

Abstract
The late stage of dry age-related macular degeneration (AMD), or geographic atrophy (GA), is characterized by extensive retinal pigment epithelial (RPE) cell death, and a cure is not available currently. We have recently demonstrated that RPE cells die from necrosis in response to oxidative stress, providing a potential novel mechanism for RPE death in AMD. In this study, we screened U.S. Food and Drug Administration-approved natural compounds and identified gossypol acetic acid (GAA) as a potent inhibitor of oxidative stress-induced RPE cell death. GAA induces antioxidative response and inhibits accumulation of excessive reactive oxygen species in cells, through which it prevents the activation of intrinsic necrotic pathway in response to oxidative stress. Sestrin2 (SESN2) is found to mediate GAA function in antioxidative response and RPE survival upon oxidative stress. Moreover, Forkhead box O3 transcription factor (FoxO3) is further found to be required for GAA-mediated SESN2 expression and RPE survival. Mechanistically, GAA promotes FoxO3 nuclear translocation and binding to the SESN2 enhancer, which in turn increases its transcriptional activity. Taken together, we have identified GAA as a potent inhibitor of oxidative stress-induced RPE necrosis by regulating the FoxO3/SESN2 pathway. This study may have significant implications in the therapeutics of age-related diseases, especially GA.
INTRODUCTION
Age-related macular degeneration (AMD) is the leading cause of severe vision loss in people aged over 50, and its prevalence increases exponentially in people over the age of 70 (1). Currently, it is estimated that 1.75 million individuals suffer from this disease in the United States, and 7 million are said to be “at risk” (2). There are two types of AMD, the “dry” and “wet” forms, respectively. Dry AMD is a chronic disease that usually causes some degree of visual impairment and sometimes progresses to severe blindness. Dry AMD accounts for 90% of AMD cases and is currently without treatment available. The late stage of dry AMD, which is also knows as geographic atrophy (GA), is characterized by scattered or confluent areas of degeneration of retinal pigment epithelium (RPE) cells and the overlying photoreceptors that rely on the RPE for trophic support (3). AMD is a multifactorial disease with unclear etiology. Age is the most consistent risk factor associated with AMD, and genetic factors, oxidative stress, and inflammation also significantly contribute to AMD pathogenesis (4). Cigarette smoking, which induces systemic oxidative stress, has been proved to be a significant risk factor for AMD. Consistently, clinical studies have shown that the progression of AMD can be slowed with antioxidant vitamins and zinc supplements (5, 6). The retina is one of the highest oxygen-consuming tissues in the human body and, in particular, RPE is vulnerable to oxidative damage (7, 8). The mechanism of RPE cell death in response to oxidative stress and in GA has been controversial. Apoptosis was suggested as a major mechanism of RPE cell death, even though several studies suggested necrosis as mechanism of RPE cell death in vitro (9, 10) and in vivo (11, 12). Necrosis used to be considered a passive and unregulated form of cell death. Recent studies found necrosis to be a regulated process mediated by receptor interacting protein (RIP) kinases, leading to its renaming as necroptosis (13). We recently conducted systematic analysis of RPE cell death in response to oxidative stress and observed cardinal features of necrosis in RPE cells upon oxidative stress, including ATP depletion, RIPK3 (receptor-interacting protein kinase 3) aggregation, and nuclear and plasma membrane leakage and breakdown (14). These studies argued against apoptosis and established necrosis as a major mechanism of RPE cell death in response to oxidative stress.
In an effort to screen for U.S. Food and Drug Administration (FDA)-approved natural products and compounds that prevent oxidative stress-induced RPE necrosis, we report here the identification of gossypol acetic acid (GAA) as an effective inhibitor of oxidative stress-induced necrosis in RPE cells. GAA exclusively inhibited the activation of intrinsic necrotic pathway induced by oxidative stress as shown by prevention of ATP depletion and RIPK3 activation. Mechanistically, GAA induced antioxidative response and inhibited reactive oxygen species (ROS) accumulation by upregulating SESN2 gene expression. Through both loss-of-function and gain-of-function studies, we show that SESN2 mediated the protective effect of GAA. Forkhead box O3 transcription factor (FoxO3) was further found to be a major regulator of SESN2 expression in RPE in response to GAA. Our study establishes GAA as a potent inhibitor of oxidative stress-induced RPE necrosis through regulating FoxO3/SESN2 pathway.
MATERIALS AND METHODS
Cell culture and treatments.
Human RPE cell line (ARPE-19, CLR-2302; American Type Culture Collection [ATCC]) was cultured in Dulbecco modified Eagle–F-12 medium (HyClone) supplemented with 10% fetal bovine serum (FBS; HyClone) and 1× penicillin-streptomycin solution (HyClone) at 37°C in 5% CO2. A human dermal fibroblast cell line (HDeF; PCS-201-012, ATCC) was cultured in Dulbecco modified Eagle medium-high glucose (HyClone) supplemented with 10% FBS (HyClone) and 1× penicillin-streptomycin solution (HyClone) at 37°C in 5% CO2. Cells were treated with GAA, gossypol (both dissolved in dimethyl sulfoxide [DMSO]; Sigma-Aldrich), ascorbic acid (dissolved in water; Sigma-Aldrich), or α-tocopherol (Sigma-Aldrich) for 24 h prior induction of oxidative stress, unless stated otherwise. To induce oxidative stress in ARPE-19 cells, the cells were treated with freshly prepared solutions of 300 μM hydrogen peroxide (Sigma-Aldrich) or 150 μM tert-butyl hydroperoxide (tBHP; Sigma-Aldrich) for 24 h. HDeFs were treated with 800 μM hydrogen peroxide (H2O2) for 24 h for oxidative stress induction. For HDeF necrosis induction, cells were treated with 20 μM pan-caspase inhibitor z-VAD (Sigma-Aldrich) and 40 ng of tumor necrosis factor alpha (TNF-α; Sigma-Aldrich)/ml.
Chemical library screening.
ARPE-19 cells were plated in 96-well plate (5,000 cells/well) 24 h prior to screening. Next, cells were treated with 5 μM concentrations of compounds from a library containing 1,840 FDA-approved drugs and natural products (The Spectrum Collection; MicroSource Discovery Systems, Inc.) (15). ARPE-19 cells were then exposed to 150 μM tBHP for 24 h, followed by an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] test to determine cell viability. Positive candidate compounds from the primary screening that showed a minimum of 50% rescue were chosen for repetitive secondary screening. Necrostatin-1 (Enzo Life Sciences) at 33 μM was used as a positive control in the screening.
Generation of SESN2 overexpressing stable RPE cell lines.
Human SESN2 was cloned into pcDNA3 vector (Life Technologies) using RNA from ARPE-19 cells. The primers used were 5′-AAAAAAAAGCTTATGATCGTGGCGGACTCCGAGTG-3′ and 5′-AAAAAAGAATTCTCAGGTCATGTAGCGGGTGAATGG-3′. To generate SESN2 overexpressing stable cell lines, ARPE-19 cells were transfected with pcDNA3-SESN2 vector. Two days after the transfections, the cell culture medium was replaced with a medium containing 800 μg of G418 (Gibco)/ml for 2 weeks to obtain cell colonies. Picked colonies were cultured in 400 μg of G418/ml, which was removed before further experiments.
Cell transfection.
