

Abstract
Defensins are a class of ubiquitously expressed cationic antimicrobial peptides (CAPs) that play an important role in innate defense. Plant defensins are active against a broad range of microbial pathogens and act via multiple mechanisms, including cell membrane permeabilization. The cytolytic activity of defensins has been proposed to involve interaction with specific lipid components in the target cell wall or membrane and defensin oligomerization. Indeed, the defensin Nicotiana alata defensin 1 (NaD1) binds to a broad range of membrane phosphatidylinositol phosphates and forms an oligomeric complex with phosphatidylinositol (4,5)-bisphosphate (PIP2) that facilitates membrane lysis of both mammalian tumor and fungal cells. Here, we report that the tomato defensin TPP3 has a unique lipid binding profile that is specific for PIP2 with which it forms an oligomeric complex that is critical for cytolytic activity. Structural characterization of TPP3 by X-ray crystallography and site-directed mutagenesis demonstrated that it forms a dimer in a “cationic grip” conformation that specifically accommodates the head group of PIP2 to mediate cooperative higher-order oligomerization and subsequent membrane permeabilization. These findings suggest that certain plant defensins are innate immune receptors for phospholipids and adopt conserved dimeric configurations to mediate PIP2 binding and membrane permeabilization. This mechanism of innate defense may be conserved across defensins from different species.
INTRODUCTION
Plant defensins are small (∼5 kDa), cysteine-rich, cationic peptides that belong to the broad class of innate defense molecules known as cationic antimicrobial peptides (CAPs). Plant defensins play a major role in plant innate immunity and have been identified in all analyzed plant species to date, as either constitutively expressed or induced defense molecules that are produced in response to pathogenic attack or environmental stress (1). The tertiary structure of all plant defensins is highly conserved, comprising a triple-stranded antiparallel β-sheet and a single α-helix stabilized by four disulfide bridges, known as the “cysteine-stabilized alpha-beta” or “CSαβ” motif (2). Despite this conserved three-dimensional structure, there is a high degree of variation in the primary sequence of plant defensins, particularly at intervening loop regions, which are typically important for activity (3).
Many plant defensins have antifungal activity, but other functions have been reported, including antibacterial activity, ion channel blocking, protein synthesis inhibition, and trypsin and α-amylase inhibition as well as roles in heavy metal tolerance, plant development, and pollen tube guidance (3–5). They can be divided into two classes based on whether or not a C-terminal propeptide (CTPP) (of ∼33 amino acids) is present (2, 6). This domain is involved in vacuolar targeting and protects the plant cells from phytotoxicity during transit through the secretory pathway (7). Defensins expressed with the additional CTPP domain are known as class II defensins and have been identified only in the Solanaceae and Poaceae families. All other plant defensins belong to class I (2).
The antifungal mechanisms employed by several class I plant defensins involve, at least in part, cell wall/membrane disruption of target cells, with reports of various membrane lipids being implicated in this process. For example, the radish defensin RsAFP2 interacts with the sphingolipid glycosylceramide (GlcCer) at the cell membrane of Candida albicans, inducing reactive oxygen species (ROS) production and cell death (8). The alfalfa defensin MsDef1 also requires the presence of GlcCer to inhibit the growth of Fusarium graminearum (9). Similarly, membrane permeabilization of Saccharomyces cerevisiae by the dahlia defensin DmAMP1 depends on expression of mannosyldiinositolphosphorylceramide (10). More recently, binding to phosphatidic acid (PA) has been implicated in the membrane-permeabilizing activity of MtDef4 on Fusarium graminearum (11). Despite the identification of these lipid ligands for defensins, the molecular basis of the interactions remains poorly defined. The identification and characterization of class II defensins, found mainly in floral tissue and in seeds, have been limited. NaD1, a solanaceous defensin from the flowers of the ornamental tobacco (Nicotiana alata), exhibits fungicidal and growth-inhibitory activities toward several plant-pathogenic fungi, including Fusarium oxysporum, Fusarium graminearum, Botrytis cinerea, Saccharomyces cerevisiae, Candida albicans, and several Aspergillus and Cryptococcus species (6, 12–15). NaD1 has a three-stage mechanism that involves cell wall binding, membrane permeabilization, and entry into the cytoplasm that results in cell death for both hyphal and yeast fungal forms (14). Furthermore, NaD1 has been reported to induce Ca2+ influx and K+ efflux, ROS production, and activation of the high-osmolarity glycerol (HOG) stress response pathway in certain fungi (13, 16). More recently, we demonstrated that dimerization of NaD1 is important for antifungal activity. The dimer has an extensive positively charged surface that may promote the interaction with negatively charged moieties within the fungal cell wall and/or membrane (15). As the ability of defensins to form dimers has been reported previously (17), these data suggest that dimeric configurations of defensins are likely to be of functional importance for their antimicrobial activity. We subsequently showed that NaD1 interacts with a broad range of phosphatidylinositol phosphates with specific binding to the plasma membrane phospholipid, phosphatidylinositol (4,5)-bisphosphate (PIP2), being critical for membrane permeabilization (18). The crystal structure of the NaD1-PIP2 complex revealed an oligomer comprising seven NaD1 dimers in a cationic grip conformation bound to 14 PIP2 molecules. A number of positively charged amino acids were identified in the β2-β3 loop region of NaD1 that mediate interaction with the negatively charged phospho-head groups of PIP2, and these residues were confirmed to play a role in both fungal and mammalian cell membrane permeabilization (18). These findings provided, for the first time, evidence of (i) a membrane phosphatidylinositol as a defensin target and (ii) lipid-mediated oligomerization of a defensin as a key mechanism of action.
The Solanaceae family includes tobacco, tomato, petunia, capsicum, eggplant, chili, and several other species. To date, only a few defensins from Nicotiana, petunia, and tomato have been isolated. The relatively high sequence identity and proposed structural conservation among class II solanaceous defensins have prompted the question of whether the observed defensin-lipid complex formation of NaD1 and its role in target cell permeabilization may be conserved within this peptide class. However, no similar structural or functional studies have been reported for any other plant defensin to address this question. The tomato pistil predominant 3 defensin (TPP3) was first identified by Milligan and Gasser in 1995 when screening for highly expressed proteins in pistils from tomato flowers (19). It shares 63% amino acid sequence identity with the mature defensin domain of NaD1 (20, 21). In this study, through several biological and protein biophysical approaches, we have demonstrated that TPP3 adopts the cationic grip dimeric configuration reported for NaD1 and binds specifically to PIP2. Lipid binding facilitates oligomerization and induces membrane permeabilization of both mammalian and fungal cells. These observations reveal a conserved mechanism of action between class II defensins and support a growing body of evidence for the requirement of highly specific defensin-lipid interactions at the membrane of target cells to elicit cytolysis within this peptide class.
MATERIALS AND METHODS
Cell lines.
Human epithelial cervical cancer (HeLa) cells and leukemia monocyte lymphoma (U937) cells were cultured in RPMI 1640 medium (Invitrogen) at 37°C and 5% CO2. All culture media were supplemented with 5 to 10% fetal calf serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Invitrogen).
Expression and purification of defensins.
The mature form of NaD1 was isolated from flowers of the ornamental tobacco Nicotiana alata as described previously (6). The mature defensin domain of TPP3 or the point mutants TPP3(K6A), TPP3(K28A), TPP3(Q40L), and TPP3(K42E) were expressed as secreted recombinant proteins in the methylotrophic yeast Pichia pastoris and purified using an SP Sepharose column (GE Healthcare Biosciences) as described previously (21).
Mammalian tumor cell viability and membrane permeability assays.
For flow cytometry-based membrane permeabilization assays, human myeloid leukemia U937 cells at 1 × 106 cells/ml in RPMI 1640 medium containing 0.1% bovine serum albumin (BSA) were treated with defensins at 37°C for 30 min prior to addition of propidium iodide (PI) (1 μg/ml) or ToPro-3 (1 μM; Life Technologies) and flow cytometry analysis. In certain experiments, cells were treated with neomycin (2.5 to 20 mM) for 30 min prior to the addition of defensins. The release of ATP and lactate dehydrogenase (LDH) (22) from permeabilized cells was measured using the ATP bioluminescence assay (Roche) and LDH cytotoxicity assay kit II (Abcam), respectively, according to the manufacturer's instructions.
