

Abstract
Recently, we demonstrated in female mice that protection against ANG II-induced hypertension and associated cardiovascular changes depend on cytochrome P-450 (CYP)1B1. The present study was conducted to determine if Cyp1b1 gene disruption ameliorates renal dysfunction and organ damage associated with ANG II-induced hypertension in female mice. ANG II (700 ng·kg−1·min−1) infused by miniosmotic pumps for 2 wk in female Cyp1b1+/+ mice did not alter water consumption, urine output, Na+ excretion, osmolality, or protein excretion. However, in Cyp1b1−/− mice, ANG II infusion significantly increased (P < 0.05) water intake (5.50 ± 0.42 ml/24 h with vehicle vs. 8.80 ± 0.60 ml/24 h with ANG II), urine output (1.44 ± 0.37 ml/24 h with vehicle vs. 4.30 ± 0.37 ml/24 h with ANG II), and urinary Na+ excretion (0.031 ± 0.016 mmol/24 h with vehicle vs. 0.099 ± 0.010 mmol/24 h with ANG II), decreased osmolality (2,630 ± 79 mosM/kg with vehicle vs. 1,280 ± 205 mosM/kg with ANG II), and caused proteinuria (2.60 ± 0.30 mg/24 h with vehicle vs. 6.96 ± 0.55 mg/24 h with ANG II). Infusion of ANG II caused renal fibrosis, as indicated by an accumulation of renal interstitial α-smooth muscle actin, collagen, and transforming growth factor-β in Cyp1b1−/− but not Cyp1b1+/+ mice. ANG II also increased renal production of ROS and urinary excretion of thiobarburic acid-reactive substances and reduced the activity of antioxidants and urinary excretion of nitrite/nitrate and the 17β-estradiol metabolite 2-methoxyestradiol in Cyp1b1−/− but not Cyp1b1+/+ mice. These data suggest that Cyp1b1 plays a critical role in female mice in protecting against renal dysfunction and end-organ damage associated with ANG II-induced hypertension, in preventing oxidative stress, and in increasing activity of antioxidant systems, most likely via generation of 2-methoxyestradiol from 17β-estradiol.
Keywords: angiotensin II, cytochrome P-450 1B1-deficient female mice, kidney dysfunction, increased oxidative stress, reduced antioxidant activity
it is well established that male subjects are more prone to developing cardiovascular diseases, including hypertension and renal dysfunction, compared with premenopausal female subjects of the same age (31, 42, 53). Sex differences in hypertension and renal disease have also been demonstrated in experimental models, including ANG II- and DOCA-salt-induced hypertension and Dahl salt-sensitive and spontaneously hypertensive rats (SHRs) (18, 37, 43, 47, 61). Female subjects are protected or have a substantially lower increase in blood pressure (BP) and associated cardiovascular and renal dysfunction than do their male counterparts in these models of hypertension (14, 18, 37, 43, 47, 56, 61). Sex differences in the development of hypertension have been attributed to differential effects of sex hormones on the renin-angiotensin system and sex chromosomes (19). Testosterone stimulates the expression of renal angiotensinogen and/or renin mRNA in SHRs (7, 13) and Dahl salt-sensitive rats (63). Estrogen also increases plasma and tissue levels of angiotensinogen and has been shown to either increase (3, 34) or decrease (49) renin levels. In additon, estrogen reduces angiotensin-converting enzyme (ACE) activity in blood and tissues and increases circulating levels of ANG(1–7) (46, 49, 55). Estrogen downregulates expression of the ANG II type 1 (AT1) receptor but upregulates expression of the ANG II type 2 (AT2) receptor in the vasculature and kidney; ovariectomy downregulates AT2 receptor expression, which is prevented by estrogen in SHRs (52). AT2 receptor blockade in mice minimizes ANG II-induced hypertension (45), and Mas receptor antagonists reverse protection against hypertension in female SHRs (55). Thus, the effect of estrogen to downregulate the prohypertensive components and upregulate the antihypertensive components of the renin-angiotensin system contribute to lower BP in female subjects.
We have previously reported that cytochrome P-450 (CYP)1B1, which is highly expressed in the cardiovascular and renal systems and is capable of metabolizing fatty acids, retinoids, and sex steroids, contributes to the development of hypertension and associated pathogenesis in male mice, mostly by generating ROS (20, 21, 23). However, in female mice, CYP1B1, which metabolizes 17β-estradiol to 2-hydroxyestradiol, which is then converted by catechol-O-methyltransferase (COMT) to 2-methoxyestradiol (2-MeE2), protects against ANG II-induced hypertension and associated cardiovascular pathophysiological changes, including oxidative stress (22). ANG II, via its actions in the kidney, plays an important role in regulating BP, and increased levels of ANG II promote renal dysfunction and end-organ damage (9, 17, 20, 21, 44). Premenopausal female subjects are protected against the progression of renal disease, and estrogen and some of its metabolites exert renoprotective effects (12, 35). These observations and the demonstration that CYP1B1 is expressed in the kidney led to the hypothesis that CYP1B1 protects against renal dysfunction and end-organ damage associated with ANG II-induced hypertension in female subjects. To test this hypothesis, we investigated the effect of Cyp1b1 gene disruption on the actions of ANG II on renal function and the underlying mechanism(s) in female mice.
MATERIALS AND METHODS
Materials.
ANG II was purchased from Bachem (Torrance, CA), and dihydroethidium (DHE) was from Invitrogen (Carlsbad, CA). CYP1B1 antibody was purchased from BD Biosciences (Franklin Lakes, NJ), and antibodies against α-smooth muscle (α-SMA), 3-nitrotyrosine (3-NT), and transforming growth factor (TGF)-β were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). RT-PCR primers for ACE, AT1A receptor, ACE2, AT2 receptor, and Mas receptor were purchased from Integrated DNA Technologies (Coralville, IA); probes were purchased from Roche Diagnostics (Indianapolis, IN). All other chemicals were purchased from Sigma (St. Louis, MO).
Animals.
All experiments were performed according to protocols approved by our Institutional Animal Care and Use Committee in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Cyp1b1−/− mice were validated as previously described (4) and backcrossed 10 generations to a C57BL/6 background, and then brother-sister mated to generate a homozygous line. Female C57BL/6 (Cyp1b1+/+, Jackson Laboratory, Bar Harbor, ME) mice were used as control animals throughout the experiments for comparison of drug effects. All animals were 20–30 g and ∼8 wk of age at the beginning of the experiments. The genotype of Cyp1b1+/+ and Cyp1b1−/− mice was routinely assessed in our laboratory by PCR as previously described (6). For PCR analysis, genomic DNA was obtained from tail snips using the Wizard SV Genomic DNA Purification System (Promega, Madison, WI) according to the manufacturer's instructions.
Metabolic cage study for analysis of renal function.