Cell transfection was performed using Lipofectamine LTX (Life Technologies). Briefly, 1 μg of HMGB1-yellow fluorescent protein (YFP), ANT1-red fluorescent protein (RFP), RIPK3-green fluorescent protein (GFP), or pcDNA3-SESN2 plasmid DNA was mixed with 5 μl of Lipofectamine LTX. The complex was added to the ARPE-19 cell cultured in a four-chamber glass slide or six-well plate 20 min later. Expression of the recombinant proteins was visualized after 24 h using a fluorescence microscope. For small interfering RNA (siRNA) transfection, 50 nM siRNA was transfected similarly with 5 μl of Lipofectamine RNAi MAX (Life Technologies). The gene expression level was analyzed by quantitative reverse transcription-PCR (qRT-PCR) after 4 days to ensure efficient gene knockdown. The sequences for the siRNAs for different targeting genes were as follows: SESN2 (sense, 5′-CCUACAAUACCAUCGCCAU-3′; antisense, 5′-AUGGCGAUGGUAUUGUAGG-3′), FoxO3 (sense, 5′-GAAUGAUGGGCUGACUGAA-3′; antisense, 5′-UUCAGUCAGCCCAUCAUUC-3′), FoxO1 (sense, 5′-GAAUUCAAUUCGUCAUAAU-3′; antisense, 5′-AUUAUGACGAAUUGAAUUC-3′), NRF2 (NF-E2-related factor 2; sense, 5′-GACUCUUAUUGGAUACAGU-3′; antisense, 5′-ACUGUAUCCAAUAAGAGUC-3′), and p53 (sense, 5′-GAAGUUGGCUCUGACUGUA-3′; antisense, 5′-UACAGUCAGAGCCAACCUC-3′).
MTT assay, ATP levels, and ROS detection.
Both MTT and ATP level tests were performed as described previously (14). ROS detection was performed using a total ROS/superoxide detection kit (Enzo Life Sciences). In brief, ARPE-19 cells were pretreated with GAA for 24 h before inducing oxidative stress. Cells were incubated with oxidative stress detection reagent for 1 h, washed three times with wash buffer, and then subjected to 300 μM H2O2 treatment for 30 min. After treatment, the growth medium was removed, and the cells were washed three times with wash buffer supplied by the manufacturer, dried briefly, overlaid with mounting medium, and analyzed under a fluorescence microscope. Captured pictures were analyzed using ImageJ software to quantify the mean gray value after background subtraction.
Immunocytochemistry.
SESN2 immunostaining was performed as described previously (16). Briefly, cells were washed three times with 1× phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 30 min at room temperature. Cells were then washed with 1× PBS for three times and permeabilized with 1× PBS containing 0.1% Triton X-100 (Sigma-Aldrich) for 30 min. After blocking with 1× PBS containing 3% horse serum (Gibco) for 30 min, anti-SESN2 antibody (Santa Cruz Biotechnology) at a 1:250 dilution was applied, and the cells were incubated at 4°C overnight. Alexa Fluor 594-conjugated rabbit anti-mouse secondary antibody (Life Technologies) was prepared in blocking buffer, followed by incubation with the cells for 30 min at room temperature. After a washing step with 1× PBS, the cells were mounted with DAPI (4′,6′-diamidino-2-phenylindole)-containing mounting medium for fluorescence (Vector) and analyzed under a fluorescence microscope.
Gene expression.
RNA was purified from ARPE-19 cells using an RNeasy minikit (Qiagen), followed by DNase I treatment (Thermo Scientific), and cDNA was synthesized with an iScript cDNA synthesis kit (Bio-Rad). qRT-PCR was performed by mixing 10 ng of cDNA with SYBR green PCR master mix (Applied Biosystems) and primer mix (0.25 μM concentrations of each primer) and then analyzed over 45 cycles. The primers used are listed in Table S2 in the supplemental material.
ChIP.
A chromatin immunoprecipitation (ChIP) assay was performed as described previously using an EZ-ChIP kit (Millipore) (16). In brief, sheared and cross-linked chromatin was isolated from GAA-treated or control ARPE-19 cells, precleared with protein G-agarose (Roche), and incubated with 10 μg of ChIP-grade antibody for FoxO3A (Abcam). The DNA pulled down was purified by the spin filter purification system supplied by the manufacturer. ChIP samples were analyzed using real-time quantitative PCR. Control primer for the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) promoter was supplied by the manufacturer, and the primers used to amplify FoxO3A binding region in the SESN2 promoter were 5′-CTTATCCACCCTCACCCCTG-3′ and 5′-ACAGTTTTCCTTCAGAGCTCC-3′. The FoxO3A binding site in SESN2 promoter was identified by using Mapper2 software (http://genome.ufl.edu/mapper/).
Western blot analysis.
Western blot analysis was performed using ARPE-19 cells as described previously (17). The antibodies used were as follows: rabbit polyclonal anti-NRF2 (1:500; Santa Cruz Biotechnology), rabbit monoclonal anti-FoxO1 (1:1,000; Cell Signaling), rabbit polyclonal anti-FoxO3A (1:400; Cell Signaling), polyclonal rabbit anti-AKT (1:1,000; Cell Signaling), polyclonal rabbit phospho-AKT (Ser473; 1:1,000; Cell Signaling), mouse monoclonal anti-β-tubulin (1:1,000; Cell Signaling), and monoclonal mouse anti-GAPDH (1:1,000; Millipore). After primary antibody incubation, membranes were probed with IRDye 800CW donkey anti-mouse IgG (LiCor) or IRDye 680RD goat anti-rabbit IgG (LiCor) secondary antibodies and then imaged and quantified using the LiCor Odyssey system.
Statistics.
Each experiment was repeated at least three times. Student t tests were used to determine the statistical significance between groups. P values of <0.05 were considered statistically significant and are annotated by asterisks in the figures.
RESULTS
Screening of FDA-approved compounds for prevention of oxidative stress-induced RPE death.
To identify novel small molecules/compounds that can block oxidative stress-induced ARPE-19 cell death, we conducted a chemical screening of a library containing 1,840 FDA-approved drugs and natural products (15). ARPE-19 cells cultured in 96-well dishes were treated with 5 μM compounds for 24 h, followed by 150 μM tBHP to induce cell death. Cell viability was measured with an MTT test 24 h later (Fig. 1A). Initial screening identified 17 positive hits that showed >50% rescue in cell viability compared to the DMSO control (Fig. 1B). As a result of the secondary screening, three compounds were identified and verified for the ability to protect RPE cells from oxidative stress-induced cell death: 4-acetoxyphenol, ebselen, and gossypol acetic acid (GAA) complex (Fig. 1C). Identification of ebselen indicated the specificity and success of the screening. Ebselen (2-phenyl-1,2-benziselenazol-3[2H]-one), a mimic of glutathione peroxidase, is a strong, selenium-based antioxidant with numerous pharmacologic activities, such as neuroprotection, reducing inflammation, scavenging ROS, and inhibiting lipid oxidation (18). Ebselen has been shown to decrease the level of malondialdehyde in cataractous lenses, and it is being tested as a possible treatment for reperfusion injury and stroke (19). 4-Acetoxyphenol has been shown to be a potent activator of the NRF2-antioxidant response element pathway and is being tested as a possible treatment for neurodegenerative disease (20). Gossypol is a naturally occurring polyphenolic compound derived from cottonseed and was initially identified as an antifertility agent for males (21, 22). Gossypol was identified as a BH3 mimetic and a potent inhibitor of Bcl-XL and, to a lesser extent, of Bcl-2 (23, 24). It induces apoptosis in numerous tumor cells with high Bcl-XL and/or Bcl-2 expression levels, leaving normal cells with low expression levels relatively unaffected (21, 25). GAA is a crystallized form of gossypol (Fig. 2A) (26). It has been shown to be a powerful inhibitor of lipid peroxidation, exhibits pro- and antioxidative behavior, and is currently being tested as an anticancer drug (27, 28) (clinical trial NCT00390403). By screening FDA-approved drugs/natural products library, we have identified compounds that can prevent oxidative stress-induced RPE cell death.