Fungal growth inhibition assay.
The ability of TPP3 and the TPP3 point mutants to inhibit the growth of the filamentous fungus Fusarium graminearum was assessed by performing fungal hyphal growth inhibition assays as described previously (13).
Confocal laser scanning microscopy.
Live imaging was performed on a Zeiss LSM-780 confocal microscope using a 63× oil immersion objective in a 37°C, 5% CO2 atmosphere. Adherent cells were cultured on coverslips prior to imaging, while nonadherent cells were immobilized onto 1% poly-l-lysine-coated coverslips. All cell types were prepared for imaging in RPMI 1640 medium containing 0.1% BSA and 1 μg/ml of PI or 1 μM To-Pro-3. Defensins were added directly to the imaging chamber via a capillary tube. In certain experiments, cells were stained with PKH67 (Sigma-Aldrich) prior to imaging, as per the manufacturer's instructions. Images were then analyzed using ImageJ software (18) or Zen software (Zeiss, Oberkochen, Germany). In certain experiments, HeLa cells were transfected with a plasmid construct for free green fluorescent protein (GFP) or GFP-PH(PLCδ1) using Xtreme Gene 9 transfection reagent (Roche) as per the manufacturer's instructions prior to imaging. To determine the effects of TPP3 on transfected HeLa cells, images were captured over a 30 min (1,800-s) time frame from the time of TPP3 addition, with number of seconds to membrane permeabilization recorded (nonpermeabilized cells were given a time score of 1,800 s).
Protein-lipid overlay assays.
Protein-lipid overlays using Membrane Strip, PIP Strip, and PIP Array (Echelon) assays were performed using 1 μg/ml of defensins as described previously (23). For quantification of all protein-lipid overlay assays by densitometry, corrected readings for individual lipid spots were calculated by subtracting a reading from a region that was directly adjacent on the strip, representing nonspecific background.
ATP release assay with ATP-encapsulated liposomes. (i) Liposome preparation.
Liposomes were prepared using l-α-phosphatidylcholine (PC, chicken egg) and l-α-phosphatidylinositol-4,5 bisphosphate (PIP2, porcine brain) purchased from Avanti Polar Lipids (Alabaster, AL). PC (dissolved in chloroform) and PIP2 (dissolved in chloroform-methanol-water, 20:9:1) were combined with the desired molar ratio of lipid components (pure PC or PC-PIP2, 95:5). The lipid mixture was dried under a stream of nitrogen gas followed by further drying under a vacuum for 16 h. The lipid films were rehydrated to 5 mg/ml in 20 mM HEPES (pH 7.4) containing 5 mg/ml of ATP, for 1 h at 37°C with periodic gentle agitation. Lipid mixtures were then freeze-thawed three times before 15 rounds of extrusion through an Avanti Polar Lipids Mini-Extruder with a 0.2-μm membrane. Liposomes were washed three times in 20 mM HEPES (pH 7.4) by centrifugation at 16,000 × g to remove nonencapsulated ATP. Liposomes were used within 24 h.
(ii) ATP release assay.
The release of ATP from liposomes treated with defensins was measured using a SpectraMax microtiter plate reader. ATP-encapsulated liposomes in 20 mM HEPES (pH 7.4) equivalent to 100 μg of dried lipid were combined with 50 μl of luciferase reagent (Roche) and added to 10 μl of defensin prepared in buffer (final concentration of 15 μM) immediately prior to recording luminescence at 30-s intervals over a 30-min time course. Corrected luminescence was then calculated by subtracting background luminescence (buffer only) for each reading. A rate of 100% lysis was determined by recording single luminescence endpoints for liposomes incubated for 5 min prior with 1% Triton X-100 and then subtracting background luminescence (buffer only). Defensin-mediated membrane lysis was then expressed as a percentage of total lysis.
Protein cross-linking.
TPP3 and related defensins in the presence or absence of PIP2 were cross-linked with bis(sulfosuccinimidyl)suberate (BS3) and analyzed by reducing SDS-PAGE as described previously (15).
Transmission electron microscopy (TEM).
TPP3 at 0.9 mg/ml and 1,2-dioctanoyl-sn-glycero-3-[phosphoinositol-4,5-bisphosphate] (Avanti Polar Lipids) at 0.23 mM in water (10 μl) were applied to 400-mesh copper grids (ProSciTech) coated with a thin layer of carbon for 2 min. Excess material was removed by blotting, and samples were negatively stained twice with 10 μl of a 2% (wt/vol) uranyl acetate solution (Electron Microscopy Services). The grids were air dried and viewed using a JEOL JEM-2010 transmission electron microscope operated at 80 kV or a FEI Tecnai TF30 transmission electron microscope operated at 300 kV.
Crystallization and data collection.
TPP3 crystals were grown using the sitting drop vapor diffusion method at 293 K in 24% (wt/vol) polyethylene glycol 4000 (PEG 4000), 0.16 M ammonium acetate, and 0.1 M sodium citrate (pH 5.1) as described previously (21). Native diffraction data were collected as described previously (21) (Table 1).
TABLE 1.
Crystallographic data collection and refinement statistics
| Parameter | Value(s)a |
|---|---|
| Data collection statistics | |
| Space group | P6122 |
| Unit-cell parameters | |
| a, b, c (Å) | 64.97, 64.97, 82.40 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| Wavelength (Å) | 0.9537 |
| Resolution (Å) | 46.46–1.70 (1.79–1.70) |
| Rmerge | 0.072 (1.073) |
| Rp.i.m.b | 0.012 (0.174) |
| I/σI | 51.4 (5.3) |
| Completeness (%) | 100 (100) |
| Multiplicity | 34.7 (38.1) |
| No. of reflections | 412,499 (64,170) |
| No. of observed reflections | 11,889 (1,684) |
| Refinement | |
| Resolution (Å) | 1.70 |
| No. of reflections | 11,848 |
| Rwork/Rfree | 0.1866/0.2079 |
| No. of atoms | |
| Protein | 826 |
| Ligand/ion | 4 |
| Water | 64 |
| B-factors | |
| Protein | 28.74 |
| Water | 35.68 |
| RMSD | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.1 |
Values in parentheses are for the highest-resolution shell.
Rp.i.m., precision-indicating merging R factor.
Model building and refinement.
The crystal structure of TPP3 was solved by molecular replacement with PHASER (24) using a monomer of NaD1 (PDB code 4aaz) (15) as a search model. Electron density was continuous within residues 2 to 49 (chain A) and 3 to 49 (chain B) with no discernible density identified outside these regions. The final model was built using Coot (25) and refined with Phenix (26) to a resolution of 1.7 Å with values of Rwork and Rfree of 18.66% and 20.79%, respectively. All figures were prepared using PyMol (DeLano Scientific).
Protein structure accession number.
Coordinates have been deposited in the Protein Data Bank under accession code 4UJ0.
RESULTS
TPP3 interacts with PIP2 in vitro.
TPP3 shares 63% identity with NaD1, and the residues that participate in dimer formation and PIP2 binding are largely conserved across both defensins (Fig. 1A). To investigate the lipid binding profile of TPP3, in particular its ability to bind membrane phosphatidylinositol phosphates, including PIP2, lipid binding assays using Membrane Strips, PIP Strips, and Sphingo Strips (Fig. 1B) were performed. A PIP Strip was probed with NaD1 and included for comparison (Fig. 1C). Quantification of the relative binding to PIP Strips was achieved by performing densitometry analysis on three independent strips for TPP3 versus NaD1 (Fig. 1D). TPP3 bound specifically to PIP2, with negligible binding to other PIPs. Notably, a lack of binding to phosphatidylinositol monophosphates as well as those containing di- and triphosphates in the 3 position was observed. As the binding to purified phosphoinositides using in vitro approaches such as lipid overlay assays is not always representative of phosphoinositides in lipid bilayer membranes, experiments with liposomes were also conducted. Thus, to determine whether the observed binding of TPP3 to PIP2 also occurred in the context of a lipid bilayer and whether this binding could induce membrane permeabilization, a liposomal leakage assay with ATP-encapsulated liposomes was performed (Fig. 1E). ATP-encapsulated liposomes containing either PC or PC-PIP2 were treated with TPP3 or NaD1 over 30 min. Both TPP3 and NaD1 induced complete lysis of PIP2-containing liposomes, whereas no lysis was observed for PC-only liposomes.