Mice were anesthetized with ketamine (87 mg/kg ip) and xylazine (13 mg/kg ip), and micro-osmotic pumps were implanted subcutaneously to infuse ANG II (700 ng·kg−1·min−1) or vehicle (0.9% saline) for 14 days (model 1002, Alzet, Cupertino, CA). Systolic BP was measured twice weekly using a noninvasive tail-cuff method (model XBP 1000, Kent Scientific). The data on systolic BP have been previously reported (22). To assess renal function, these mice were housed on day 12 of ANG II infusion in metabolic cages for 24 h for the measurement of water consumption and separation of urine from fecal material and food waste. Urine was collected in tubes that contained a small volume of mineral oil to prevent evaporation. Animals were euthanized on day 14, and kidneys were collected for biochemical and histological analysis. After the calculation of volume, urine was aliquoted and stored at −80°C for further analysis. Urine was analyzed for osmolality using a Vapro vapor pressure osmometer (model 5520, Wescor, South Logan, UT), protein content by the standard Bradford method, and Na+ concentration by a flame photometer (model 443, Instrumentation Laboratory, Lexington, MA). Albumin concentration in urine samples was measured by a mouse albumin ELISA kit (Bethyl Laboratroies, Montgomery, TX) according to the manufacturer's instructions. Creatinine concentration in plasma samples was determined by HPLC/MS/MS at the Mouse Metabolic Phenotyping Center at Yale School of Medicine (New Haven, CT). For plasma collection, animals were anesthetized, and blood was withdrawn directly from the abdominal aorta and transferred to K+-EDTA tubes (BD Microtainer, BD Biosciences). Blood was centrifuged at 1,500 g for 15 min at 4°C, and plasma was collected and stored at −80°C until further analysis.
CYP1B1 activity assay.
CYP1B1 activity was determined using the P450-Glo Assay Kit (Promega) as previously described (20). At the completion of the experiments, animals were euthanized as described above, the left ventricle was punctured, and blood was flushed by perfusion with cold saline (3 min). Kidneys were dissected free, cleaned of surrounding tissue, snap frozen in liquid N2, and stored at −80°C until use. Kidney samples were homogenized in ice-cold 0.1 M potassium phosphate buffer (pH 7.4) using a TissueLyser II (2 × 3 min). After homogenization, samples were centrifuged at 10,000 g for 20 min at 4°C, and the supernatant was collected and stored at −80°C until further use. Protein content in the samples was determined by the Bradford method, and 500 μg protein was added to a reaction mixture containing 20 μM L-CEE substrate and 0.1 M potassium phosphate buffer (pH 7.4) and incubated at 37°C for 10 min. NADPH (final concentration: 100 μM) was added, and the solution was further incubated at 37°C for 45 min. Finally, a 1:1 volume of luciferin detection reagent was added to the samples, and they were mixed for 10 s, after which they were incubated at room temperature for 20 min. Luminescence was measured with a luminometer (model TD-20/20, Turner Designs, Sunnyvale, CA). Potassium phosphate buffer was used as a blank and subtracted from each reading; activity was expressed as relative luminescence units.
Western blot analysis.
Mice were euthanized and kidneys were removed as described above. Kidney samples were homogenized in lysis buffer, and protein content was determined by the Bradford method. Approximately 10 μg protein was loaded and resolved on 8% SDS-polyacrylamide gels and processed for Western blot analysis as previously described (20, 62). Blots were probed with different primary antibodies and the corresponding secondary antibodies, and the intensity of the bands was measured with ImageJ 1.42 software (http://rsb.info.nih.gov/nih-image; NIH). Protein expression of CYP1B1 was calculated as a ratio of expression of β-actin.
Immunohistochemical analysis.
At the completion of the experiments, animals were anesthetized as described above, the carotid artery was cannulated, and animals were perfused with saline (3 min). The kidney was dissected free and placed in OCT compound (Sakura Finetek USA, Torrance, CA). Sections (10 μm) were processed α-SMA (measure of myofibroblasts), TGF-β, and Masson's trichrome staining (collagen deposition) as previously described (33). Additional sections were stained for 3-NT [an indicator of peroxynitrite, the resultant compound of the reaction between nitric oxide (NO) and superoxide] as previously described (2). Stained sections were viewed with an Olympus inverted system microscope (model BX41, Olympus America) and photographed with a SPOT Insight digital camera (model Insight 2MP Firewire, Diagnostic Instruments, Sterling Heights, MI). Staining of TGF-β, collagen, 3-NT, and α-SMA was quantitated by taking the average of three fields from each slide to calculate the mean values for three animals using ImageJ (rsb.info.nih.gov/ij/). Data are presented as the percent area of positive staining.
Measurements of renal mRNA expression of ACE, AT1A receptor, ACE2, AT2 receptor, and Mas receptor.
RNA was extracted from snap-frozen kidneys using the TRIzol method (8). Reverse transcription was performed using the Transcriptor First Strand cDNA Synthesis Kit (Roche) with 1 μg total RNA. ACE, AT1A receptor, ACE2, AT2 receptor, and Mas receptor expressions were analyzed by real-time RT-PCR. Quantitative real-time PCR was performed in 384-well plates with a LightCycler (LC)480 (Roche) using LC480 Master Mix and a Universal Probe Library (UPL) probe (Roche) at a concentration of 10 μM with a final reaction volume of 10 μl under the following conditions: 95°C for 5 min for activation and 45 cycles of 95°C for 10 s, 60°C for 60 s, and 72°C for 10 s for amplification. After six endogenous control genes were tested, β-actin was used as an endogenous control. The sequences of primers and the relevant probes (UPL) for ACE, AT1A receptor, ACE2, AT2 receptor, Mas receptor, and β-actin are shown in Table 1. All samples were analyzed in triplicate. Relative mRNA levels were normalized to the housekeeping gene mRNA in the same sample of cDNA. Expression of ACE, AT1A receptor, ACE2, AT2 receptor, and Mas receptor relative to β-actin in each sample was calculated on the basis of the ΔΔCt method (where Ct is threshold cycle) as previously described (48).
Table 1.
Primer sequences for measuring renal RNA expression from genes associated with the ANG II pathway
| Sequence |
||||
|---|---|---|---|---|
| Identification | Forward | Reverse | Amplicon Length | UPL Probe Number |
| AT1A | 5′-GGCTTGAGTCCTGGTCCAC-3′ | 5′-CAGCCATTTTATACCAATCTTTCA-3′ | 131 | 41 |
| AT2 | 5′-GGAGCTCGGAACTGAAAGC-3′ | 5′-CTGCAGCAACTCCAAATTCTT-3′ | 131 | 41 |
| Mas | 5′-GCATCGTCTTTCAGCTACTTCTC-3′ | 5′-TCTGGCAGGATCACAGAGTTT-3′ | 101 | 52 |
| ACE | 5′-TCTGCTTCCCCAACAAGACT-3′ | 5′-AGGATGTTGGTGAGTCTGG-3′ | 61 | 62 |
| ACE2 | 5′-TGTAGAACGTACCTTCGCAGAG-3′ | 5′-GTAGGAAGGGTAGGTATCCATCAA-3′ | 91 | 101 |
| β-Actin | 5′-AAGGCCAACCGTGAAAAGAT-3′ | 5′-GTGGTACGACCAGAGGCATAC-3′ | 110 | 56 |
UPL, Universal Probe Library; AT1A, ANG II type 1A receptor; AT2, ANG II type 2 receptor; ACE, ANG-converting enzyme.