FIG 1.
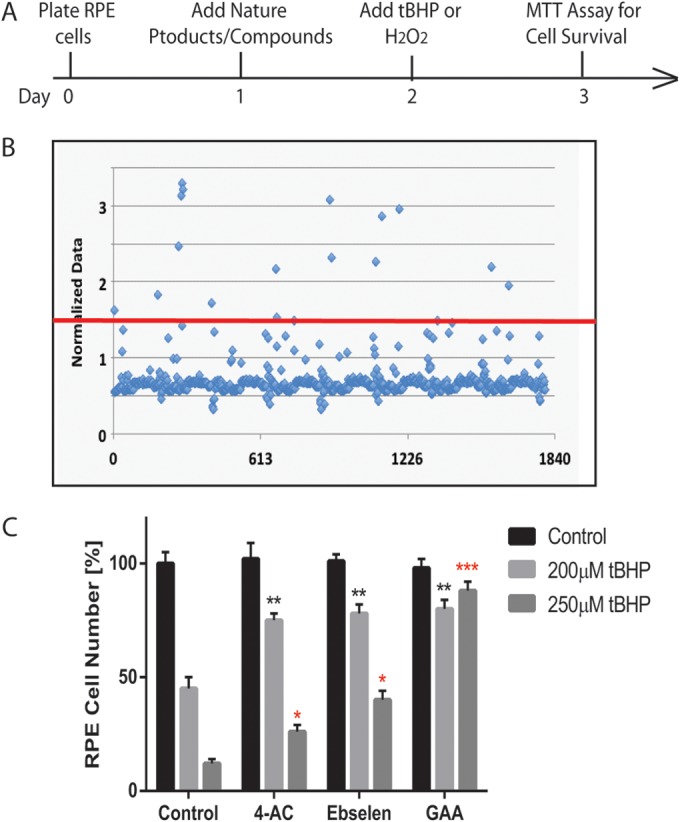
Identification of natural compounds that protect ARPE-19 cells from oxidative stress-induced cell death. (A) Time frame of the screening. (B) Dot graph showing the effect of the compounds in RPE cell survival. The x axis represents compounds screened; the y axis represents cell survival after the induction of oxidative stress. The red line is a cutoff point for the compounds that protected >50% of the cells. (C) Three compounds identified in secondary screening showing protection of RPE death under two different concentrations: 4-acetoxyphenol (4-AC), ebselen, and gossypol acetic acid (GAA). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FIG 2.

GAA protects ARPE-19 cells from oxidative stress-induced cell death. (A) Chemical structure of gossypol acetic acid (GAA). (B) Representative MTT pictures showing GAA protects ARPE-19 cells from H2O2-induced cell death. (C) Comparison of ARPE-19 cell survival upon pretreatment with GAA and then with 300 μM H2O2 or 150 μM tBHP. (D) Comparison of cell protective abilities of gossypol acetic acid (GAA), gossypol (GOS), and acetic acid (AA). Cells treated with 5 μM concentrations of each compound were exposed to H2O2 or tBHP. Cell survival was evaluated by MTT assay. (E) Analysis of GAA protective effect on RPE cells under different GAA concentrations. Cells were pretreated for 24 h with different concentrations of GAA and treated with H2O2. (F) Comparison of GAA protective ability with other established indirect antioxidants: vitamin E (α-tocopherol) and vitamin C (ascorbic acid). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
GAA protects ARPE-19 cell from oxidative stress induced cell death.
To confirm the ability of GAA in protecting ARPE-19 cells from oxidative stress induced by both tBHP and H2O2, ARPE-19 cells were pretreated with 5 μM GAA for 24 h and then exposed to 300 μM H2O2 or 150 μM tBHP. Without the GAA pretreatment, H2O2 and tBHP caused 80% ± 7% and 87% ± 9% decreases, respectively, in cell survival. Pretreatment with GAA prior to oxidative stress significantly increased ARPE-19 cells survival rates to 76% ± 7% and 63% ± 4% for H2O2 and tBHP, respectively (Fig. 2B and C). GAA is a crystalline complex consisting of equimolar quantities of gossypol and acetic acid (29). We sought to determine whether gossypol is sufficient to prevent RPE death induced by H2O2 or tBHP and found that gossypol is comparable to GAA in preventing H2O2- or tBHP-induced RPE cell death. We also tested whether acetic acid itself has protective property and found acetic acid failed to protect RPE cells from H2O2- or tBHP-induced cell death, supporting that gossypol is the active component in GAA for its activity (Fig. 2D). To test whether GAA can directly neutralize oxidants by chemical redox reaction, we added 5 μM GAA to RPE cells at the time of inducing oxidative stress. Under these conditions, GAA was not able to protect RPE cells, suggesting that it is not a direct antioxidant (see Fig. S1 in the supplemental material). We also tested the effect of different concentration of GAA in protecting H2O2-induced RPE death and found that GAA at higher concentrations started to exert toxicity to the RPE cells and that lower concentrations of GAA were less effective in preventing oxidative stress-induced RPE death, whereas ∼5 μM GAA was most effective with a minimum toxic effect (Fig. 2E). These results indicate that the optimal protective activity of GAA biological activity on RPE cells is limited to a low concentration range, which is in contrast to the high concentration that is used to kill cancer cells (30). This is also consistent with the documented hormetic effect of GAA (31). To further characterize the potency of GAA, we compared its activity to other commonly used antioxidants that were included in the Age-Related Eye Disease Studies (AREDS), such as α-tocopherol (vitamin E) and ascorbic acid (vitamin C) (5, 6, 32). Both antioxidants at their published concentrations (100 μM) rescued ARPE-19 at a level similar to that observed for 5 μM GAA. However, when they were used at the same concentration as GAA (5 μM), they both lost the ability to protect ARPE-19 cells from oxidative stress-induced cell death (Fig. 2F). Based on these results, we have identified GAA as a potent inhibitor of RPE cell death in response to oxidative stress.
GAA inhibits the activation of intrinsic necrotic pathway in response to oxidative stress.
