5504–5514. doi: 10.1128/JB.00518-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidales LE, Cardenas LC, Robleto E, Yasbin RE, Pedraza-Reyes M. 2009. Defects in the error prevention oxidized guanine system potentiate stationary-phase mutagenesis in Bacillus subtilis. J Bacteriol 191:506–513. doi: 10.1128/JB.01210-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee H, Lee Y, Kim H, Choi J, Hassan H, Chung M. 1998. Mechanism of regulation of 8-hydroxyguanine endonuclease by oxidative stress: role of FNR, ArcA, and Fur. Free Radic Biol Med 24:1193–1201. [DOI] [PubMed] [Google Scholar]
- 15.Yasbin RE, Miehl-Lester R, Love PE. 1987. Mutagenesis in Bacillus subtilis, p 73–84. In Alacevic M, Hranueli D, Tomen Z (ed), Genetics of industrial microorganisms. Proceedings of the 5th International Symposium, GIM-86. Ognjen Prica Printing Works, Karlovac, Yugoslavia. [Google Scholar]
- 16.Sung HM, Yasbin RE. 2000. Transient growth requirement in Bacillus subtilis following the cessation of exponential growth. Appl Environ Microbiol 66:1220–1222. doi: 10.1128/AEM.66.3.1220-1222.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasbin RE, Fields PI, Andersen BJ. 1980. Properties of Bacillus subtilis 168 derivatives freed of their natural prophages. Gene 12:155–159. doi: 10.1016/0378-1119(80)90026-8. [DOI] [PubMed] [Google Scholar]
- 18.Debora BN, Vidales LE, Ramirez R, Ramirez M, Robleto E, Yasbin RE, Pedraza-Reyes M. 2011. Mismatch repair modulation of MutY activity drives Bacillus subtilis stationary-phase mutagenesis. J Bacteriol 193:236–245. doi: 10.1128/JB.00940-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vagner V, Dervyn E, Ehrlich SD. 1998. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology 144:3097–3104. doi: 10.1099/00221287-144-11-3097. [DOI] [PubMed] [Google Scholar]
- 20.Kaltwasser M, Wiegert T, Schumann W. 2002. Construction and application of epitope- and green fluorescent protein-tagging integration vectors for Bacillus subtilis. Appl Environ Microbiol 68:2624–2628. doi: 10.1128/AEM.68.5.2624-2628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicholson WL, Setlow P. 1990. Sporulation, germination and outgrowth, p 391–431. In Harwood CR, Cutting SM (ed), Molecular biological methods for Bacillus, 2nd ed John Wiley and Sons, Sussex, England. [Google Scholar]
- 22.Pedraza-Reyes M, Yasbin RE. 2004. Contribution of the mismatch DNA repair system to the generation of stationary-phase-induced mutants of Bacillus subtilis. J Bacteriol 186:6485–6491. doi: 10.1128/JB.186.19.6485-6491.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sung H, Yasbin RE. 2002. Adaptive, or stationary-phase, mutagenesis, a component of bacterial differentiation in Bacillus subtilis. J Bacteriol 184:5641–5653. doi: 10.1128/JB.184.20.5641-5653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosche WA, Foster PL. 2000. Determining mutation rates in bacterial populations. Methods 20:4–17. doi: 10.1006/meth.1999.0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 26.Storz G, Imlay JA. 1999. Oxidative stress. Curr Opin Microbiol 2:188–194. doi: 10.1016/S1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- 27.Lu AL, Li X, Gu Y, Wright PM, Chang DY. 2001. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem Biophys 35:141–170. doi: 10.1385/CBB:35:2:141. [DOI] [PubMed] [Google Scholar]
- 28.Tajiri T, Maki H, Sekiguchi M. 1995. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat Res 336:257–267. doi: 10.1016/0921-8777(94)00062-B. [DOI] [PubMed] [Google Scholar]