Measurement of NADPH oxidase activity.
NADPH oxidase activity was determined in tissue homogenates by measuring lucigenin (N,N′-dimethyl-9,9′-biacridinium dinitrate)-enhanced chemiluminescence as previously described (64) with some modifications (20). After anesthesia (described above), the kidney was isolated, cleaned of surrounding tissue, snap frozen in liquid N2, and stored at −80°C until use. Tissue samples were homogenized (2 × 3 min) in ice-cold lysis buffer containing protease inhibitors (20 mmol/l phosphate buffer, 1 mmol/l EGTA, 10 μg/ml aprotinin, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, 0.5 mmol/l PMSF, and 150 mmol/l sucrose) using a TissueLyser II (Qiagen). After homogenization, samples were sonicated and centrifuged at 10,000 g for 20 min at 4°C, and the supernatant was collected and stored at −80°C until further use. Protein content in the samples was determined by the Bradford method, and equal amounts of protein were combined 1:1 with a reaction mixture containing 5 μmol/l lucigenin (final concentration) and 100 μmol/l NADPH (final concentration). Luminescence was measured every minute for 10 min using a luminometer. Lysis buffer was used as a blank and subtracted from each reading, and activity was expressed as arbitrary units.
Measurement of renal superoxide production.
To measure superoxide production, kidney sections were exposed to DHE following previously described and validated methods (32). Fresh, unfixed kidney and artery samples were placed in OCT compound (Sakura Finetek USA) and frozen at −80°C. Kidney and vascular ring segments were cut into 30-μm sections using a cryostat (model CM1850, Leica Microsystems, Bannockburn, IL) and placed on a glass slide. Sections were incubated in PBS for 30 min at 37°C, and DHE (10 μmol/l) was then topically applied (59). Coverslips were applied, and sections were further incubated at 37°C in a light-protected, humidified chamber for 30 min. Sections were then rinsed in PBS, and fluorescence was detected using a 585-nm filter and an Olympus inverted system microscope (model IX50, Olympus America). Images were photographed with an Olympus digital camera (model DP71, Olympus America) and analyzed using ImageJ (version 1.42).
Measurement of urinary levels of thiobarbituric acid-reactive substances.
Urine levels of thiobarbituric acid-reactive substances (TBARS), an indicator of lipid peroxidation, were measured using a TBARS assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions.
Measurement of renal activity of antioxidant enzymes.
The renal activity of antioxidant enzymes (SOD and catalase) was measured. Total SOD activity was measured in kidney tissue using a SOD assay kit (Cayman Chemical) according to the manufacturer's instructions. Catalase activity was measured in kidney tissue using a Catalase assay kit (Cayman Chemical) according to the manufacturer's instructions.
Measurement of urinary levels of nitrate/nitrite.
Urine levels of nitrate/nitrite (NOx), an indicator of NO bioavailability, were measured using a NOx colorimetric assay kit (Cayman Chemical) according to the manufacturer's instructions.
Measurement of urinary levels of 2-MeE2.
Urine levels of 2-MeE2 were determined using a 2-MeE2 enzyme immunoassay kit (Cayman Chemical) according to the manufacturer's instructions.
Statistical analysis.
Data were analyzed by one-way ANOVA followed by a Neuman-Keuls post hoc test or Student's t-test. Values of a minimum of three different experiments are expressed as means ± SE. P values of <0.05 were considered statistically significant.
RESULTS
Infusion of ANG II increased renal CYP1B1 activity and expression in female mice.
CYP1B1 activity and protein expression were increased in kidneys of ANG II-infused Cyp1b1+/+ mice (Fig. 1, A and B, respectively). CYP1B1 activity and protein expression were absent in Cyp1b1−/− mice (Fig. 1, A and B, respectively).
Fig. 1.

ANG II-induced hypertension is associated with increased renal cytochrome P-450 (CYP)1B1 activity and expression in female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk, as described in materials and methods. Animals were euthanized, and renal tissue was collected to measure CYP1B1 activity using the P450-Glo assay, as described in materials and methods. A: activity of CYP1B1 expressed as relative luminescence units (RLU). B: CYP1B1 protein expression was measured by Western blot analysis in renal tissue from vehicle- and ANG II-infused Cyp1b1+/+ and Cyp1b1−/− mice using ∼10 μg protein for loading, as described in materials and methods. CYP1B1 protein expression was normalized against β-actin, which was used as a loading control. au, Arbitrary units. Data are expressed as means ± SE; n = 3–5 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle.
ANG II-induced hypertension was associated with increased thirst and renal dysfunction in Cyp1b1−/− but not Cyp1b1+/+ mice.
Basal water intake and urine output were not different between Cyp1b1+/+ and Cyp1b1−/− mice (Table 2). Infusion of ANG II did not alter 24-h water consumption or urine output in Cyp1b1+/+ mice; however, in Cyp1b1−/− mice, infusion of ANG II caused a marked increase in daily water consumption and urine output (Table 2). Plasma levels of creatinine were not altered by ANG II in Cyp1b1+/+ or Cyp1b1−/− mice. ANG II infusion decreased urine osmolality, increased urinary excretion of Na+, and caused proteinuria and albuminuria in Cyp1b1−/− but not Cyp1b1+/+ mice (Table 2).
Table 2.
Renal dysfunction caused by infusion of ANG II is observed in Cyp1b1−/− but not Cyp1b1+/+ female mice
|
Cyp1b1+/+ Mice |
Cyp1b1−/− Mice |
|||
|---|---|---|---|---|
| Parameter | Vehicle | ANG II | Vehicle | ANG II |
| Water intake, ml/24 h | 5.20 ± 0.12 | 5.40 ± 0.94 | 5.50 ± 0.42 | 8.80 ± 0.60* |
| Urine output, ml/24 h | 1.44 ± 0.26 | 2.46 ± 0.74 | 1.44 ± 0.37 | 4.30 ± 0.37* |
| Plasma creatinine, mg/dl | 0.078 ± 0.002 | 0.077 ± 0.005 | 0.072 ± 0.002 | 0.075 ± 0.010 |
| Urinary osmolality, mosM/kg | 2720 ± 115 | 2140 ± 262 | 2630 ± 79 | 1281 ± 205* |
| Urinary Na+ excretion, mmol/24 h | 0.045 ± 0.018 | 0.045 ± 0.021 | 0.031 ± 0.016 | 0.099 ± 0.010* |
| Proteinuria, mg/24 h | 2.49 ± 0.47 | 2.64 ± 0.50 | 2.60 ± 0.30 | 6.96 ± 0.55* |
| Albuminuria, mg/24 h | 0.23 ± 0.13 | 0.30 ± 0.10 | 0.13 ± 0.03 | 1.63 ± 0.33* |
| Body weight, g | 20.1 ± 0.3 | 20.6 ± 1.2 | 21.0 ± 0.6 | 22.3 ± 0.7 |
| Food intake, g/24 h | 5.03 ± 0.21 | 4.62 ± 0.84 | 5.83 ± 0.18 | 5.31 ± 1.13 |
Data are expressed as means ± SE; n = 5 for all experiments. Cytochrome P-450 (Cyp)1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk and placed in metabolic cages for 24 h before the experiments were completed. The parameters listed above were determined as described in materials and methods.