We have shown previously that ARPE-19 cells die from necrosis in response to oxidative stress (14). We sought to determine whether GAA protects oxidative stress-induced RPE necrosis. Consistent with our published results, transfected RIPK3-GFP forms discrete punctuation, indicating activation of RIPK3 and induction of necrosis (Fig. 3Aa to c). Signs of RIPK3 activation were not observed in GAA pretreated RPE cells at 1 to 2 h after exposure to 300 μM H2O2 (Fig. 3Ad to f). Increased permeability of nuclear membrane and passive release of high mobility group protein B1 (HMGB1) from the nucleus to the cytoplasm is another hallmark of necrosis (17). When HMGB1-YFP-transfected ARPE-19 cells were treated with 300 μM H2O2, HMGB1 was released to the cytoplasm 2 h later (Fig. 3Ba). However, GAA pretreatment inhibited this process (Fig. 3Bb). GAA also protected mitochondrial clumping resulting from necrosis, as indicated by ANT1-RFP visualization (Fig. 3Bc and d). ATP depletion is considered an early event of necrotic cells. We analyzed the effect of GAA on intracellular ATP level in cells exposed to oxidative stress. ARPE-19 cells exposed to H2O2 showed drastic ATP decreases within the first 3 h. GAA pretreatment only mildly decreased cellular ATP levels when cells were treated with H2O2, supporting the view that GAA prevents oxidative stress-induced RPE necrosis (Fig. 3C). Similar to apoptosis, necrosis can be triggered by intrinsic or extrinsic stimuli. We sought to determine whether GAA prevents necrosis induced by extrinsic stimuli. ARPE-19 cells are insensitive to TNF-α-induced cell death (33). We therefore used human dermal fibroblasts (HDeFs) that are sensitive to TNF-α-induced necrosis in the presence of pan-caspase inhibitor z-Vad. Pretreatment with GAA did not protect HDeFs from TNF-α-induced necrosis, although it protected HDeFs from oxidative stress-induced cell death (Fig. 3D and E). To conclude, GAA prevents the activation of the intrinsic necrotic pathway in RPE cells in response to oxidative stress.
FIG 3.

GAA inhibits induction of necrosis by oxidative stress in ARPE-19 cells. (A) Induction of RIPK3 activation H2O2, as visualized by the distinct punctuations of transfected RIPK3-GFP reporter (a to c), which was inhibited by GAA pretreatment (d to f). Bar, 25 μm. (B) GAA pretreatment inhibited H2O2-induced HMGB1 passive release to cytoplasm as visualized by transfected HMGB1-YFP reporter (a and b) and mitochondrial clump formation as visualized by transfected ANT1-RFP reporter (c and d). (C) H2O2 rapidly inhibited ATP production in RPE cells, which was rescued by GAA pretreatment. (D) Extrinsic necrotic pathway triggered by z-Vad pretreatment and TNF-α stimulation was not inhibited by GAA pretreatment in HDeF cells. (E) GAA pretreatment prevented H2O2-induced in HDeF cell death. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns or N.S., not significant.
GAA acts as an inducer of sestrin2 (SENS2) expression.
GAA has been shown to have pro- and antioxidative properties (27). To define the mechanism through which GAA protects ARPE-19 cells from oxidative stress-induced necrosis, we first tested whether GAA affects the level of cellular ROS upon oxidative stress. Exposure to 300 μM H2O2 for 30 min increased the ROS level in ARPE-19 by ∼2-fold, as shown by ROS staining (Fig. 4A) and subsequent quantification (Fig. 4B). However, GAA pretreatment blunted the increase in the ROS level induced by H2O2.
FIG 4.

SESN2 is required for regulating ROS level and RPE survival by GAA. (A) Inhibition of intracellular ROS accumulation in response to oxidative stress by GAA pretreatment. ROS green fluorescence was analyzed under a fluorescence microscope at 488 nm. Bar, 25 μm. (B) Quantification of the ROS accumulation in panel A. (C) Regulation of SESN expression level by 24 h pretreatment with 5 μM GAA and/or H2O2 exposure, as measured by qRT-PCR. (D) Regulation of SESN2 protein level by pretreatment with GAA and/or H2O2 exposure visualized by SESN2 immunostaining. Bar, 25 μm. (E) qRT-PCR showing knockdown of SESN2 by a specific siRNA. (F) SESN2 knockdown abolished the ability of GAA to protect RPE cells from necrosis induced by 300 μM H2O2 as analyzed by MTT assay. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Having established that GAA blunts ROS increase induced by oxidative stress, we sought to determine further whether GAA regulates the expression of genes that may regulate the ROS level. We screened a panel of oxidative stress-related genes by qRT-PCR (see Table S1 in the supplemental material). ARPE-19 cells were pretreated with GAA and then exposed to 300 μM H2O2, and RNA samples were collected 30 min later. As determined by qRT-PCR, SESN2 expression was significantly upregulated in cells pretreated with GAA alone and was further upregulated when GAA-pretreated cells were exposed to H2O2 (Fig. 4C).
The sestrin (SESN) family of proteins was originally discovered as targets of p53 protein (34). Together with sulfiredoxins (Srx), SESNs function to repair overoxidized peroxiredoxins (Prx, a family of antioxidant enzymes that controls cytokine-induced peroxide levels) (35). There are three isoforms of SESN proteins: SESN1, SESN2, and SESN3, which are expressed ubiquitously in all adult tissues, although to different extents (36). SESN2 was identified as a stress-responsive gene involved in the regulation of cell viability (37) in response to prolonged hypoxia, DNA-damaging treatments (gamma or UV irradiation, doxorubicin), or oxidative stress (34). SESN1 and SESN2 overexpression has been shown to decrease level of ROS after H2O2 treatment by substantially increasing the recovery rate of the overoxidized Prx (38). Among the SESN family members, the expression of SESN1 and SESN3 was not upregulated by GAA (Fig. 4C). The upregulation of SESN2 protein was also confirmed by immunostaining (Fig. 4D).
To test whether SESN2 is required to mediate GAA function in protecting RPE cells from oxidative stress-induced necrosis, RPE cell survival was analyzed after SESN2 knockdown by siRNA transfection and H2O2 treatment. SESN2 silencing was confirmed by qRT-PCR (Fig. 4E). As shown in Fig. 4F, SESN2 knockdown did not affect RPE survival at baseline. However, SESN2 silencing rendered RPE cells susceptive to oxidative stress induced by H2O2. About 80% of the cells died when the SESN2-knockdown cells were subjected to H2O2 treatment. Moreover, the protective effect of GAA in oxidative-stress induced RPE death was lost when SESN2 was silenced. Taken together, these results establish a critical function for SESN2 in protecting RPE cells from oxidative stress-induced necrosis and for GAA as an effective inducer of SESN2 activity.
SESN2 regulates intracellular ROS level and protects RPE cells from oxidative stress-induced cell death.
We asked further whether SESN2 overexpression can mimic the effect of GAA in RPE necrosis. To do so, SESN2 expression plasmid was transfected into ARPE-19 cells, and the SESN2-overexpressing cells were selected by using G418. Overexpression of SESN2 in RPE cells was confirmed by immunostaining (Fig. 5A). Overexpression of SESN2 in RPE cell did not affect cell survival at baseline. However, overexpression of SESN2 itself rescued up to 93% of the cells when RPE cells were treated with H2O2, significantly more than treatment with GAA itself (Fig. 5B).
FIG 5.