FIG 1.

The tomato defensin TPP3 interacts specifically with phosphatidylinositol 4,5-bisphosphate (PIP2) in vitro. (A) Amino acid sequence alignment of TPP3 (NCBI database accession no. U20591) and NaD1 (NCBI database accession no. AF509566). NaD1 residues and putative TPP3 residues participating in PIP2 binding are annotated with a bold dash; residues participating in cationic grip dimer formation are annotated with a double dash. An additional alanine residue is present at the N terminus of TPP3, generated through recombinant processing, described previously (21). (B and C) Detection of TPP3 binding to cellular lipids by protein-lipid overlay assays using Membrane Lipid Strips, PIP Strips, and Sphingo Strips (B) compared with PIP Strip of NaD1 (C). Data in panels B and C are representative of at least two independent experiments. (D) Densitometry analysis of PIP Strip probed with TPP3 compared with NaD1, normalized for binding to PIP2. (E) Ability of NaD1 and TPP3 to permeabilize ATP-encapsulated liposomes. ATP-encapsulated liposomes containing either PC or PC-PIP2 were treated with 15 μM defensin in the presence of luciferase reagent. “Percentage total lysis” represents the degree of ATP release from cells expressed as a percentage of 100% lysis (as determined by Triton X-100 treatment). Error bars in panels D and E represent SEMs from three independent experiments.
Crystal structure of TPP3.
To gain structural insight into the molecular mechanism of action of TPP3, we determined its crystal structure. Similar to other plant defensins, TPP3 adopts a CSαβ motif, harboring three canonical disulfide bonds, with a fourth disulfide bond connecting the N and C termini, thus generating a pseudocyclic molecule (Fig. 2A). The asymmetric unit contains a dimer of TPP3, which leads to the loss of 396 Å2 of solvent accessible surface in the interface. The TPP3 dimer is formed by antiparallel alignment of the β1 strands and is stabilized by two K6–K6 hydrogen bonds and two C49–K47 salt bridge interactions. The overall topology of the dimer is reminiscent of the cationic grip observed in the NaD1-PIP2 oligomeric complex, and superimposition of the TPP3 dimer with a NaD1-PIP2 dimer using Dali-Lite (27) yielded a Z-score of 17.1 with a root mean square deviation (RMSD) of 1.2 Å over 93 Cα atoms. Inspection of the superimposed structures identified residues K6, H35, and K42 in TPP3, which closely match critical PIP2 binding residues in the NaD1-PIP2 dimer (K4, H33, and R40), suggesting a potential lipid binding site in the TPP3 dimer (Fig. 2B).
FIG 2.

Crystal structure of TPP3. (A) Cartoon representation of the TPP3 structure (orange) shown as a dimer in the asymmetric unit with the molecular surface shown in pale orange. (B) Surface representations of TPP3 and NaD1, displaying the “cationic grip” formed between the two TPP3 monomers (orange and pale yellow), the NaD1 cationic grip (teal and aqua) in which two PIP2 molecules are present (magenta), and the superimposition of the TPP3 dimer (orange) over the NaD1 dimer containing PIP2 molecules (aqua and magenta). (C) Qualitative electrostatic surface representation of TPP3 (blue is positive, red is negative, and white is uncharged or hydrophobic). This figure was generated using Pymol.
Electrostatic surface analysis of the TPP3 dimer.
Given that the membrane permeabilization ability of NaD1 may be initiated by interactions with negatively charged glycoproteins on the plasma membrane surface (14), the electrostatic surface potential of the TPP3 dimer was investigated. The majority of the dimer surface is charged positively, both in the dimer cavity and sides, with a relatively small area near the dimer interface displaying negative charge (Fig. 2C).
It has been shown previously that antimicrobial peptides, including defensins (of plant and animal origin), may form dimers or higher-order oligomers to elicit target cell membrane destabilization (15, 28, 29). PISA (proteins, interfaces, structures, and assemblies) analysis revealed that four salt bridges are formed between residues R23 and D19 from a single monomer between adjacent dimers, forming an extended tetrameric configuration in the crystal (Fig. 3). Despite the equivalent residues on the second monomer being accessible, they did not participate in salt bridges with a neighboring dimer, and thus no continuous oligomer was formed within the crystal.
FIG 3.
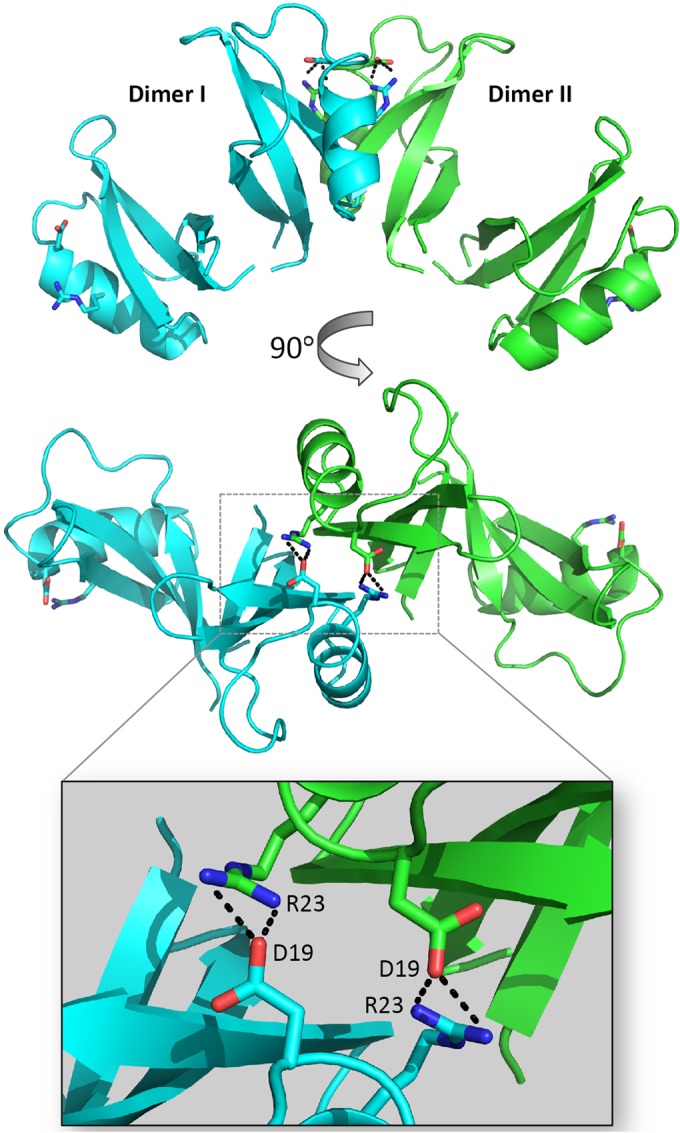
TPP3 dimers form tetramers in the crystal unit. Two TPP3 dimers interact to form a tetramer through salt bridge formations between D19 and R23 residues in the α1-helices. An enlarged view of the dimer-dimer interface (inset) displays side chains of salt bridge-forming residues. This figure was generated using Pymol.
TPP3 is cytolytic to mammalian tumor cells and induces plasma membrane blebs.
To determine whether TPP3 can induce cytolysis of mammalian tumor cells, flow cytometry-based PI uptake as well as ATP release assays were performed on U937 cells. TPP3 induced membrane permeabilization in a concentration-dependent manner, as represented by the percentage of PI-positive cells (Fig. 4A). At the highest concentration of 10 μM, TPP3 induced 69.6% cell death compared with 62.3% cell death in the NaD1 positive control. Similarly, ATP was released rapidly from cells following treatment with TPP3 in a concentration-dependent manner (Fig. 4B). Furthermore, release of LDH from U937 cells following TPP3 treatment also occurred in a concentration-dependent manner, with 10 μM TPP3 inducing 35% cytotoxicity (Fig. 4C), compared with 65% in NaD1 (data not shown). Collectively, these data suggest that TPP3 can induce cytolysis of mammalian tumor cells, including the release of large (≤140 kDa) cytoplasmic proteins.