P < 0.05, vehicle versus the corresponding value from ANG II-infused animals.
Cyp1b1 gene disruption resulted in renal damage caused by infusion of ANG II.
Infusion of ANG II was associated with increased interstitial fibrosis, as indicated by increased staining of interstitial α-SMA and TGF-β in kidneys of Cyp1b1−/− but not Cyp1b1+/+ mice (Figs. 2 and 3, respectively). Staining of kidney sections with Masson's trichrome revealed collagen deposition in the interstitial spaces of kidneys from ANG II-treated Cyp1b1−/− mice; collagen deposition was reduced in Cyp1b1+/+ mice (Fig. 4).
Fig. 2.

Renal interstitial accumulation of α-smooth muscle actin (α-SMA) caused by ANG II infusion in Cyp1b1−/− but not Cyp1b1+/+ female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk, as described in materials and methods. A: ANG II infusion increased renal interstitial staining of α-SMA in Cyp1b1−/− mice, which was minimized in Cyp1b1+/+ mice. B: graph showing quantified data. Data are expressed as means ± SE; n = 3 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice.
Fig. 3.

Renal deposition of transforming growth factor (TGF)-β caused by ANG II infusion in Cyp1b1−/− but not Cyp1b1+/+ female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk, as described in materials and methods. A: ANG II infusion increased interstitial staining of TGF-β in kidneys of Cyp1b1−/− but not Cyp1b1+/+ mice. B: graph showing quantified data. Data are expressed as means ± SE; n = 3 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice.
Fig. 4.

Renal collagen deposition caused by ANG II infusion in Cyp1b1−/− but not Cyp1b1+/+ female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk, as described in materials and methods. A: ANG II infusion increased collagen deposition, as revealed by blue Masson's trichrome staining in kidneys of ANG II-treated Cyp1b1−/− but not Cyp1b1+/+ mice. B: graph showing quantified data. Data are expressed as means ± SE; n = 3 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice.
Infusion of ANG II increased renal NADPH oxidase activity, superoxide production, and urinary excretion of TBARS in Cyp1b1−/− but not Cyp1b1+/+ mice.
Infusion of ANG II was associated with increased renal NADPH oxidase activity in Cyp1b1−/− but not Cyp1b1+/+ mice (Fig. 5A). The increase in NADPH oxidase activity in Cyp1b1−/− mice correlated with an increase in renal superoxide production, as indicated by increased 2-hydroxyethidium fluorescence intensity within the kidney sections, specifically in the glomerulus; there was no increase in 2-hydroxyethidium fluorescence in ANG II-infused Cyp1b1+/+ mice (Fig. 5, B and C). ANG II infusion was also associated with increased urinary excretion of TBARS, an indicator of lipid peroxidation, in Cyp1b1−/− but not Cyp1b1+/+ mice (Fig. 5D).
Fig. 5.

ANG II infusion increased renal NADPH oxidase activity, superoxide production, and urinary excretion of thiobarbituric acid-reactive substances (TBARS) in Cyp1b1−/− female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk. A: NADPH oxidase activity in renal homogenates. B: superoxide production, as determined by fluorescence intensity of 2-hydroxyethidium, in the kidney. Representative photomicrographs of kidneys from mice in each of the different treatment groups after incubation with dihydroethidium are shown (Arrows indicate glomeruli). C: graph showing quantified data. D: at the completion of the experiments, urine was collected, and TBARS excretion was determined using a commercially available kit. Data are expressed as means ± SE; n = 4–5 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice.
Infusion of ANG II increased renal SOD activity in Cyp1b1+/+ but not Cyp1b1−/− mice and decreased renal catalase activity in Cyp1b1−/− but not Cyp1b1+/+ mice.
In Cyp1b1+/+ mice, infusion of ANG II was associated with an increase in total SOD activity in the kidney; this increase was absent in ANG II-infused Cyp1b1−/− mice (Fig. 6A). In contrast, renal catalase activity was decreased in Cyp1b1−/− mice infused with ANG II but was unchanged in ANG II-infused Cyp1b1+/+ mice (Fig. 6).
Fig. 6.
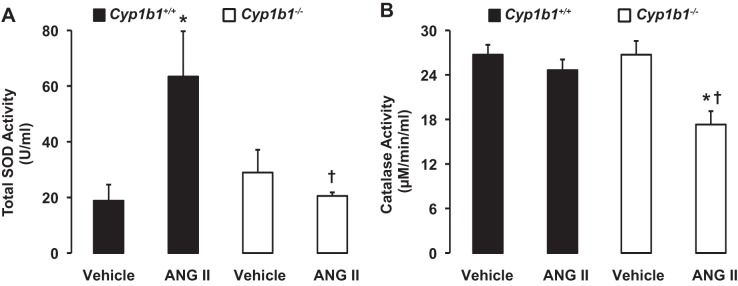
ANG II infusion differentially regulates activities of antioxidant defense enzymes in kidneys of Cyp1b1+/+ and Cyp1b1−/− female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk. At the completion of the experiments, renal tissue was collected to measure SOD activity (A) and catalase activity (B) using commercially available kits. Data are expressed as means ± SE; n = 5 for all experiments. *P < 0.05 ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice.
Infusion of ANG II was associated with decreased urinary excretion of NOx and increased renal expression of 3-NT in Cyp1b1−/− but not Cyp1b1+/+ mice.
Urinary excretion of NOx, an indicator of NO bioavailability, was not altered in Cyp1b1+/+ mice infused with ANG II; however, in Cyp1b1−/− mice, infusion of ANG II resulted in a dramatic decrease in urinary excretion of NOx (Fig. 7A). The decreased urinary excretion of NOx in Cyp1b1−/− mice correlated with an increase in 3-NT-positive staining in the kidney, an indicator of peroxynitrite formation, which resulted from the reaction between NO and superoxide; this increase was minimized in Cyp1b1+/+ mice (Fig. 7, B and C).
Fig. 7.
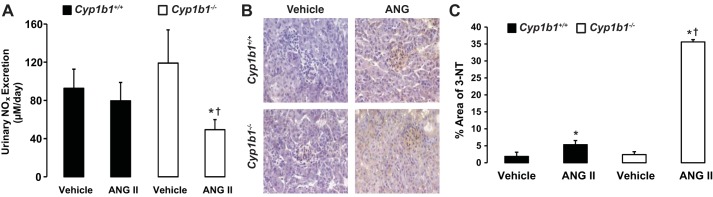
Infusion of ANG II decreased nitric oxide (NO) bioavailability and increased production of 3-nitrotyrosine (3-NT) in kidneys of Cyp1b1−/− but not Cyp1b1+/+ female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk. A: at the completion of the experiments, urine was collected, and excretion of nitrite/nitrate (NOx) was determined using a commercially available kit. B: increased staining of 3-NT, an indicator of peroxynitrite formation, was observed in kidney sections from ANG II-treated Cyp1b1−/− mice but not Cyp1b1+/+ mice. C: graph showing quantified data of 3-NT-positive staining. Data are expressed as means ± SE; n = 3 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice.