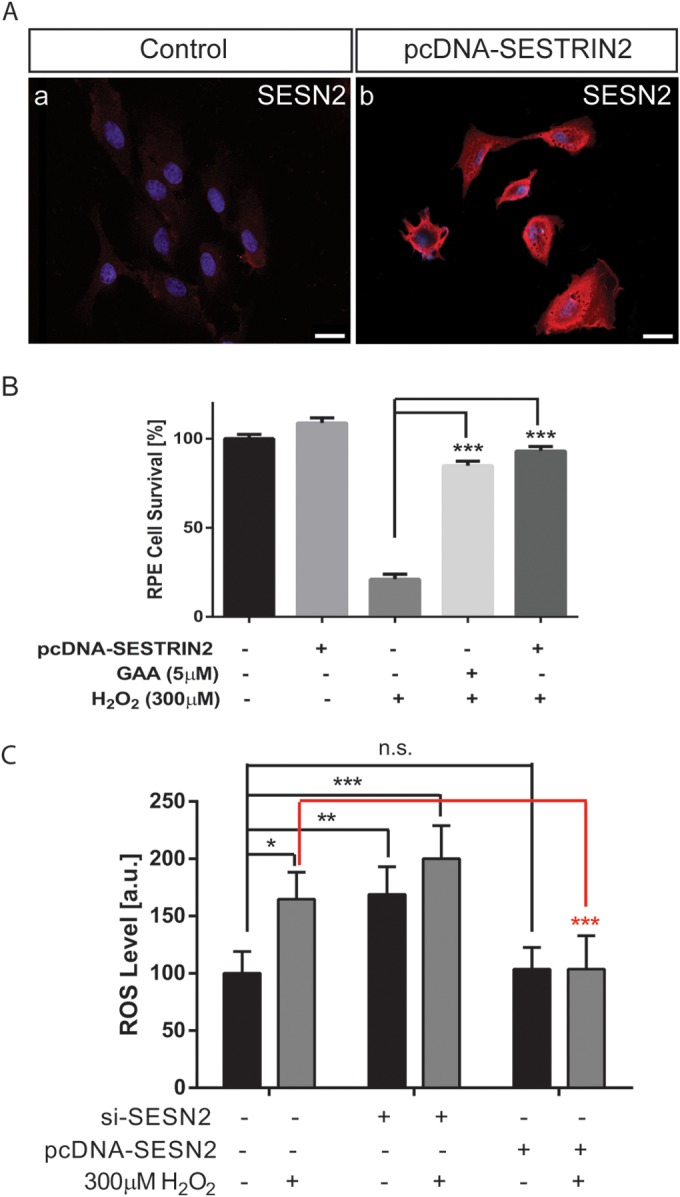
SESN2 prevents RPE ROS accumulation and promotes RPE survival upon oxidative stress. (A) SESN2 protein expression as visualized by immunostaining in nontransfection control cells (a) compared to a SESN2 overexpressing RPE stable cell line (b). Bars, 25 μm. (B) SESN2 overexpression showed comparable protective capability in RPE cells treated with H2O2 to GAA, as revealed by an MTT assay. (C) Effect of SESN2 overexpression or knockdown on ROS accumulation in control and 300 μM H2O2-treated RPE cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant.
We further examined whether SESN2 is responsible for the ROS level changes regulated by GAA in RPE cells. RPE cellular ROS level was analyzed after SESN2 knockdown or overexpression in RPE cells. In the control cells, H2O2 (300 μM) increased ROS level significantly at 30 min after treatment (Fig. 5C). Downregulation of SESN2 alone resulted in significantly increased ROS level in the control cells, which was further increased, although insignificantly, by H2O2 treatment. However, although SESN2 upregulation did not affect RPE ROS level at baseline, it blunted the ROS level increase in response to H2O2 treatment. Altogether, our results establish that GAA is an effective inducer of SESN2 activity, which effectively prevents cell killing activity induced by elevated cellular ROS level.
Requirement for FoxO3 in SESN2 expression and GAA-mediated RPE survival in response to oxidative stress.
To further determine the regulation mechanism of SESN2 by GAA, we focused on NRF2, p53, and FoxO transcription factors, which have been independently shown to control SESN2 gene expression (34, 38–41). To analyze the role of p53 in mediating GAA induced expression of SESN2, p53 was silenced by specific siRNA. The knockdown of p53 and its target gene p21 was confirmed by qRT-PCR (Fig. 6A). p53 knockdown did not affect the ability of GAA to upregulate SESN2 expression with or without H2O2 treatment (Fig. 6B). When RPE cell survival was analyzed, p53 knockdown did not significantly affect the ability of GAA to protect RPE cells from oxidative stress-induced necrosis or RPE cell survival under basal conditions or under H2O2 treatment (Fig. 6C). This suggests that p53 is not a major player regulating SESN2 expression in response to GAA. Consistent with these results, p21 promoter activity that is responsive to p53 was not affected by H2O2 and/or GAA treatment based on luciferase assay (see Fig. S2 in the supplemental material) (42). To test the involvement of NRF2 in mediating SESN2 expression by GAA, a specific siRNA to NRF2 was used to knockdown NRF2 expression in RPE cells. Knockdown of NRF2 and its target gene NQO1 were confirmed by qRT-PCR (Fig. 6D) and by Western blot with anti-NRF2 antibody (Fig. 6E). NRF2 knockdown failed to reduce SESN2 expression in response to GAA with or without H2O2 treatment (Fig. 6F). When RPE survival was measured, NRF2 knockdown slightly reduced RPE survival at baseline. Upon H2O2 treatment, RPE survival was not significantly changed in si-NRF2-treated samples compared to the control samples. Moreover, with NRF knockdown, GAA still rescued similar percentage of cells as in control samples (Fig. 6G). These data argue against NRF2 as a major regulator of SESN2 expression in RPE cells in response to GAA. We then hypothesized the FoxO genes may be responsible for SESN2 regulation in RPE cells in response to GAA. By qRT-PCR, FoxO1 and FoxO3 are the major members of FoxO genes expressed in the RPE cells (see Fig. S3 in the supplemental material). When FoxO3 was knocked down in RPE cells, SESN2 expression was dramatically reduced with or without GAA or H2O2 treatment (Fig. 7A and B). Accordingly, upon FoxO3 knockdown, RPE survival was significantly reduced at baseline, and GAA no longer protected RPE survival in response to H2O2 treatment (Fig. 7C). We also independently silenced FoxO1 or FoxO4 in RPE cells and found FoxO1 knockdown did not significantly affect SESN2 upregulation but blunted the protective response of GAA in response to H2O2 (see Fig. S4 in the supplemental material). When FoxO4 was knocked down, upregulation of SESN2 by GAA was decreased, but GAA still significantly protected oxidative stress-induced RPE cell death. These suggest differential mechanisms for FoxO genes in regulating SESN2 expression and GAA response.
FIG 6.

Analysis of the p53 and NRF2 requirement to regulate GAA-mediated SESN2 expression in RPE cells. (A) Knockdown of p53 and downregulation of its target gene p21 by specific siRNA to p53, as analyzed by qRT-PCR. (B) SESN2 expression level measured by qRT-PCR after pretreatment with GAA in p53-knockdown cells. (C) Knockdown of p53 did not affect the ability of GAA to protect RPE cells from oxidative stress-induced cell death. (D) NRF2 knockdown and downregulation its target gene NQO1 by a specific siRNA to NRF2, as shown by qRT-PCR. (E) Confirmation of NRF2 protein knockdown by siRNA, as determined by Western blotting. (F) NRF2 knockdown did not affect SESN2 upregulation by GAA-H2O2, as measured by qRT-PCR. (G) NRF2 knockdown did not affect GAA in protecting ability to protecting H2O2-iduced RPE cell death. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FIG 7.