FIG 4.

TPP3 permeabilizes U937 tumor cells in a concentration-dependent manner and induces plasma membrane blebbing. (A) Flow cytometry-based analysis of cell viability determined by uptake of PI. Cells were incubated for 30 min at 37°C with TPP3 at 10, 5, 2.5, or 1.25 μM, NaD1 at 10 μM, or PBS; the percentage of PI-positive cells representing the level of defensin-mediated membrane permeabilization is shown. A similar degree of membrane permeabilization was observed for TPP3-treated PC3 cells (data not shown). (B) ATP release of TPP3-treated U937 cells. TPP3 at 20, 10, 5, or 2.5 μM, NaD1 at 20 μM, or PBS only was added to U937 cells in the presence of luciferase reagent. Readings at 562 nm were then recorded at 30-s intervals over 30 min, with bioluminescence representing the degree of ATP release from cells. (C) LDH release by TPP3-treated U937 cells. Cytotoxicity was calculated as a percentage of total LDH released by TPP3-treated cells, based on cell lysis buffer control (100% lysis). (D) CLSM live cell imaging of PKH67-stained HeLa and U937 cells treated with TPP3 (10 μM) in the presence of PI, 24 min after addition of TPP3. Error bars in panels A and C represent SEMs (n = 3). Scale bars in panel D represent 10 μm; the measurement for the arrow in panel D is 22 μm. Data are representative of at least two independent experiments.
Live confocal laser scanning microscopy (CLSM) was used to examine changes in cell morphology upon TPP3 treatment (Fig. 4D). In both adherent (HeLa) and nonadherent (U937) cell types, large plasma membrane blebs were formed in affected cells, coinciding with PI uptake. The bleb size of TPP3-treated cells was large (>20 μm) and did not retract over periods of 30 min.
TPP3 oligomerizes with PIP2 in vitro.
The ability of TPP3 to interact with PIP2 was examined using transmission electron microscopy (Fig. 5A), revealing that long, string-like fibrils of TPP3 were formed only in the presence of PIP2. Multiple fibrils with a diameter of approximately 10 nm were observed. At times, fibrils stacked adjacently or in bundles, suggesting lateral association. The propensity of the TPP3 to oligomerize in solution was also investigated using the chemical cross-linker BS3 (Fig. 5B). In the absence of PIP2, both monomeric and dimeric forms of TPP3 were detected. However, in the presence of PIP2, the formation of larger oligomeric assemblies of various sizes was detected, appearing as “laddering” and suggesting that TPP3 binds PIP2 to form higher-order oligomers.
FIG 5.

TPP3 interacts with PIP2. (A) Negative-staining TEM images of TPP3 showing the formation of fibrils in the presence of PIP2. Scale bars represent 200 nm. (B) Cross-linking analysis of TPP3 with PIP2. The ability of TPP3 to form oligomers in the presence of PIP2 was investigated using the amine reactive cross-linker BS3, followed by SDS-PAGE and Coomassie staining. (C) Ability of TPP3 to permeabilize tumor cells in GFP-PH-overexpressing cells. HeLa cells overexpressing either GFP-PH or free GFP were treated with TPP3 and imaged with CLSM to determine the mean time to membrane permeabilization. Statistical analysis was performed using an unpaired, two-tailed t test (n = 26 for GFP-PH and n = 21 for free GFP-expressing cells; P = 0.001). (D) Ability of neomycin to inhibit TPP3-mediated U937 cell lysis in a concentration-dependent manner, as determined by flow cytometry. (E) Ability of neomycin to inhibit LL-37-mediated U937 cell lysis. For panels D and E, error bars represent SEMs (n = 3); results are representative of those from three independent experiments.
TPP3 interacts with plasma membrane PIP2 to mediate membrane permeabilization.
To determine whether the interaction between TPP3 and PIP2 occurs in cells, the level of available PIP2 at the inner leaflet of HeLa cells was modified using the GFP-tagged pleckstrin homology domain of phospholipase C(δ1) (GFP-PH), a biological PIP2 sensor (30). HeLa cells overexpressing either GFP-PH or free GFP were treated with TPP3 and visualized using CLSM over a 30-min time course. GFP-PH-expressing cells exhibited an average time of 1,210 s taken to membrane permeabilization (or 2-fold delay), compared with an average of 570 s for free-GFP-expressing cells (Fig. 5C).
To further examine the effects of inner leaflet PIP2 sequestration on the ability of TPP3 to induce tumor cell lysis, U937 cells were pretreated with various concentrations of neomycin (0 to 20 mM) (Fig. 5D). Neomycin binds to PIP2 in biological membranes (31, 32) and can be used as a sequestering agent to block interactions with PIP2. Using a flow cytometry-based PI uptake assay, U937 cells pretreated with neomycin were then treated with 10 μM TPP3. A concentration-dependent inhibition of TPP3-mediated cell membrane permeabilization was observed for neomycin-treated cells. In contrast, cells treated with LL-37, an amphipathic α-helical cationic cathelicidin that induces target cell lysis through a pore-forming mechanism not dependent on the presence of PIP2 in target cell membranes (33), showed no neomycin-mediated inhibition of membrane permeabilization (Fig. 5E). Together, these data indicate that sequestration of inner leaflet PIP2 can decrease the ability of TPP3 to induce membrane permeabilization.
PIP2 binding can be disrupted by specifically targeting conserved residues in the proposed lipid-binding pocket.
In NaD1, residue K4 is crucial for dimer formation and fungal cell killing (15). Furthermore, the structure of the NaD1-PIP2 complex revealed that K4 also participates in binding to the 5-phosphate moiety of PIP2 in the cationic grip dimer pocket, thus contributing to lipid-mediated oligomerization (18). Structural analysis of the TPP3 dimer identified K6 as the equivalent of K4 in the cationic grip configuration of NaD1 (Fig. 6A). To determine whether TPP3(K6) is also important for lipid-mediated oligomerization and membrane permeabilization, we generated a TPP3(K6A) mutant for functional analysis. Cross-linking experiments with BS3 indicated a loss in the ability of TPP3(K6A) to form higher-order oligomers in the presence of PIP2 (Fig. 6B). Furthermore, PIP Strip analysis revealed a complete loss of binding to PIP2 [antibody cross-reactivity of TPP3(K6A) was determined by Western blotting (data not shown)] (Fig. 6C), which corresponded with the inability of TPP3(K6A) to induce membrane permeabilization of tumor cells (Fig. 6D) and PIP2-containing liposomes (Fig. 6E). These observations coincided with an approximately 3-fold reduction in the ability of TPP3(K6A) to inhibit hyphal growth of the filamentous fungus Fusarium graminearum compared with that of wild-type TPP3 (Fig. 7). Together, these data suggest that K6 of TPP3 is required for PIP2 binding and oligmerization, which contribute to the cytolytic activity of this defensin. It should be noted that although K4 of NaD1 was important for both dimer formation and antifungal activity (15), the K6A mutation in TPP3 did not abrogate dimer formation. Since the NaD1 dimer solved in the absence of PIP2 did not adopt the more stable cationic grip configuration, it is possible that the K4A mutation in NaD1 was adequate to disrupt dimerization (15). Examination of the TPP3 dimer structure suggests that the interactions mediated by K47 and C49 in TPP3(K6A) may be sufficient for maintaining dimer formation by this mutant (Fig. 6A). These data suggest distinct roles for K6 of TPP3 and K4 of NaD1 in dimer and oligomer formation.
FIG 6.