ANG II infusion decreased renal mRNA expression of Mas receptor but not AT1A receptor, AT2 receptor, ACE, or ACE2 in Cyp1b1+/+ mice and decreased expression of AT2 receptor, Mas receptor, and ACE2 in Cyp1b1−/− mice.
Cyp1b1 gene disruption did not alter basal mRNA encoding AT1A receptor, AT2 receptor, ACE, ACE2, or Mas receptor (Fig. 8, A–E). Infusion of ANG II did not alter mRNAs encoding AT1A receptor, AT2 receptor, ACE, or ACE2 compared with basal levels of these mRNAs in Cyp1b1+/+ and Cyp1b1−/− mice (Fig. 8, A–D). However, infusion of ANG II decreased mRNA expression of Mas receptor in Cyp1b1+/+ and Cyp1b1−/− mice (Fig. 8E). Comparison of the expression of renal mRNA in these two mouse phenotypes showed that, during ANG II infusion, AT2 receptor mRNA expression and ACE2 mRNA expression were decreased in Cyp1b1−/− mice compared with Cyp1b1+/+ mice (Fig. 8, B and D, respectively).
Fig. 8.

Infusion of ANG II is not associated with changes in renal ANG II type 1A (AT1A) receptor or angiotensin-converting enzyme (ACE)2 expression in Cyp1b1+/+ and Cyp1b1−/− mice but decreased ANG II type 2 (AT2) receptor and ACE2 expression in Cyp1b1−/− mice and Mas receptor expression in Cyp1b1+/+ and Cyp1b1−/− female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk. A–E: renal tissue was collected to measure mRNA expression of AT1A receptor (A), AT2 receptor (B), ACE (C), ACE2 (D), and Mas receptor (E), as described in materials and methods. mRNA gene expression was normalized against β-actin, which was used as a housekeeping gene, and calculated on the basis of the ΔΔCt method (where Ct is threshold cycle). Data are expressed as means ± SE; n = 3 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice.
Measurement of urinary levels of 2-MeE2.
Basal urinary excretion of 2-MeE2 was decreased in Cyp1b1−/− mice (Fig. 9). Infusion of ANG II increased urinary excretion of 2-MeE2 in Cyp1b1+/+ but not Cyp1b1−/− mice (Fig. 9).
Fig. 9.
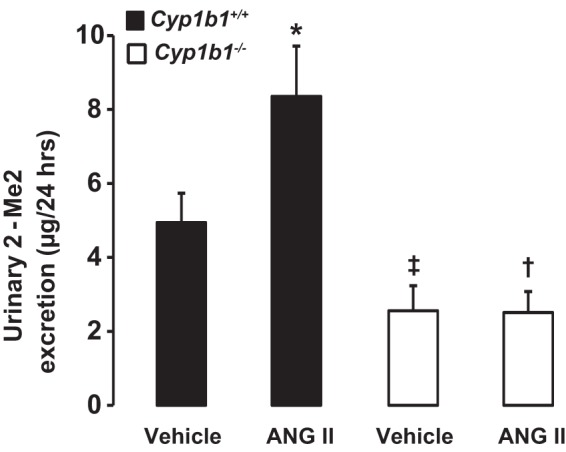
Infusion of ANG II increased 2-methoxyestradiol (2-MeE2) production in Cyp1b1+/+ but not Cyp1b1−/− female mice. Cyp1b1+/+ and Cyp1b1−/− mice were infused with vehicle or ANG II for 2 wk. At the completion of the experiments, urine was collected, and excretion of 2-MeE2 was determined using a commercially available kit. Data are expressed as means ± SE; n = 4 for all experiments. *P < 0.05, ANG II vs. the corresponding value with vehicle; †P < 0.05, ANG II-infused Cyp1b1−/− mice vs. ANG II-infused Cyp1b1+/+ mice; ‡P < 0.05, vehicle-infused Cyp1b1−/− mice vs. vehicle-infused Cyp1b1+/+ mice.
DISCUSSION
The major finding of the present study is that CYP1B1, which contributes to renal dysfunction and organ damage in male mice associated with ANG II-induced hypertension (20, 21), had an opposite role in female mice. CYP1B1 protects against ANG II-induced renal dysfunction and end-organ damage in female mice by promoting decreased oxidative stress and increased activity of antioxidant systems, most likely as a result of the effect of 2-MeE2 from 17β-estradiol. Infusion of ANG II for 2 wk did not alter water intake or urine output or promote renal dysfunction in Cyp1b1+/+ mice, but in Cyp1b1−/− mice, ANG II increased water intake, urine output, and Na+ excretion, decreased urine osmolality, and caused marked proteinuria and albuminuria. These observations, together with our demonstration that ANG II increased CYP1B1 activity and expression in Cyp1b1+/+ mice, suggest that CYP1B1 plays a protective role against ANG II-induced renal dysfunction in female mice. Furthermore, the finding that ANG II increased water intake in Cyp1b1−/− but not Cyp1b1+/+ mice indicated that Cyp1b1 could also be involved in regulating the central action of ANG II on thirst in female mice. Alternatively, the increased urine output in Cyp1b1−/− mice could be due to increased water intake. Another interesting finding was that ANG II increased Na+ excretion in Cyp1b1−/− mice, which could be due to an increased effect of ANG II on BP that overrides the direct tubular effects of ANG II. In contrast to female Cyp1b1−/− mice, in male Cyp1b1+/+ mice, where ANG II markedly increases BP, it also increased Na+ excretion but minimized the increase in both BP and Na+ excretion in Cyp1b1−/− mice (20). Further studies to determine fractional Na+ excretion and its time course and total body Na+ are required to address the role of CYP1B1 on the actions of ANG II on Na+ balance in female and male mice. Our demonstration that ANG II in Cyp1b1−/− but not Cyp1b1+/+ mice caused proteinuria and albuminuria, as well as renal fibrosis, as indicated by interstitial α-smooth muscle actin, TGF-β, and collagen accumulation, suggests that CYP1B1 also protects against ANG II-induced organ damage in female mice. The degree of these changes produced by ANG II in Cyp1b1−/− mice appeared to be insufficient to alter glomerular filtration rate, as indicated by the lack of change in plasma levels of creatinine.
The mechanism by which ANG II produces renal dysfunction and end-organ damage in Cyp1b1−/− mice could be due to increased BP. Although we cannot exclude this possibility, ANG II may also produce cardiovascular or renal pathophysiological changes independent of increased BP (15, 25, 30, 54). For example, treatment with triple therapy (hydralazine, reserpine, and hydrochlorothiazide) prevents increased BP but not end-organ damage, inflammation, or cellular growth in the kidney of transgenic rats carrying both human renin and angiotensinogen genes (30). ANG II is known to increase production of ROS, which contributes to the development of hypertension and end-organ damage in male rats and mice (20, 21, 23, 41). However, ROS do not appear to contribute to the development of hypertension in various models of experimental hypertension in female subjects (27). The progression of renal disease caused by renal artery pseudoaneurysm, which is minimized in female rats by 17β-estradiol, is associated with decreased NADPH oxidase activity and expression of its subunit p22phox (24). ANG II infusion does not alter the expression of components of NADPH oxidase or urinary excretion of TBARS and reduces NOx in the female mouse kidney (50). In the present study, infusion of ANG II did not alter NADPH oxidase activity, production of ROS, or urinary excretion of NOx, an index of NO production, but increased SOD activity and did not affect catalase activity in Cyp1b1+/+ mice. However, in Cyp1b1−/− mice, ANG II infusion increased renal NADPH oxidase activity and ROS production, reduced urinary excretion of NOx, and increased 3-NT deposition and activity of SOD and catalase. These observations suggest that CYP1B1 plays a protective role in the female kidney against ANG II-induced oxidative stress by increasing the activity of antioxidants and minimizing an increase in the activity of oxidants and consequently preventing end-organ damage.