- 29.Delaney S, Jarem DA, Volle CB, Yennie CJ. 2012. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic Res 46:420–441. doi: 10.3109/10715762.2011.653968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michaels ML, Pham L, Cruz C, Miller JH. 1991. MutM, a protein that prevents G·C-T·A transversions, is formamidopyrimidine-DNA glycosylase. Nucleic Acids Res 19:3629–3632. doi: 10.1093/nar/19.13.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chetsanga CJ, Lindahl T. 1979. Release of 7-methylguanine residues whose imidazole rings have been opened from damaged DNA by a DNA glycosylase from Escherichia coli. Nucleic Acids Res 6:3673–3684. doi: 10.1093/nar/6.11.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sidorkina O, Dizdaroglu M, Laval J. 2001. Effect of single mutations on the specificity of Escherichia coli FPG protein for excision of purine lesions from DNA damaged by free radicals. Free Radic Biol Med 31:816–823. doi: 10.1016/S0891-5849(01)00659-1. [DOI] [PubMed] [Google Scholar]
- 33.Barajas-Ornelas RDC, Ramírez-Guadiana FH, Juárez-Godínez R, Ayala-García VM, Robleto E, Yasbin RE, Pedraza-Reyes M. 2014. Error-prone processing of apurinic/apyrimidinic (AP) sites by PolX underlies a novel mechanism that promotes adaptive mutagenesis in Bacillus subtilis. J Bacteriol 196:3012–3022. doi: 10.1128/JB.01681-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alhama J, Ruiz-Laguna J, Rodriguez-Ariza A, Toribio F, López-Barea J, Pueyo C. 1998. Formation of 8-oxoguanine in cellular DNA of Escherichia coli strains defective in different antioxidant defences. Mutagenesis 13:589–594. doi: 10.1093/mutage/13.6.589. [DOI] [PubMed] [Google Scholar]
- 35.Jain R, Kumar P, Varshney U. 2007. A distinct role of formamidopyrimidine DNA glycosylase (MutM) in down-regulation of accumulation of G, C mutations and protection against oxidative stress in mycobacteria. DNA Repair (Amst) 6:1774–1785. doi: 10.1016/j.dnarep.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Sanders LH, Sudhakaran J, Sutton MD. 2009. The GO system prevents ROS-induced mutagenesis and killing in Pseudomonas aeruginosa. FEMS Microbiol Lett 294:89–96. doi: 10.1111/j.1574-6968.2009.01550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki M, Yonemura Y, Kurusu Y. 2000. Genetic analysis of Bacillus subtilis mutator genes. J Gen Appl Microbiol 46:183–187. doi: 10.2323/jgam.46.183. [DOI] [PubMed] [Google Scholar]
- 38.Cabrera M, Nghiem Y, Miller JH. 1988. mutM, a second mutator locus in Escherichia coli that generates G·C-T·A transversions. J Bacteriol 170:5405–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nghiem Y, Cabrera M, Cupples C, Miller J. 1988. The mutY gene: a mutator locus in Escherichia coli that generates G.C-T.A transversions. Proc Natl Acad Sci U S A 85:2709–2713. doi: 10.1073/pnas.85.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunningham RP, Weiss B. 1985. Endonuclease III (nth) mutants of Escherichia coli. Proc Natl Acad Sci U S A 82:474–478. doi: 10.1073/pnas.82.2.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collier C, Machón C, Briggs GS, Smits WK, Soultanas P. 2012. Untwisting of the DNA helix stimulates the endonuclease activity of Bacillus subtilis Nth at AP sites. Nucleic Acids Res 40:739–750. doi: 10.1093/nar/gkr785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki T, Yamamoto K, Harashima H, Kamiya H. 2008. Base excision repair enzyme endonuclease III suppresses mutagenesis caused by 8-hydroxy-dGTP. DNA Repair (Amst) 7:88–94. doi: 10.1016/j.dnarep.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 43.Pybus C, Pedraza-Reyes M, Ross CA, Martin H, Ona K, Yasbin RE, Robleto E. 2010. Transcription-associated mutation in Bacillus subtilis cells under stress. J Bacteriol 192:3321–3328. doi: 10.1128/JB.00354-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saumaa S, Tover A, Kasak L, Kivisaar M. 2002. Different spectra of stationary-phase mutations in early-arising versus late-arising mutants of Pseudomonas putida: involvement of the DNA repair enzyme MutY and the stationary-phase sigma factor RpoS. J Bacteriol 184:6957–6965. doi: 10.1128/JB.184.24.6957-6965.