K6 in TPP3 is required for oligomerization, PIP2 binding, and membrane permeabilization. (A) Superimposition of TPP3 (orange) and NaD1-PIP2 complex (wheat, green, and aqua) crystal structures. Putative ionic interactions between NaD1(K4) and PIP2 are indicated in black. Residues proposed to be involved in dimerization of TPP3 are indicated in the right panel; the boxed area in the left panel is enlarged and rotated 90° in the right panel. Interatomic distances are measured in angstroms. The images were generated using Pymol, and superimposition was generated using SSM (49). (B) The ability of the TPP3(K6A) mutant to dimerize in solution and oligomerize in the presence of PIP2 was determined using the BS3 cross-linker. (C) The ability of TPP3(K6A) to bind PIP2 was determined by PIP Strip analysis. (D) The ability of TPP3(K6A) to induce membrane permeabilization of mammalian tumor cells was examined by flow cytometry. Error bars represent SEMs (n = 3). Data in panels B to D are representative of at least two independent experiments. (E) The ability of TPP3 (K6A) to permeabilize ATP-encapsulated liposomes. ATP-encapsulated liposomes containing either PC or PC-PIP2 were treated with 15 μM defensin in the presence of luciferase reagent. “Percentage total lysis” represents the degree of ATP release from cells expressed as a percentage of 100% lysis (as determined by Triton X-100 treatment). Error bars in panel E represent SEMs from three independent experiments.
FIG 7.
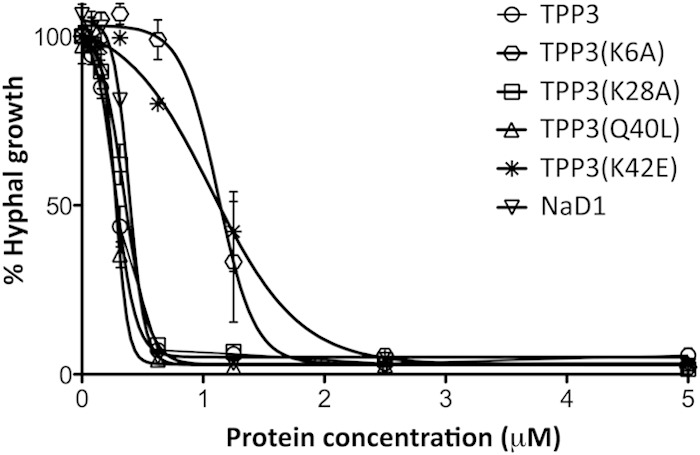
Antifungal activity of TPP3 and TPP3 point mutants. The ability of TPP3, TPP3 point mutants, and NaD1 to kill the filamentous fungus Fusarium graminearum was examined using a fungal growth inhibition assay in which fungal hyphae were incubated with increasing concentrations of defensin for 24 h. Data are expressed as percentage of total growth (untreated control). Error bars represent SDs and are representative of three independent experiments.
In addition to K4, R40 in NaD1 is required for cooperative PIP2 binding and oligomerization, with the NaD1(R40E) mutant having lost the ability to oligomerize and bind phosphatidylinositol 4-phosphate (18). Analysis of the TPP3 dimer indicated that K42 in TPP3 is the structural equivalent to R40 in NaD1 and is likely to be in a position to engage the PIP2 4-phosphate moiety (Fig. 8A). To test whether K42 is required for TPP3-PIP2 complex formation, we generated a TPP3(K42E) mutant. TPP3(K42E) displayed a loss in the ability to form higher-order oligomers in the presence of PIP2 (Fig. 8B) as well as a loss of binding to PIP2, as determined by PIP Strip analysis (Fig. 8C). Furthermore, this mutant displayed a reduction in the ability to induce fungal hyphal growth inhibition to a similar degree to that observed for TPP3(K6A) (Fig. 7) as well as complete abrogation of mammalian tumor cell membrane permeabilization (Fig. 8D). Thus, K42 in TPP3 is likely to have the same role as its structural equivalent R40 in NaD1, that being the binding to the 4-phosphate moiety of PIP2 within the cationic grip dimer, to mediate cooperative higher-order oligomerization and subsequent membrane permeabilization.
FIG 8.

K42E mutation in TPP3 results in a loss of oligomerization, PIP2 binding, and tumor cell killing. (A) SSM superimposition of TPP3 (orange) and NaD1-PIP2 complex (wheat, green, and aqua) crystal structures. Ionic interactions between R40 and PIP2 (aqua) are indicated in black (measured in angstroms). This figure was generated using Pymol. (B) The ability of loss-of-function mutant TPP3(K42E) to oligomerize in the presence of PIP2 was determined using the BS3 cross-linker. (C) The ability of TPP3(K42E) to interact with biologically active lipids was determined using a PIP Strip. (D) A PI uptake assay was performed to determine the ability of TPP3(K42E) to permeabilize U937 cells. Error bars represent SEMs (n = 3). Data in panels B to D represent means ± SEMs and are representative of results from at least two independent experiments.
In contrast to these observations, residue K28, which is not situated within the cationic grip and was not predicted to participate in lipid binding, induced no change in PIP2-mediated oligomerization, PIP2 binding, or mammalian or fungal cell killing when it was mutated to TPP3(K28A) (Fig. 7 and 9).
FIG 9.

Disruption of residue K28 through loss-of-function mutation does not significantly affect activity. (A) The ability of TPP3(K28A) to bind biologically active lipids was determined using the PIP Strip lipid binding assay. (B) The ability of TPP3(K28A) to oligomerize in the presence of PIP2 was determined using the BS3 cross-linker. (C) A PI uptake assay was performed to determine the ability of TPP3(K28A) to permeabilize U937 cells. Error bars represent SEMs (n = 3). Data in panels A to C are representative of results from at least two independent experiments. (D) TPP3 dimer (orange and pale yellow) in which the two PIP2 molecules from the NaD1-PIP2 complex (green and aqua) are overlaid and the position of residue K28 in each monomer is shown. The image was generated using Pymol.
TPP3 has a distinct lipid binding pattern.
The unique specificity of TPP3 for PIP2 is distinct from that of NaD1, which has a broader specificity and binds to several mono-, di-, and triphosphatidylinositol phosphates. Notably, di- and triphosphatidylinsoitol phosphates containing the 3-phosphate moiety interact with NaD1 but not TPP3. We therefore sought to identify the specific residues within TPP3 that could contribute to its ability to discriminate against 3-phosphate-containing phosphatidylinositol phosphates. Superimposition of the TPP3 and NaD1-PIP2 complex dimers revealed that Q40 in TPP3 has the potential to sterically hinder binding to the 3-phosphate moiety of a phosphatidylinositol ring (Fig. 10A) and that this could be sufficient to block binding to phosphatidylinositol phosphates containing a 3-phosphate moiety. To test this hypothesis, a TPP3(Q40L) mutant was generated. Lipid binding analysis using PIP arrays (Fig. 10C) and PIP Strips (data not shown) revealed that TPP3(Q40L) exhibited a broadened lipid binding specificity compared with that of the wild type. Densitometry analysis of lipid binding assays at 100 pmol were then performed (Fig. 10D); the greatest increase in relative binding for the TPP3(Q40L) mutant was observed with phosphatidylinositol-3,4,5-triphosphate (PIP3). In contrast, no change in lipid binding specificity occurred with the control mutant, TPP3(K28A) (Fig. 9A). In addition, the ability of TPP3 to undergo PIP2-mediated oligomerization was not affected by the Q40L mutation, as indicated with BS3 cross-linking (Fig. 10B).
FIG 10.

Q40 in TPP3 sterically hinders binding to the phosphoinositol ring 3-phosphate moiety. (A) Superimposition of TPP3 (orange) and NaD1-PIP2 complex (wheat, green, and aqua) crystal structures, highlighting relative side chain positions of L38 in NaD1 (boxed in main image) and Q40 in TPP3 (inset). Images were generated using Pymol. (B) The ability of TPP3(Q40L) to oligomerize in the presence of PIP2 was determined using the BS3 cross-linker. (C) Comparison between the ability of TPP3 and TPP3(Q40L) to bind biologically active lipids was determined using the PIP Array lipid binding assay. (D) Densitometry analysis of three independent lipid binding assays with 100 pmol of lipids. Relative binding is normalized to PIP2 and expressed as relative densitometry (grayscale pixel units).
Collectively, these data strongly suggest that TPP3 interacts specifically with PIP2 at the membrane of cells to induce membrane disruption. Furthermore, the adoption of the cationic grip configuration by TPP3, previously identified in the NaD1-PIP2 complex, represents a conserved defensin structure for lipid recognition that mediates oligomerization to effectively induce target cell lysis.