The protection against ANG II-induced hypertension in female subjects compared with male subjects has been attributed to the action of 17β-estradiol to downregulate AT1 receptors (10, 45) and upregulate AT2 receptors (52). Expression of ACE2 and ANG(1–7) levels, which are increased in female SHRs compared with male SHRs, has been shown to be responsible for the lower BP in female SHRs than in male SHRs (55). ANG II, via AT2 receptor activation, and ANG(1–7), generated via increased ACE2 activity through Mas and/or AT2 receptors, produce cardiovascular and renoprotective effects and counteract the actions of ANG II mediated via AT1 receptors (5). Although the renal expression of AT2 receptors in female rats, including SHRs, is higher than in male rats, infusion of ANG II does not alter their expression (46, 52). In the present study, Cyp1b1 gene deletion did not alter basal mRNA expression of AT1A receptor, AT2 receptor, Mas receptor, ACE, or ACE2 compared with Cyp1b1+/+ mice, indicating that basal expression of these components of the renin-angiotensin system in the kidney is independent of the Cyp1b1 gene. Infusion of ANG II did not alter renal mRNA expression of AT2 receptor or ACE2 in Cyp1b1+/+ or Cyp1b1−/− mice. ANG II produces a small, insignificant decrease in Mas receptor mRNA expression but an increase in protein expression in female SHRs (55). In our study, infusion of ANG II decreased Mas receptor mRNA expression in both Cyp1b1+/+ and Cyp1b1−/− mice, suggesting that it is likely to contribute to renal protection against ANG II in Cyp1b1+/+ mice. We, however, have no explanation for the decreased mRNA expression of Mas receptor in Cyp1b1−/− mice by ANG II.
In the present study, Cyp1b1 gene disruption abolished both basal and ANG II-induced increases in its activity; however, it did not alter basal renal function or cause kidney damage but allowed ANG II to cause renal dysfunction, oxidative stress, and end-organ damage. Therefore, it appears that CYP1B1 is not required to maintain basal renal function but is required to suppress the deleterious effect of ANG II in female mice. Since CYP1B1 is constitutively expressed, it appears that ANG II stimulates the production of a product that protects against the renal actions of this peptide in Cyp1b1+/+ mice. The protection against ANG II-induced hypertension in female rats and mice has been attributed to estrogen, which also protects against the progression of renal disease in female subjects (18, 37, 43, 61). Lack of estrogen produced by ovariectomy promotes renal dysfunction, and 17β-estradiol replacement improves renal function and associated pathogenesis in streptozotocin-induced diabetic rats (28).
CYP1B1 metabolizes 17β-estradiol into hydroxyestradiols (OHEs), namely, 2-OHE and 4-OHE, which are subsequently metabolized by COMT into 2-MeE2 and 4-MeE2 (40, 65). 2-MeE2 exerts cardiovascular protective effects (12). Recently, we showed that ANG II stimulates the production of 2-MeE2, which protects against ANG II-induced hypertension in female Cpy1b1+/+ mice (22). 17β-Estradiol, 2-OHE, and/or 2-MeE2 also exert several renoprotective effects, including inhibition of mesangial cell proliferation and synthesis of collagen (11), expression and activity of TGF-β, expression of collagen types I and IV (29, 36, 51), and increased expression of extracellular matrix-degrading metalloproteinase (39). 2-OHE also slows the progression of renal dysfunction in an experimental model of polycystic kidney disease (1). These observations, together with the demonstration that ANG II increased urinary levels of 2-MeE2 in Cyp1b1+/+ mice and decreased its level in Cyp1b1−/− mice, suggest that protection against ANG II-induced renal dysfunction and end-organ damage in Cyp1b1+/+ female mice is mediated by 2-MeE2. Since ANG II-induced changes in urinary levels of 2-MeE2 followed the same pattern in Cyp1b1−/− and Cyp1b1+/+ female mice to that observed in plasma levels, the urinary 2-MeE2 could be derived from extrarenal sources. However, in view of the expression of both CYP1B1 and COMT in the kidney, it is possible that urinary 2-MeE2 could also be derived from the kidney. Further studies are required to determine the effect of 2-OHE, MeE2, and other estrogen metabolites (26) on ANG II-induced renal dysfunction and end-organ damage in Cyp1b1−/− female and Cyp1b1+/+ male mice and their relationship to oxidative stress and antioxidant systems. Moreover, in view of an essential role of T cells in ANG II-induced hypertension in male mice (16), which is minimized in female mice and enhanced by ovariectomy (38), the contribution of T cells to17β-estradiol metabolites in modulating the actions of ANG II needs to be investigated. Furthermore, in light of the paradoxical effects of estrogen and its CYP1B1-derived metabolite 2-MeE2 to attenuate (57, 60) and 16α-hydroxyesterone (58) to promote hypoxia-induced pulmonary arterial hypertension, it is not known if Cyp1b1 gene disruption modifies any action of ANG II on the pulmonary circulation and right ventricular function in female mice.
In conclusion, the present study showed that CYP1B1 plays a critical role in female mice in protecting against ANG II-induced renal dysfunction and end-organ damage associated with hypertension by minimizing oxidative stress and by increasing activity of antioxidant systems. These renoprotective effects of CYP1B1 against ANG II in female mice are most likely mediated by the 17β-estradiol metabolite 2-MeE2. In contrast, in male mice, CYP1B1 contributes to ANG II-induced hypertension (20). Our preliminary observations indicate that in male mice, 6β-hydroxytestosterone, which can be generated by CYP1B1 from testosterone, contributes to ANG II-induced hypertension and its pathogenesis (Pingili A. K. et al., unpublished observations). Recently, it has been demonstrated that, in addition to gonadal hormones, sex chromosome complement plays an important role in the development of ANG II-induced hypertension (19). Therefore, further studies are required to elucidate the relationship between sex chromosome complement and CYP1B1 in the action of ANG II on renal function in both male and female subjects. The present study has important clinical implications: agents that inhibit activity of Cyp1b1 could be detrimental to renal function in female subjects by reducing the production of the Cyp1b1 metabolite 2-OHE and, subsequently, 2-MeE2, which have renoprotective effects.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-19134-38-39 (to K. U. Malik) and K01-HL-96410-04 (to A. Kanu). J. A. Moore was supported by a summer student fellowship from the American Society of Pharmacology and Experimental Therapeutics.