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blom EJ, Ridder ANJA, Lulko AT, Roerdink JBTM, Kuipers OP. 2011. Time-resolved transcriptomics and bioinformatic analyses reveal intrinsic stress responses during batch culture of Bacillus subtilis. PLoS One 6:e27160. doi: 10.1371/journal.pone.0027160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisicchia P, Botella E, Delumeau O, Doherty G, Denham E, Fogg M, Fromion V, Goelzer A, Hansen A, Härtig E, Harwood C, Homuth G, Jarmer H, Jules M, Klipp E, Le Chat L, Lecointe F, Lewis P, Liebermeister W, March A, Mars R, Nannapaneni P, Noone D, Pohl S, Rinn B, Rügheimer F, Sappa P, Samson F, Schaffer M, Schwikowski B, Steil L, Stülke J, Wiegert T, Devine K, Wilkinson A, van Dijl J, Hecker M, Völker U, Bessières P, Noirot P. 2012. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- 47.Fuangthong M, Herbig AF, Bsat N, Helmann JD. 2002. Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J Bacteriol 184:3276–3286. doi: 10.1128/JB.184.12.3276-3286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Love PE, Yasbin RE. 1984. Genetic characterization of the inducible SOS-like system of Bacillus subtilis. J Bacteriol 160:910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benson AK, Haldenwang WG. 1992. Characterization of a regulatory network that controls sigma B expression in Bacillus subtilis. J Bacteriol 174:749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benson AK, Haldenwang WG. 1993. The sigma B-dependent promoter of the Bacillus subtilis sigB operon is induced by heat shock. J Bacteriol 175:1929–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boylan SA, Rutherford A, Thomas SM, Price CW. 1992. Activation of Bacillus subtilis transcription factor sigma B by a regulatory pathway responsive to stationary-phase signals. J Bacteriol 174:3695–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lovett CM, Love PE, Yasbin RE. 1989. Competence-specific induction of the Bacillus subtilis RecA protein analog: evidence for dual regulation of a recombination protein. J Bacteriol 171:2318–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Völker U, Engelmann S, Maul B, Riethdorf S, Völker A, Schmid R, Mach H, Hecker M. 1994. Analysis of the induction of general stress proteins of Bacillus subtilis. Microbiology 140:741–752. doi: 10.1099/00221287-140-4-741. [DOI] [PubMed] [Google Scholar]
- 54.Lovett CM, Love PE, Yasbin RE, Roberts JW. 1988. SOS-like induction in Bacillus subtilis: induction of the RecA protein analog and a damage-inducible operon by DNA damage in Rec and DNA repair-deficient strains. J Bacteriol 170:1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hecker M, Pané-Farré J, Völker U. 2007. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev Microbiol 61:215–236. doi: 10.1146/annurev.micro.61.080706.093445. [DOI] [PubMed] [Google Scholar]
- 56.Au N, Kuester-schoeck E, Mandava V, Bothwell LE, Canny SP, Chachu K, Colavito SA, Fuller SN, Groban ES, Hensley LA, Brien TCO, Shah A, Tierney JT, Tomm LL, Gara TMO, Goranov AI, Grossman AD, Lovett CM. 2005. Genetic composition of the Bacillus subtilis SOS system. J Bacteriol 187:7655–7666. doi: 10.1128/JB.187.22.7655-7666.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim HS, Park YW, Kasai H, Nishimura S, Park CW, Choi KH, Chung MH. 1996. Induction of E. coli oh8Gua endonuclease by oxidative stress: its significance in aerobic life. Mutat Res 363:115–124. doi: 10.1016/0921-8777(96)00006-7. [DOI] [PubMed] [Google Scholar]
- 58.Yoon S-H, Lee H-S, Choi J-Y, Kang H-K, Lee J-J, Hyun J-W, Choi J, Ye S-K, Chung M-H. 2003. MutY is down-regulated by oxidative stress in E. coli. Free Radic Res 37:873–879. doi: 10.1080/1071576031000150760. [DOI] [PubMed] [Google Scholar]
- 59.Matsumoto Y, Zhang QM, Takao M, Yasui A, Yonei S. 2001. Escherichia coli Nth and human hNTH1 DNA glycosylases are involved in removal of 8-oxoguanine from 8-oxoguanine/guanine mispairs in DNA. Nucleic Acids Res 29:1975–1981. doi: 10.1093/nar/29.9.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross C, Pybus C, Pedraza-Reyes M, Sung HM, Yasbin RE, Robleto EA. 2006. Novel role of mfd: effects on stationary-phase mutagenesis in Bacillus subtilis. J Bacteriol 188:7512–7520. doi: 10.1128/JB.00980-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.