DISCUSSION
The tertiary structure of plant defensins is well conserved; however, the primary amino acid sequence is highly variable and has been proposed to reflect the broad range of functions for this protein family. It has been postulated that CAPs, including defensins, may form higher-order oligomers as a means of destabilizing target cell membranes (34–36). However, the structural basis of CAP-mediated membrane permeabilization remains poorly defined. Our recent study (18) on the solanaceous defensin from ornamental tobacco, NaD1, reported the first description of the structural basis for defensin-mediated membrane permeabilization and implicated PIP2 as the ligand required to induce oligomerization. In this study, we have characterized the cytolytic activity of the closely related tomato defensin TPP3, determined its crystal structure, and defined its interaction with the plasma membrane phosphatidylinositol phospholipid, PIP2.
TPP3 forms a cationic grip dimer.
The crystal structure of TPP3 revealed a dimeric configuration in the asymmetric unit, which bears a striking resemblance to the PIP2-bound NaD1 dimer that forms the fundamental building block in the NaD1-PIP2 complex (18). Whereas the NaD1-PIP2 dimer is likely to represent the active dimer conformation, TPP3 was not crystallized in the presence of a ligand, yet it adopts a very similar configuration. The ability of NaD1 to dimerize was first reported by Lay et al. (15), who identified two different crystal dimer forms, one of which was formed by two opposing β1 strands in an antiparallel configuration in the absence of a lipid ligand. Furthermore, the dimerization was important for the antifungal activity of NaD1 (15). The subsequent elucidation of the NaD1-PIP2 oligomeric structure (18) revealed an NaD1 dimeric unit that forms a cationic grip, where a deep cationic pocket at the interface of a NaD1 dimer accommodates the head groups of two PIP2 molecules (15). The cooperative binding of PIP2 in a single dimer by adjacent NaD1 dimers mediates the oligomeric assembly. A remarkably similar dimeric form is adopted in the crystal structure of TPP3, suggesting a conserved mechanism for binding to PIP2.
PISA analysis revealed that K6, K47, and C49 of TPP3 are involved in dimer-stabilizing hydrogen bonds and ionic interactions, while the superimposition of the TPP3 dimer over the NaD1-PIP2 complex suggested that K6 is likely to interact with the 4- and/or 5-phosphate moieties of PIP2 molecules within the cationic grip dimer pocket in a configuration similar to that in the NaD1-PIP2 complex (18). Indeed, a loss of PIP2-mediated oligomerization, PIP2 binding, and liposomal permeabilization was observed for TPP3(K6A). Furthermore, a loss of membrane-permeabilizing activity of both mammalian tumor and fungal hyphal cells was reported for this mutant. Thus, this structurally conserved residue is likely to be important for lipid-mediated oligomerization and membrane lysis across species. Whereas the NaD1(K4A) mutant lost the ability to dimerize in solution, examination of the TPP3 structure suggests that the TPP3(K6A) mutant can maintain the cationic grip dimer by forming additional ionic interactions at the dimer interface between the K47 and C49 residues. These interactions were not apparent in the previously described NaD1 dimer (15), suggesting that TPP3 forms a more stable dimeric structure than NaD1.
Evidence of dimer formation has been reported for several other defensins. For example, X-ray crystallography revealed that the antifungal Mexican turnip defensin SPE10 forms a dimer, with dimerization suggested to play a role in its antifungal activity (17). Dimer formation was also observed in the crystal structures of human α-defensins 4, 5, and 6 (HN4, HN5, and HN6, respectively) (37). Furthermore, HN6 forms tetramers (which comprised two homodimers) that oligomerize and trap bacteria by forming fibril-like nanonets (38). Human β-defensins 2 and 3, which are active against several Gram-positive and Gram-negative bacteria as well as C. albicans, have been shown through nuclear magnetic resonance (NMR) spectroscopy to form dimers (34, 35). This suggests that the role of dimerization, and possibly oligomerization, may be important in defensin function across various mechanisms of antimicrobial action.
PIP2 is required for the formation of higher-order TPP3 oligomers.
TEM revealed that TPP3 can form long, fibril-like structures in the presence of PIP2, and protein cross-linking analyses showed that that TPP3 forms higher-order oligomers in the presence of PIP2. Neither of these events occurred with defensin alone, indicating an absolute requirement for PIP2 in the formation of these fibrils. Interestingly, the formation of a tetramer comprised of two adjacent TPP3 dimers interacting through α1-helices was observed in the crystal. This dimer-dimer interface differs from that observed in the NaD1-PIP2 oligomer, which enabled formation of an extended arch of dimers. Although only two dimers were seen to interact in the TPP3 crystal, one can envisage that if such an interaction was repeated it might form an extended, fibril-like structure, albeit in a different configuration from the NaD1 oligomer. Since TPP3 did not form fibrils in the absence of PIP2, it is likely that the ionic interactions in the dimer-dimer interface on their own are insufficient to result in higher-order oligomerization and fibril formation. It is tempting to speculate that the presence of PIP2 would enable the formation of additional interactions between PIP2 head groups and TPP3 to reinforce and extend such a tetrameric structure, in a similar manner to that described for the NaD1 interaction with PIP2. However, the precise role that PIP2 plays in oligomerization of TPP3 remains to be defined, and this may require the structure determination of a TPP3-PIP2 complex.
In the NaD1-PIP2 oligomeric complex, the NaD1(R40) residue is crucial for PIP2-mediated oligomerization, with the loss-of-function NaD1(R40E) mutant displaying both a loss of PIP2-mediated cross-linking and decreased efficiency of mammalian tumor and fungal cell killing. Since the loss-of-function mutant of the equivalent residue in TPP3 [TPP3(K42E)] also displayed both a loss of PIP2-mediated oligomerization and inhibition of mammalian tumor and fungal cell killing, it could be postulated that this residue has a similar role across defensin species in facilitating oligomer formation that leads to target cell membrane destabilization.
With the exception of NaD1, ligand-mediated fibril formation of a defensin has not been reported previously. There is, however, some evidence that defensins or defensin-related peptides can form fibrils in the absence of a ligand. For example, a 19-amino-acid fragment of the radish seed defensin RsAFP19, comprising the β2-β3 strand region, forms amyloid-like fibrils at pH 8.0 and a temperature of 60°C in vitro (39). As mentioned above, oligomerization of HN6 was recently reported (38) in which nanonets comprised of HN6 fibrils form around and engulf Salmonella enterica serovar Typhimurium (38). This mechanism did not utilize a lipid or any other ligand to initiate fibril formation, although the requirement for a lipid was not explicitly tested. It was postulated that contiguous tetramers made up of dimer pairs could form “elongated helical structures” through a series of hydrogen bonds. Interestingly, our TEM images revealed that the string-like fibrils formed by TPP3 and PIP2 tended to laterally associate by stacking against each other horizontally, resulting in “bundles” of fibrils. While the net-forming mechanism of HN6 is highly dissimilar to the membrane-permeabilizing action of TPP3, it is worth noting that hydrogen bond-mediated associations between adjacent defensin oligomers have been observed previously (38). It could be speculated that such interactions could occur between TPP3-PIP2 fibrils on the target cell membrane and contribute to membrane destabilization. Finally, it should be noted that although TPP3 can form oligomers in the presence of PIP2 in vitro, whether TPP3-PIP2 oligomers form in the context of PIP2 containing phospholipid bilayers or on cell membranes remains to be determined.
TPP3 interacts with plasma membrane PIP2 through a conserved mechanism.
In this report, we have shown that TPP3 interacts with PIP2, both in the context of an artificial lipid bilayer and at the plasma membrane of mammalian tumor cells. The ability of any defensin to induce target cell membrane permeabilization through interaction with PIP2 has been reported only for NaD1, although a number of other plant defensins have been reported to interact with different lipids at the membrane of target cells to elicit antifungal activity. These include the RsAFP2 and MtDef1 defensins, which interact with GluCer (8, 9); DmAMP1, which requires M(IP)2C (10); and MtDef4, which binds to phosphatidic acid (11). In addition to these, the fungal defensin plectasin from Pseudoplectania nigrella, which shares the same plant defensin CSαβ architecture, exhibits growth-inhibitory activity against Gram-positive bacteria through direct binding to the cell wall precursor lipid II (40). It has also been reported that the effector protein from the fungus-like oomycete pathogen binds to outer membrane PI(3)P through a conserved cationic motif to enable entry into host cells (41). Taken together, these observations support the hypothesis that the lipid-binding fungal and plant defensins target a wide range of lipids across different species and that this lipid-targeting mechanism extends back into the early evolution of eukaryotes.