FoxO3 is required to mediate SESN2 expression and RPE survival upon oxidative stress. (A) FoxO3 knockdown as measured by qRT-PCR. (B) Repression of SENS2 expression by FoxO3 knockdown in RPE cells with or without 5 μM GAA treatment for 24 h and exposure to 300 μM H2O2. (C) FoxO3 knockdown reduced RPE viability at baseline and abolished the ability of GAA in protecting oxidative stress-induced RPE death. (D) Subcellular localization of FoxO3 was investigated by immunostaining after pretreatment with 5 mM GAA and with or without the induction of oxidative stress with 300 mM H2O2 for 30 min. (E) Diagram of SESN2 enhancer highlighting the FoxO binding site. The conserved sequence was highlighted in black, and the approximate location of primers for PCR is indicated by a pair of arrows. (F) ChIP-qPCR analysis of FoxO3A binding to FoxO binding site in the enhancer region of SESN2. RPE cells were pretreated with 5 μM GAA and/or treated with 300 μM H2O2. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Having established that silencing of FoxO genes, especially FoxO3 gene, prevented GAA-mediated SESN2 upregulation, we tested further the mechanism how FoxO3 regulates SESN2 expression. First, we examined the FoxO3 subcellular localization in response to GAA and H2O2 treatment. RPE cells were transfected with FLAG-FoxO3A expression plasmid and pretreated them for 24 h with 5 μM GAA with or without 300 μM H2O2 treatment (43). In nontreated cells FoxO3A is localized mainly in the cytoplasm (Fig. 7Da). Pretreatment with GAA for 24 h induced translocation of the majority of FoxO3A to the nucleus regardless of H2O2 treatment (Fig. 7Db and d). Treatment with 300 μM H2O2 alone for 30 min only induced the nuclear transport of a very small portion of FoxO3A (Fig. 7Dc). To further test whether FoxO3 binds to the SESN2 enhancer, we identified the FoxO binding site in the enhancer region of SESN2 gene and performed a ChIP assay to test the direct binding of FoxO3 to SESN2 enhancer (Fig. 7E). As shown by qRT-PCR with primers specific to the FoxO binding site, pretreatment with GAA with or without H2O2 significantly enhanced the binding of FoxO3A to SESN2 enhancer (Fig. 7F), indicating that FoxO3 directly binds to the SESN2 regulatory region to enhance SESN2 expression. Taken together, our data indicate that GAA regulates the FoxO/SESN2 pathway to prevent RPE necrosis in response to H2O2.
DISCUSSION
Inhibition of oxidative stress-induced RPE death represents a viable approach for treating dry AMD and GA. By screening a library of FDA-approved natural products and compounds, we identified GAA as a potent natural compound that protects ARPE-19 cells from oxidative stress-induced necrosis. GAA protects up to 80% of ARPE-19 cells from death when they are exposed to 300 μM H2O2. Mechanistically, GAA prevents the activation of intrinsic necrotic pathway induced by oxidative stress but not extrinsic necrotic pathway induced by TNF-α, likely due to its inhibition of ROS accumulation in cells exposed to oxidative stress. SESN2 was identified as an effector for GAA. SESN2 regulates RPE ROS level and GAA-mediated RPE survival in response to oxidative stress. Furthermore, FoxO3 transcription factor shuttles to the nucleus in response to GAA and is critical for SESN2 regulation and RPE survival.
GAA possesses potent antioxidative property in RPE cells.
By screening a FDA-approved chemicals and natural compounds, we identified GAA as a potent inducer of antioxidant response that prevents oxidative stress-induced RPE necrosis. We found that GAA is not toxic to RPE cells when used at 5 μM, a concentration 10 times lower than that used in cancer cells. Our findings that both gossypol and GAA protects RPE from oxidative stress-induced RPE necrosis seem to be paradoxical compared to the previous studies showing that they can kill Bcl2 overexpressed cancer cells, but these are consistent with the documented hormetic effect of gossypol (31). GAA was shown to be powerful inhibitor of lipid peroxidation and exhibits pro- and antioxidant behavior (27). We found that GAA does not affect ROS production at the baseline, and it does not directly neutralize the oxidants. However, GAA pretreatment blunts ROS level increase induced by oxidative stress. Additionally, we established that GAA is a more potent in protecting RPE cells from H2O2-induced oxidative stress than well-established antioxidants such as vitamin C and E, the main ingredients for the AREDS studies that have been shown to slow the progression of dry AMD. We found that GAA prevents oxidative stress-induced RPE death when it is used at a concentration that is 20 times lower than vitamin C or vitamin E, suggesting potent antioxidative property of GAA in RPE cells.
Probing oxidative stress and intrinsic necrotic pathway.
RPE cells are postmitotic cells and have limited regeneration potential and restricted apoptotic potential (44, 45). Consistent with its low caspase-8 level, we have recently established that RPE cells die from necrosis rather than apoptosis in response to oxidative stress (14, 33). Necrotic pathways can be classified as an extrinsic necrotic pathway that is stimulated by external stimuli through receptors and intrinsic necrotic pathway that is not triggered through the cell surface receptors. The mechanism that triggers intrinsic necrotic pathway is still unclear. RPE cells treated with oxidative stress represent an ideal model to study the mechanism of intrinsic necrotic pathway. The compound GAA from our screening that prevents oxidative stress-induced RPE death provides an excellent opportunity to probe the intrinsic necrosis process. We found the blunted ROS increase by GAA correlates with the lack of activation of necrosis, which is evidenced by the absence of RIPK3 activation, HMGB1 release, or ATP depletion. This places the ROS increase upstream of the initiation of intrinsic necrotic pathway. Interestingly, GAA only inhibits intrinsic necrotic pathway induced by oxidative stress but not extrinsic pathway triggered by TNF-α/z-Vad treatment, highlighting the different mechanism between intrinsic and extrinsic necrotic pathways. How the increased ROS level leads to RIPK3 activation and necrosome formation warrants future investigations.
FoxO3/SESN2 axis in RPE survival in response to oxidative stress.
Regarding the mechanism of GAA in preventing oxidative stress-induced RPE cell death, we found that SESN2, but not SESN1 or SESN3, is upregulated by GAA. SESNs act to repair overoxidized Prxs, therefore regenerating functional Prxs (38). We found that silencing of SESN2 blunts the protective effect of GAA in RPE cells in response to oxidative stress, while overexpression of SESN2 shows outstanding capability in protecting RPE cells from oxidative stress-induced death. These results are consistent with increased ROS level upon SESN2 knockdown and the blunted ROS increase in response to oxidative stress upon SESN2 overexpression. Similar studies in macrophages have shown that upregulation of SESN2 protects against H2O2-induced Prx overoxidation (38, 46). In the nervous system SESN2 has been found to control ROS-dependent neuropathic pain signaling after peripheral nerve injury (47) and putatively p53-mediated antioxidant function in the retina (48). SESN2 is also needed for resveratrol, an active component in red wine, to inhibit LXRα-mediated hepatic lipogenesis (49). Particularly in Drosophila, SESN2 has been shown to prevent age-related pathology (50).
p53, FoxO, and NRF2 transcription factors have been independently shown to regulate SESN2 expression. We found that FoxO3 is required for SESN2 expression in RPE cells and mediates the regulation of SESN2 expression by GAA. Moreover, FoxO3 is required for RPE survival at baseline and also GAA-mediated RPE survival in response to oxidative stress. This establishes the importance of FoxO3/SESN2 axis in regulating RPE survival in response to oxidative stress. FOXO3a has been shown to be involved in protection from oxidative stress by upregulating antioxidants such as catalase and manganese superoxide dismutase (51). Variants of FoxO3 have been associated with longevity in humans (52). FoxO3 homologous genes, including daf-16 in the nematode Caenorhabditis elegans and dFOXO in the fruit fly, are also associated with longevity in those organisms (53, 54). Mechanistically, we showed that GAA promotes FoxO3 nuclear translocation and binding to the SESN2 enhancer, which in turn increases its transcriptional activity. This is consistent with the in silico analysis showing the existence of FoxO-binding site in the proximate enhancer of SESN2 gene. The mechanism how GAA controls FoxO3 nuclear transport is still unclear, but phosphorylation by phosphatidylinositol-3 kinase/AKT has been shown to control FoxO cytoplasmic-nuclear shuttling and its activity (55, 56). Taken together, our findings show that in RPE cells GAA induces nuclear translocation of FoxO3a, which in turn increases SESN2 expression and protection against oxidative stress-induced necrosis.