DISCLAIMER
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Heart, Lung, and Blood Institute.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.L.J. and K.U.M. conception and design of research; B.L.J., J.A.M., A.K.P., A.M.E., and X.R.F. performed experiments; B.L.J., J.A.M., and A.K.P. analyzed data; B.L.J., J.A.M., A.K.P., and K.U.M. interpreted results of experiments; B.L.J. and J.A.M. prepared figures; B.L.J. and K.U.M. drafted manuscript; B.L.J., A.K.P., A.K., F.J.G., and K.U.M. edited and revised manuscript; B.L.J., J.A.M., A.K.P., A.M.E., X.R.F., A.K., F.J.G., and K.U.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. David L. Armbruster for editorial assistance. Creatinine concentration in plasma samples was determined by HPLC/MS/MS at the Mouse Metabolic Phenotyping Center at Yale School of Medicine (New Haven, CT).
REFERENCES
- 1.Anderson S, Oyama TT, Lindsley JN, Schutzer WE, Beard DR, Gattone VHII, Komers R. 2-Hydroxyestradiol slows progression of experimental polycystic kidney disease. Am J Physiol Renal Physiol 302: F636–F645, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bank N, Kiroycheva M, Singhal PC, Anthony GM, Southan GJ, Szabo C. Inhibition of nitric oxide synthase ameliorates cellular injury in sickle cell mouse kidneys. Kidney Int 58: 82–89, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Brosnihan KB, Senanayake PS, Li P, Ferrario CM. Bi-directional actions of estrogen on the renin-angiotensin system. Braz J Med Biol Res 32: 373–381, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Buters JT, Sakai S, Richter T, Pineau T, Alexander DL, Savas U, Doehmer J, Ward JM, Jefcoate CR, Gonzalez FJ. Cytochrome P450 CYP1B1 determines susceptibility to 7,12-dimethyl[a]anthracene-induced lymphomas. Proc Natl Acad Sci USA 96: 1977–1982, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carey RM, Wang ZQ, Siragy HM. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 35: 155–163, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Castro DJ, Baird WM, Pereira CB, Giovanini J, Löhr CV, Fischer KA, Yu Z, Gonzalez FJ, Krueger SK, Williams DE. Fetal mouse Cyp1b1 and transplacental carcinogenesis from maternal exposure to dibenzo(a,l)pyrene. Cancer Prev Res (Phila) 1: 128–134, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen YF, Naftilan AJ, Oparil S. Androgen-dependent angiotensinogen and renin messenger RNA expression in hypertensive rats. Hypertension 19: 456–463, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Chomczynski P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 15: 532–537, 1993. [PubMed] [Google Scholar]
- 9.Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest 115: 1092–1099, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dean SA, Tan J, O'Brien ER, Leenen FH. 17β-Estradiol downregulates tissue angiotensin-converting enzyme and ANG II type 1 receptor in female rats. Am J Physiol Regul Integr Comp Physiol 288: R759–R766, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Dubey RK, Gillespie DG, Keller PJ, Imthurn B, Zacharia LC, Jackson EK. Role of methoxyestradiols in the growth inhibitory effects of estradiol on human glomerular mesangial cells. Hypertension 39: 418–424, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Dubey RK, Jackson EK. Potential vascular actions of 2-methoxyestradiol. Trends Endocrinol Metab 20: 374–379, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellison KE, Ingelfinger JR, Pivor M, Dzau VJ. Androgen regulation of rat renal angiotensinogen messenger RNA expression. J Clin Invest 83: 1941–1945, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fortepiani LA, Yanes L, Zhang H, Racusen LC, Reckelhoff JF. Role of androgens in mediating renal injury in aging SHR. Hypertension 42: 952–955, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Griffin SA, Brown WC, MacPherson F, McGrath JC, Wilson VG, Korsgaard N, Mulvany MJ, Lever AF. Angiotensin II causes vascular hypertrophy in part by a non-pressor mechanism. Hypertension 17: 626–635, 1991. [DOI] [PubMed] [Google Scholar]
- 16.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II-induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall JE. Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol Regul Integr Comp Physiol 250: R960–R972, 1986. [DOI] [PubMed] [Google Scholar]
- 18.Hinojosa-Laborde C, Lange DL, Haywood JR. Role of female sex hormones in the development and reversal of Dahl hypertension. Hypertension 35: 484–489, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Hong J, Zheng W, Wu X, Liu J, Acelbarger CM, Watkins R, Arbold AP, Sandberg K. Sex chromosome effects unmasked in angiotensin II-induced hypertension. Hypertension 55: 1275–1282, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jennings BL, Anderson LJ, Estes AM, Fang XR, Song CY, Campbell WB, Malik KU. Involvement of cytochrome P-450 1B1 in renal dysfunction, injury, and inflammation associated with angiotensin II-induced hypertension in rats. Am J Physiol Renal Physiol 302: F408–F420, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jennings BL, Anderson LJ, Estes AM, Yaghini FA, Fang XR, Porter J, Gonzalez FJ, Campbell WB, Malik KU. Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension 59: 348–354, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jennings BL, George LW, Pingili AK, Khan NS, Estes AM, Fang XR, Gonzalez FJ, Malik KU. Estrogen metabolism by cytochrome P450 1B1 modulates the hypertensive effect of angiotensin II in female mice. Hypertension 64: 134–140, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jennings BL, Sahan-Firat S, Estes AM, Das K, Farjana N, Fang XR, Gonzalez FJ, Malik KU. Cytochrome P450 1B1 contributes to angiotensin II-induced hypertension and associated pathophysiology. Hypertension 56: 667–674, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji H, Zheng W, Menini S, Pesce C, Kim J, Wu X, Mulroney SE, Sandberg K. Female protection in progressive renal disease is associated with estradiol attenuation of superoxide production. Gend Med 4: 56–71, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Letavernier E, Perez J, Bellocq A, Mesnard L, de Castro Keller A, Haymann JP, Baud L. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res 102: 720–728, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Lindsey SH. Importance of estrogen metabolites. Hypertension 64: 21–22, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Ruiz A, Sartori-Valinotti J, Yanes LL, Iliescu R, Reckelhoff JF. Sex differences in control of blood pressure: role of oxidative stress in hypertension in females. Am J Physiol Heart Circ Physiol 295: H466–H474, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mankhey RW, Bhatti F, Maric C. 17β-Estradiol replacement improves renal function and pathology associated with diabetic nephropathy. Am J Physiol Renal Physiol 288: F399–F405, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Matsuda T, Yamamoto T, Muraguchi A, Saatcioglu F. Cross-talk between transforming growth factor-β and estrogen receptor signaling through Smad3. J Biol Chem 276: 42908–42914, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Mervaala E, Müller DN, Schmidt F, Park JK, Gross V, Bader M, Breu V, Ganten D, Haller H, Luft FC. Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension 35: 587–594, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Messerli FH, Garavaglia GE, Schmieder RE, Sundgaard-Riise K, Nunez BD, Amodeo C. Disparate cardiovascular findings in men and women with essential hypertension. Ann Intern Med 107: 158–161, 1987. [DOI] [PubMed] [Google Scholar]