As mentioned previously, NaD1 binds a variety of phosphatidylinositols, including PI(3,4)P2, PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3 (18), whereas TPP3 is clearly selective for PI(4,5)P2. This difference in the lipid binding specificity of TPP3 was not correlated with any change in the ability to induce membrane permeabilization of mammalian tumor cells. Nor did it change the plasma membrane morphology of TPP3-treated mammalian tumor cells compared with NaD1-treated cells, namely, the appearance of large, nonretracting plasma membrane blebs that accompanied membrane permeabilization. While the consequences of this apparent variation in lipid binding specificity remain to be determined, it is possible NaD1 has additional roles in targeting intracellular membranes such as the endolysosomes/vacuoles of yeast which are relatively abundant in PI(3)P and PI(3,5)P2 (42). Indeed, many defensins and other CAPs act on intracellular pathways of target organisms to induce growth inhibition or cell death (16, 43, 44). However, the observation that exclusivity for PIP2 binding is sufficient to endow TPP3 with potent antifungal and cytolytic activities suggests that PIP2 is a major target molecule for defensin-mediated membrane attack as part of innate defense.
A role for PIP2 in pathogen-host interactions at the target cell membrane has also been reported in other systems. For example, effector proteins from the Gram-negative bacteria Vibrio parahaemolyticus and Shigella flexneri display phosphatase activity and can dephosphorylate PIP2 at the membrane of target cells, resulting in blebbing, membrane disruption, and cell lysis (45, 46). The sea anemone equinatoxin from Actinia equina also induces membrane blebbing and cell lysis, by inducing Ca2+-mediated hydrolysis of PIP2 in target cell membranes (47). PIP2 is an important component of the cell membrane, with major roles in both cell signaling and membrane-cytoskeletal attachment. It is not surprising that the modification or sequestration of this lipid, which is present in the plasma membranes of both eukaryotic and prokaryotic cells, has been exploited by a wide variety of organisms, through different mechanisms.
Cell membrane disruption of invading microbial pathogens is an evolutionarily conserved form of immune defense reported for many antimicrobial peptides. Until now, the solanaceous defensin NaD1 was the only CAP shown to utilize the membrane lipid PIP2 to form large oligomeric complexes that lead to membrane permeabilization and cell death (18). However, as NaD1 also bound a number of other phosphatidylinositol phosphates present in different subcellular locations, the relative importance of PIP2 binding for the overall activity of this defensin was unclear. Here, we have demonstrated that the tomato defensin TPP3 exclusively binds PIP2 and shares strikingly similar structural and functional features with NaD1. Through biophysical analyses, we have shown that TPP3 forms fibrils with PIP2 and that TPP3, like NaD1, adopts a cationic grip dimer conformation that mediates binding of PIP2. In addition, we have shown that TPP3 interacts with PIP2 at the membrane of target cells to elicit membrane destabilization. We propose that this interaction facilitates membrane disruption and represents a conserved mechanism of PIP2-mediated antimicrobial action within class II solanaceous defensins. It remains of significant interest whether this PIP2 targeting mechanism is also utilized by defensins from other species.
ACKNOWLEDGMENTS
We thank members of the Hulett and Kvansakul laboratories for their suggestions. We also thank the Bio21 Collaborative Crystallisation Centre for assistance with crystallization, MX2 staff at the Australian Synchrotron for assistance with X-ray diffraction data collection, SBGrid consortium for software support, and P. Lock for assistance with CLSM.
This work was supported by The Australian Research Council (ARC Discovery Grant DP120102694; fellowship to M.K. [FT130101349]), the National Health and Medical Research Council of Australia (Project Grant APP1007918; fellowship to M.K. [637372]), Balmoral Australia Pty Ltd., and Hexima Ltd.
REFERENCES
- 1.Broekaert WF, Terras FR, Cammue BP, Osborn RW. 1995. Plant defensins: novel antimicrobial peptides as components of the host defense system. Plant Physiol 108:1353–1358. doi: 10.1104/pp.108.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lay F, Anderson M. 2005. Defensins—components of the innate immune system in plants. Curr Protein Pept Sci 6:85–101. doi: 10.2174/1389203053027575. [DOI] [PubMed] [Google Scholar]
- 3.Van der Weerden NL, Anderson MA. 2013. Plant defensins: common fold, multiple functions. Fungal Biol Rev 26:121–131. doi: 10.1016/j.fbr.2012.08.004. [DOI] [Google Scholar]
- 4.Kushmerick C, de Souza Castro M, Santos Cruz J, Bloch C Jr, Beirao PS. 1998. Functional and structural features of gamma-zeathionins, a new class of sodium channel blockers. FEBS Lett 440:302–306. doi: 10.1016/S0014-5793(98)01480-X. [DOI] [PubMed] [Google Scholar]
- 5.Pelegrini PB, Lay FT, Murad AM, Anderson MA, Franco OL. 2008. Novel insights on the mechanism of action of alpha-amylase inhibitors from the plant defensin family. Proteins 73:719–729. doi: 10.1002/prot.22086. [DOI] [PubMed] [Google Scholar]
- 6.Lay FT, Brugliera F, Anderson MA. 2003. Isolation and properties of floral defensins from ornamental tobacco and petunia. Plant Physiol 131:1283–1293. doi: 10.1104/pp.102.016626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lay FT, Poon S, McKenna JA, Connelly AA, Barbeta BL, McGinness BS, Fox JL, Daly NL, Craik DJ, Heath RL. 2014. The C-terminal propeptide of a plant defensin confers cytoprotective and subcellular targeting functions. BMC Plant Biol 14:41. doi: 10.1186/1471-2229-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thevissen K, de Mello Tavares P, Xu D, Blankenship J, Vandenbosch D, Idkowiak-Baldys J, Govaert G, Bink A, Rozental S, De Groot PW. 2012. The plant defensin RsAFP2 induces cell wall stress, septin mislocalization and accumulation of ceramides in Candida albicans. Mol Microbiol 84:166–180. doi: 10.1111/j.1365-2958.2012.08017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramamoorthy V, Cahoon EB, Li J, Thokala M, Minto RE, Shah DM. 2007. Glucosylceramide synthase is essential for alfalfa defensin-mediated growth inhibition but not for pathogenicity of Fusarium graminearum. Mol Microbiol 66:771–786. doi: 10.1111/j.1365-2958.2007.05955.x. [DOI] [PubMed] [Google Scholar]
- 10.Aerts AM, François IE, Bammens L, Cammue B, Smets B, Winderickx J, Accardo S, De Vos DE, Thevissen K. 2006. Level of M(IP)2C sphingolipid affects plant defensin sensitivity, oxidative stress resistance and chronological life-span in yeast. FEBS Lett 580:1903–1907. doi: 10.1016/j.febslet.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 11.Sagaram US, El-Mounadi K, Buchko GW, Berg HR, Kaur J, Pandurangi RS, Smith TJ, Shah DM. 2013. Structural and functional studies of a phosphatidic acid-binding antifungal plant defensin MtDef4: identification of an RGFRRR motif governing fungal cell entry. PLoS One 8:e82485. doi: 10.1371/journal.pone.0082485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes BM, Bleackley MR, Wiltshire JL, Anderson MA, Traven A, van der Weerden NL. 2013. Identification and mechanism of action of the plant defensin NaD1 as a new member of the antifungal drug arsenal against Candida albicans. Antimicrob Agents Chemother 57:3667–3675. doi: 10.1128/AAC.00365-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Weerden NL, Lay FT, Anderson MA. 2008. The plant defensin, NaD1, enters the cytoplasm of Fusarium oxysporum hyphae. J Biol Chem 283:14445–14452. doi: 10.1074/jbc.M709867200. [DOI] [PubMed] [Google Scholar]