Therapeutic implications.
We established that GAA, an FDA-approved natural compound, is a potent inducer of antioxidative response and protects RPE cells from oxidative stress-induced necrosis. The action of GAA reflects its regulation of FoxO3/SESN2 pathway. These findings have important therapeutic implications. Oxidative stress has been suggested to be a critical component of pathology associated with age-related diseases, especially AMD (57). GAA, as an FDA-approved natural compound, can readily be repurposed for the therapy of dry AMD and age-related diseases. We also showed that SESN2 overexpression has remarkable capability in protecting RPE death in response to oxidative stress. SESN2, as well as FoxO3, may serve as an ideal therapeutic target for drug screening for AMD and other age-related diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alexander Kolkin for help with the experiments. We thank Hua Lu from Tulane University for providing p21-luc plasmid. We thank Yiping Chen for critical comments on the manuscript and all of the lab members for discussions.
S.W. was supported by a Startup fund from Tulane University, National Institutes of Health grant EY021862, a career development award from Research to Prevent Blindness Foundation, and a Bright Focus Foundation Award in Age-Related Macular Degeneration. D.H.C. was a summer research student from the University of Southern California.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00178-15.
REFERENCES
- 1.Damico FM, Gasparin F, Scolari MR, Pedral LS, Takahashi BS. 2012. New approaches and potential treatments for dry age-related macular degeneration. Arq Bras Oftalmol 75:71–76. doi: 10.1590/S0004-27492012000100016. [DOI] [PubMed] [Google Scholar]
- 2.Bok D. 2005. Evidence for an inflammatory process in age-related macular degeneration gains new support. Proc Natl Acad Sci U S A 102:7053–7054. doi: 10.1073/pnas.0502819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambati J, Fowler BJ. 2012. Mechanisms of age-related macular degeneration. Neuron 75:26–39. doi: 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, Kariko K, Yoo JW, Lee DK, Hadziahmetovic M, Song Y, Misra S, Chaudhuri G, Buaas FW, Braun RE, Hinton DR, Zhang Q, Grossniklaus HE, Provis JM, Madigan MC, Milam AH, Justice NL, Albuquerque RJ, Blandford AD, Bogdanovich S, Hirano Y, Witta J, Fuchs E, Littman DR, Ambati BK, Rudin CM, Chong MM, Provost P, Kugel JF, Goodrich JA, Dunaief JL, Baffi JZ, Ambati J. 2011. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471:325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Age-Related Eye Disease Study 2 Research Group. 2013. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 309:2005–2015. doi: 10.1001/jama.2013.4997. [DOI] [PubMed] [Google Scholar]
- 6.Age-Related Eye Disease Study Research Group. 2001. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beatty S, Koh H, Phil M, Henson D, Boulton M. 2000. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Survey Ophthalmol 45:115–134. doi: 10.1016/S0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 8.Khandhadia S, Lotery A. 2010. Oxidation and age-related macular degeneration: insights from molecular biology. Expert Rev Mol Med 12:e34. doi: 10.1017/S146239941000164X. [DOI] [PubMed] [Google Scholar]
- 9.Li GY, Fan B, Zheng YC. 2010. Calcium overload is a critical step in programmed necrosis of ARPE-19 cells induced by high-concentration H2O2. Biomed Environ Sci 23:371–377. doi: 10.1016/S0895-3988(10)60078-5. [DOI] [PubMed] [Google Scholar]
- 10.Kim MH, Chung J, Yang JW, Chung SM, Kwag NH, Yoo JS. 2003. Hydrogen peroxide-induced cell death in a human retinal pigment epithelial cell line, ARPE-19. Korean J Ophthalmol 17:19–28. doi: 10.3341/kjo.2003.17.1.19. [DOI] [PubMed] [Google Scholar]
- 11.Hashizume K, Hirasawa M, Imamura Y, Noda S, Shimizu T, Shinoda K, Kurihara T, Noda K, Ozawa Y, Ishida S, Miyake Y, Shirasawa T, Tsubota K. 2008. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am J Pathol 172:1325–1331. doi: 10.2353/ajpath.2008.070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollyfield JG, Perez VL, Salomon RG. 2010. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol 41:290–298. doi: 10.1007/s12035-010-8110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christofferson DE, Yuan J. 2010. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanus J, Zhang H, Wang Z, Liu Q, Zhou Q, Wang S. 2013. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis 4:e965. doi: 10.1038/cddis.2013.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qurashi A, Liu H, Ray L, Nelson DL, Duan R, Jin P. 2012. Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum Mol Genet 21:2068–2075. doi: 10.1093/hmg/dds024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Zhou Q, Hanus J, Anderson C, Zhang H, Dellinger M, Brekken R, Wang S. 2013. Inhibition of multiple pathogenic pathways by histone deacetylase inhibitor SAHA in a corneal alkali-burn injury model. Mol Pharmaceutics 10:307–318. doi: 10.1021/mp300445a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Jiang H, Chen S, Du F, Wang X. 2012. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 18.Matsushita T, Fukuda K, Yamamoto H, Yamazaki K, Tomiyama T, Oh M, Hamanishi C. 2004. Effect of ebselen, a scavenger of reactive oxygen species, on chondrocyte metabolism. Mod Rheumatol 14:25–30. doi: 10.3109/s10165-003-0261-6. [DOI] [PubMed] [Google Scholar]
- 19.Parnham M, Sies H. 2000. Ebselen: prospective therapy for cerebral ischaemia. Expert Opin Invest Drugs 9:607–619. doi: 10.1517/13543784.9.3.607. [DOI] [PubMed] [Google Scholar]
- 20.Shaw P, Mead R, Higginbottom A, Barber S. October 2009. Therapeutics for neurological disorders. US patent application 13/125,097.
- 21.Zerp SF, Stoter R, Kuipers G, Yang D, Lippman ME, van Blitterswijk WJ, Bartelink H, Rooswinkel R, Lafleur V, Verheij M. 2009. AT-101, a small molecule inhibitor of antiapoptotic Bcl-2 family members, activates the SAPK/JNK pathway and enhances radiation-induced apoptosis. Radiat Oncol 4:47. doi: 10.1186/1748-717X-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalla NR, Vasudev M. 1980. Studies on the male antifertility agent gossypol acetic acid: in vitro studies on the effect of gossypol acetic acid on human spermatozoa. IRCS J Med Sci 8:375–376. [PubMed] [Google Scholar]
- 23.Yeow WS, Baras A, Chua A, Nguyen DM, Sehgal SS, Schrump DS, Nguyen DM. 2006. Gossypol, a phytochemical with BH3-mimetic property, sensitizes cultured thoracic cancer cells to Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Thorac Cardiovasc Surg 132:1356–1362. doi: 10.1016/j.jtcvs.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 24.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. 2003. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem 46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 25.Oliver CL, Bauer JA, Wolter KG, Ubell ML, Narayan A, O'Connell KM, Fisher SG, Wang S, Wu X, Ji M, Carey TE, Bradford CR. 2004. In vitro effects of the BH3 mimetic, (−)-gossypol, on head and neck squamous cell carcinoma cells. Clin Cancer Res 10:7757–7763. doi: 10.1158/1078-0432.CCR-04-0551. [DOI] [PubMed] [Google Scholar]
- 26.Pons WA, Pominski J, King WH. November 1962. Process for recovery of gossypol from cottonseed gums. US patent 3,062,876.