- 32.Miller FJ Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res 82: 1298–1305, 1998. [DOI] [PubMed] [Google Scholar]
- 33.Mori T, Cowley AW Jr. Role of pressure in angiotensin II-induced renal injury. Chronic servo-control of renal perfusion pressure in rats. Hypertension 43: 752–759, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Nasjletti A, Mason GM. Studies on angiotensinogen formation in a liver perfusion system. Circ Res 31, Suppl 2: 187–200, 1972. [PubMed] [Google Scholar]
- 35.Neugarten J. Gender and the progression of renal disease. J Am Soc Nephrol 13: 2807–2809, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Neugarten J, Medve I, Lei J, Silbiger SR. Estradiol suppresses mesangial cell type I collagen synthesis via activation of the MAP kinase cascade. Am J Physiol Renal Physiol 277: F875–F881, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Ouchi Y, Share L, Crofton JT, Iitake K, Brooks DP. Sex difference in the development of deoxycorticosterone-salt hypertension in the rat. Hypertension 9: 172–177, 1987. [DOI] [PubMed] [Google Scholar]
- 38.Pollow DP, Uhrlaub J, Romero-Aleshire MJ, Sandberg K, Nikolich-Zugich J, Brooks HL, Hay M. Sex differences in T-lymphocyte tissue infiltration and development of angiotensin II hypertension. Hypertension 64: 384–390, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Potier M, Elliot SJ, Tack I, Lenz O, Striker GE, Striker LJ, Karl M. Expression and regulation of estrogen receptors in mesangial cells: influence on matrix metalloproteinase-9. J Am Soc Nephrol 12: 241–251, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Rahman M, Sutter CH, Emmert GL, Sutter TR. Regioselective 2-hydroxylation of 17β-estradiol by rat cytochrome P4501B1. Toxicol Appl Pharmacol 216: 469–478, 2006. [DOI] [PubMed] [Google Scholar]
- 41.Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916–1923, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension 37: 1199–1208, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Reckelhoff JF, Zhang H, Granger JP. Testosterone exacerbates hypertension and reduces pressure-natriuresis in male spontaneously hypertensive rats. Hypertension 31: 435–439, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Remuzzi G, Ruggenenti P, Perico N. Chronic renal disease: renoprotective benefits of renin-angiotensin system inhibition. Ann Intern Med 136: 604–615, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Rogers JL, Mitchell AR, Maric C, Sandberg K, Myers A, Mulroney SE. Effect of sex hormones on renal estrogen and angiotensin type 1 receptors in female and male rats. Am J Physiol Regul Integr Comp Physiol 292: R794–R799, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Sampson AK, Moritz KM, Jones ES, Flower RL, Widdop RE, Denton KM. Enhanced angiotensin II type 2 receptor mechanisms mediate decreases in arterial pressure attributable to chronic low-dose angiotensin II in female rats. Hypertension 52: 666–671, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Sandberg K, Hong Ji. Sex differences in primary hypertension. Biol Sex Diff 3: 7, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3: 1101–1108, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Schunkert H, Danser AH, Hense HW, Derkx FH, Kürzinger S, Riegger GA. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation 95: 39–45, 1997. [DOI] [PubMed] [Google Scholar]
- 50.Sebastiaan W, Isholar DA Jr, Joles JA, Bluyssen HA, Koomans HA, Braam B. Resistance to oxidative stress by chronic infusion of angiotensin II in mouse kidney is not mediated by the AT2 receptor. Am J Physiol Renal Physiol 288: F1191–F1200, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Silbiger S, Lei J, Neugarten J. Estradiol suppresses type I collagen synthesis in mesangial cells via activation of activator protein-1. Kidney Int 55: 1268–1276, 1999. [DOI] [PubMed] [Google Scholar]
- 52.Silva-Antonialli MM, Tostes RCA, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MH, Fortes ZB, Nigro D. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res 62: 587–593, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Stampfer MJ, Colditz GA, Willett WC, Manson JE, Rosner B, Speizer FE, Hennekens CH. Postmenopausal estrogen therapy and cardiovascular disease. Ten-year follow-up from the nurses' health study. N Engl J Med 325: 756–762, 1991. [DOI] [PubMed] [Google Scholar]
- 54.Su EJ, Lombardi DM, Siegal J, Schwartz SM. Angiotensin II induces vascular smooth muscle cell replication independent of blood pressure. Hypertension 31: 1331–1337, 1998. [DOI] [PubMed] [Google Scholar]
- 55.Sullivan JC, Bhatia K, Yamamoto T, Elmarakby AA. Angiotensin(1–7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension 56: 658–666, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sullivan JC, Semprun-Prieto L, Boesen EI, Pollock DM, Pollock JS. Sex and sex hormones influence the development of albuminuria and renal macrophage infiltration in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol 293: R1573–R1579, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Tofovic SP. Estrogens and development of pulmonary hypertension: interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol 56: 696–708, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, Campbell A, Morecroft I, Loughlin L, Mcclure JD, Thomas M, Mair KM, MacLean MR. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 126: 1087–1098, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Wu CC, Lu KC, Chen JS, Hsieh HY, Lin SH, Chu P, Wang JY, Sytwu HK, Lin YF. HO-1 induction ameliorates experimental murine membranous nephropathy: anti-oxidative, anti-apoptotic and immunomodulatory effects. Nephrol Dial Transplant 23: 3082–3090, 2008. [DOI] [PubMed] [Google Scholar]
- 60.Xu D, Niu W, Luo Y, Zhang B, Liu M, Dong H, Liu Y, Li Z. Endogenous estrogen attenuates hypoxia-induced pulmonary hypertension by inhibiting pulmonary arterial vasoconstriction and pulmonary arterial smooth muscle cells proliferation. Int J Med Sci 10: 771–781, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xue B, Pamidimukkala J, Hay M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol 288: H2177–H2184, 2005. [DOI] [PubMed] [Google Scholar]
- 62.Yaghini FA, Li F, Malik KU. Expression and mechanism of spleen tyrosine kinase activation by angiotensin II and its implication in protein synthesis in rat vascular smooth muscle cells. J Biol Chem 282: 16878–16890, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Yanes LL, Sartori-Valiinotti JC, Iliescu R, Romero DG, Racusen LC, Zhang H, Reckelhoff JF. Testosterone-dependent hypertension and upregulation of intrarenal angiotensinogen in Dahl salt-sensitive rats. Am J Physiol Renal Physiol 296: F771–F779, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoshida K, Kobayashi N, Ohno T, Fukushima H, Matsuoka H. Cardioprotective effect of angiotensin II type 1 receptor antagonist associated with bradykinin-endothelial nitric oxide synthase and oxidative stress in Dahl salt-sensitive hypertensive rats. J Hypertens 25: 1633–1642, 2007. [DOI] [PubMed] [Google Scholar]
- 65.Zhu BT, Liehr JG. Inhibition of the catechol-O-methyltransferase-catalyzed O-methylation of 2- and 4-hydroxyestradiol by catecholamine: implications for the mechanism of estrogen-induced carcinogenesis. Arch Biochem Biophys 304: 248–256, 1993. [DOI] [PubMed] [Google Scholar]