- 14.van der Weerden NL, Hancock RE, Anderson MA. 2010. Permeabilization of fungal hyphae by the plant defensin NaD1 occurs through a cell wall-dependent process. J Biol Chem 285:37513–37520. doi: 10.1074/jbc.M110.134882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lay FT, Mills GD, Poon IK, Cowieson NP, Kirby N, Baxter AA, van der Weerden NL, Dogovski C, Perugini MA, Anderson MA, Kvansakul M, Hulett MD. 2012. Dimerization of plant defensin NaD1 enhances its antifungal activity. J Biol Chem 287:19961–19972. doi: 10.1074/jbc.M111.331009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayes BM, Anderson MA, Traven A, van der Weerden NL, Bleackley MR. 2014. Activation of stress signalling pathways enhances tolerance of fungi to chemical fungicides and antifungal proteins. Cell Mol Life Sci 71:2651–2666. doi: 10.1007/s00018-014-1573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song X, Zhang M, Zhou Z, Gong W. 2011. Ultra-high resolution crystal structure of a dimeric defensin SPE10. FEBS Lett 585:300–306. doi: 10.1016/j.febslet.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 18.Poon IK, Baxter AA, Lay FT, Mills GD, Adda CG, Payne JA, Phan TK, Ryan GF, White JA, Veneer PK, van der Weerden NL, Anderson MA, Kvansakul M, Hulett MD. 2014. Phosphoinositide-mediated oligomerization of a defensin induces cell lysis. eLife 3:e01808. doi: 10.7554/eLife.01808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milligan SB, Gasser CS. 1995. Nature and regulation of pistil-expressed genes in tomato. Plant Mol Biol 28:691–711. doi: 10.1007/BF00021194. [DOI] [PubMed] [Google Scholar]
- 20.Lay FT, Schirra HJ, Scanlon MJ, Anderson MA, Craik DJ. 2003. The three-dimensional solution structure of NaD1, a new floral defensin from Nicotiana alata and its application to a homology model of the crop defense protein alfAFP. J Mol Biol 325:175–188. doi: 10.1016/S0022-2836(02)01103-8. [DOI] [PubMed] [Google Scholar]
- 21.Lay FT, Veneer PK, Hulett MD, Kvansakul M. 2012. Recombinant expression and purification of the tomato defensin TPP3 and its preliminary X-ray crystallographic analysis. Acta Crystallogr Sect F Struct Biol Cryst Commun 68:314–316. doi: 10.1107/S1744309112001510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Dijk A, Veldhuizen EJ, Haagsman HP. 2008. Avian defensins. Vet Immunol Immunopathol 124:1–18. doi: 10.1016/j.vetimm.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poon IK, Hulett MD, Parish CR. 2010. Histidine-rich glycoprotein is a novel plasma pattern recognition molecule that recruits IgG to facilitate necrotic cell clearance via FcgammaRI on phagocytes. Blood 115:2473–2482. doi: 10.1182/blood-2009-07-234013. [DOI] [PubMed] [Google Scholar]
- 24.Storoni LC, McCoy AJ, Read RJ. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr Sect D Biol Crystallogr 60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schibli DJ, Hunter HN, Aseyev V, Starner TD, Wiencek JM, McCray PB, Tack BF, Vogel HJ. 2002. The solution structures of the human β-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J Biol Chem 277:8279–8289. doi: 10.1074/jbc.M108830200. [DOI] [PubMed] [Google Scholar]
- 29.Wommack AJ, Robson SA, Wanniarachchi YA, Wan A, Turner CJ, Wagner G, Nolan EM. 2012. NMR solution structure and condition-dependent oligomerization of the antimicrobial peptide human defensin 5. Biochemistry 51:9624–9637. doi: 10.1021/bi301255u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, Petrova V, McLaughlin S, Rebecchi MJ. 1995. The pleckstrin homology domain of phospholipase C- delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry 34:16228–16234. doi: 10.1021/bi00049a039. [DOI] [PubMed] [Google Scholar]
- 31.Arbuzova A, Martushova K, Hangyás-Mihályné G, Morris AJ, Ozaki S, Prestwich GD, McLaughlin S. 2000. Fluorescently labeled neomycin as a probe of phosphatidylinositol-4,5-bisphosphate in membranes. Biochim Biophys Acta 1464:35–48. doi: 10.1016/S0005-2736(99)00243-6. [DOI] [PubMed] [Google Scholar]
- 32.Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. 2000. GAP43, MARCKS, and CAP23 modulate PI (4, 5) P2 at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol 149:1455–1472. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandamme D, Landuyt B, Luyten W, Schoofs L. 2012. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol 280:22–35. doi: 10.1016/j.cellimm.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, Chertov O, Lubkowski J. 2000. The structure of human β-defensin-2 shows evidence of higher order oligomerization. J Biol Chem 275:32911–32918. doi: 10.1074/jbc.M006098200. [DOI] [PubMed] [Google Scholar]
- 35.Dhople V, Krukemeyer A, Ramamoorthy A. 2006. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim Biophys Acta 1758:1499–1512. doi: 10.1016/j.bbamem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Mani R, Cady SD, Tang M, Waring AJ, Lehrer RI, Hong M. 2006. Membrane-dependent oligomeric structure and pore formation of a β-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc Natl Acad Sci U S A 103:16242–16247. doi: 10.1073/pnas.0605079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szyk A, Wu Z, Tucker K, Yang D, Lu W, Lubkowski J. 2006. Crystal structures of human α-defensins HNP4, HD5, and HD6. Protein Sci 15:2749–2760. doi: 10.1110/ps.062336606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu H, Pazgier M, Jung G, Nuccio S-P, Castillo PA, de Jong MF, Winter MG, Winter SE, Wehkamp J, Shen B. 2012. Human α-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 337:477–481. doi: 10.1126/science.1218831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garvey M, Meehan S, Gras SL, Schirra HJ, Craik DJ, Van der Weerden NL, Anderson MA, Gerrard JA, Carver JA. 2013. A radish seed antifungal peptide with a high amyloid fibril-forming propensity. Biochim Biophys Acta 1834:1615–1623. doi: 10.1016/j.bbapap.2013.04.030. [DOI] [PubMed] [Google Scholar]
- 40.Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, Pag U, Jansen A, Nielsen AK, Mygind PH, Ravent√≥s DS. 2010. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328:1168–1172. doi: 10.1126/science.1185723. [DOI] [PubMed] [Google Scholar]
- 41.Kale SD, Gu B, Capelluto DG, Dou D, Feldman E, Rumore A, Arredondo FD, Hanlon R, Fudal I, Rouxel T. 2010. External lipid PI3P mediates entry of eukaryotic pathogen effectors into plant and animal host cells. Cell 142:284–295. doi: 10.1016/j.cell.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 42.Dove SK, Cooke FT, Douglas MR, Sayers LG, Parker PJ, Michell RH. 1997. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature 390:187–192. doi: 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- 43.Okumura K, Itoh A, Isogai E, Hirose K, Hosokawa Y, Abiko Y, Shibata T, Hirata M, Isogai H. 2004. C-terminal domain of human CAP18 antimicrobial peptide induces apoptosis in oral squamous cell carcinoma SAS-H1 cells. Cancer Lett 212:185–194. doi: 10.1016/j.canlet.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Ghavami S, Asoodeh A, Klonisch T, Halayko AJ, Kadkhoda K, Kroczak TJ, Gibson SB, Booy EP, Naderi-Manesh H, Los M. 2008. Brevinin-2R1 semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J Cell Mol Med 12:1005–1022. doi: 10.1111/j.1582-4934.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broberg CA, Zhang L, Gonzalez H, Laskowski-Arce MA, Orth K. 2010. A Vibrio effector protein is an inositol phosphatase and disrupts host cell membrane integrity. Science 329:1660–1662. doi: 10.1126/science.1192850. [DOI] [PubMed] [Google Scholar]
- 46.Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B. 2002. Conversion of PtdIns(4, 5)P2 into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J 21:5069–5078. doi: 10.1093/emboj/cdf522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.García-Sáez AJ, Buschhorn SB, Keller H, Anderluh G, Simons K, Schwille P. 2011. Oligomerization and pore formation by equinatoxin II inhibit endocytosis and lead to plasma membrane reorganization. J Biol Chem 286:37768–37777. doi: 10.1074/jbc.M111.281592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reference deleted.
- 49.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr Sect D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]