- 27.El-Sharaky AS, Wahby MM, Bader El-Dein MM, Fawzy RA, El-Shahawy IN. 2009. Mutual anti-oxidative effect of gossypol acetic acid and gossypol-iron complex on hepatic lipid peroxidation in male rats. Food Chem Toxicol 47:2735–2741. doi: 10.1016/j.fct.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Van Poznak C, Seidman AD, Reidenberg MM, Moasser MM, Sklarin N, Van Zee K, Borgen P, Gollub M, Bacotti D, Yao TJ, Bloch R, Ligueros M, Sonenberg M, Norton L, Hudis C. 2001. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: a phase I/II clinical trial. Breast Cancer Res Treat 66:239–248. doi: 10.1023/A:1010686204736. [DOI] [PubMed] [Google Scholar]
- 29.Dowd MK, Pelitire SM. 2001. Recovery of gossypol acetic acid from cottonseed soapstock. Industrial Crops Products 14:113–123. doi: 10.1016/S0926-6690(00)00094-7. [DOI] [Google Scholar]
- 30.Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ, Cohen GM. 2009. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ 16:1030–1039. doi: 10.1038/cdd.2009.48. [DOI] [PubMed] [Google Scholar]
- 31.Celorio-Mancera Mde L, Ahn SJ, Vogel H, Heckel DG. 2011. Transcriptional responses underlying the hormetic and detrimental effects of the plant secondary metabolite gossypol on the generalist herbivore Helicoverpa armigera. BMC Genomics 12:575. doi: 10.1186/1471-2164-12-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanneken A, Lin FF, Johnson J, Maher P. 2006. Flavonoids protect human retinal pigment epithelial cells from oxidative-stress-induced death. Investig Ophthalmol Vis Sci 47:3164–3177. doi: 10.1167/iovs.04-1369. [DOI] [PubMed] [Google Scholar]
- 33.Yang P, Peairs JJ, Tano R, Zhang N, Tyrell J, Jaffe GJ. 2007. Caspase-8-mediated apoptosis in human RPE cells. Investig Ophthalmol Vis Sci 48:3341–3349. doi: 10.1167/iovs.06-1340. [DOI] [PubMed] [Google Scholar]
- 34.Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, Skaliter R, Gudkov AV, Chumakov PM, Feinstein E. 2002. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 21:6017–6031. doi: 10.1038/sj.onc.1205877. [DOI] [PubMed] [Google Scholar]
- 35.Jonsson TJ, Lowther WT. 2007. The peroxiredoxin repair proteins. Subcell Biochem 44:115–141. doi: 10.1007/978-1-4020-6051-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchis-Gomar F. 2013. Sestrins: novel antioxidant and AMPK-modulating functions regulated by exercise? J Cell Physiol 228:1647–1650. doi: 10.1002/jcp.24338. [DOI] [PubMed] [Google Scholar]
- 37.Peeters H, Debeer P, Bairoch A, Wilquet V, Huysmans C, Parthoens E, Fryns JP, Gewillig M, Nakamura Y, Niikawa N, Van de Ven W, Devriendt K. 2003. PA26 is a candidate gene for heterotaxia in humans: identification of a novel PA26-related gene family in human and mouse. Hum Genet 112:573–580. doi: 10.1007/s00439-003-0917-5. [DOI] [PubMed] [Google Scholar]
- 38.Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. 2004. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- 39.Budanov AV, Karin M. 2008. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greer EL, Brunet A. 2005. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 41.Shin BY, Jin SH, Cho IJ, Ki SH. 2012. Nrf2-ARE pathway regulates induction of sestrin-2 expression. Free Radic Biol Med 53:834–841. doi: 10.1016/j.freeradbiomed.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 42.Liao JM, Lu H. 2013. ChIP for identification of p53 responsive DNA promoters. Methods Mol Biol 962:201–210. doi: 10.1007/978-1-62703-236-0_17. [DOI] [PubMed] [Google Scholar]
- 43.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. 2004. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 44.Kaldarar-Pedotti S. 1979. Mitotic activity of the pigment epithelium during embryonic and postembryonic development. Adv Ophthalmol 39:37–58. (In German.) [PubMed] [Google Scholar]
- 45.Wright KM, Deshmukh M. 2006. Restricting apoptosis for postmitotic cell survival and its relevance to cancer. Cell Cycle 5:1616–1620. doi: 10.4161/cc.5.15.3129. [DOI] [PubMed] [Google Scholar]
- 46.Essler S, Dehne N, Brune B. 2009. Role of sestrin2 in peroxide signaling in macrophages. FEBS Lett 583:3531–3535. doi: 10.1016/j.febslet.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 47.Kallenborn-Gerhardt W, Lu R, Syhr KM, Heidler J, von Melchner H, Geisslinger G, Bangsow T, Schmidtko A. 2013. Antioxidant activity of sestrin 2 controls neuropathic pain after peripheral nerve injury. Antioxid Redox Signal 19:2013–2023. doi: 10.1089/ars.2012.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Connor JC, Wallace DM, O'Brien CJ, Cotter TG. 2008. A novel antioxidant function for the tumor-suppressor gene p53 in the retinal ganglion cell. Investig Ophthalmol Vis Sci 49:4237–4244. doi: 10.1167/iovs.08-1963. [DOI] [PubMed] [Google Scholar]
- 49.Jin SH, Yang JH, Shin BY, Seo K, Shin SM, Cho IJ, Ki SH. 2013. Resveratrol inhibits LXRα-dependent hepatic lipogenesis through novel antioxidant sestrin2 gene induction. Toxicol Appl Pharmacol 271:95–105. doi: 10.1016/j.taap.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 50.Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, Karin M. 2010. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327:1223–1228. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. 2002. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 52.Rizki G, Iwata TN, Li J, Riedel CG, Picard CL, Jan M, Murphy CT, Lee SS. 2011. The evolutionarily conserved longevity determinants HCF-1 and SIR-2.1/SIRT1 collaborate to regulate DAF-16/FOXO. PLoS Genet 7:e1002235. doi: 10.1371/journal.pgen.1002235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. 2004. Drosophila dFOXO controls lifespan and regulates insulin signaling in brain and fat body. Nature 429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 54.Honda Y, Honda S. 1999. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13:1385–1393. [PubMed] [Google Scholar]
- 55.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 56.Clavel S, Siffroi-Fernandez S, Coldefy AS, Boulukos K, Pisani DF, Derijard B. 2010. Regulation of the intracellular localization of Foxo3a by stress-activated protein kinase signaling pathways in skeletal muscle cells. Mol Cell Biol 30:470–480. doi: 10.1128/MCB.00666-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai X, McGinnis JF. 2012. Oxidative stress: the achilles' heel of neurodegenerative diseases of the retina. Front Biosci 17:1976–1995. doi: 10.2741/4033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.