

Abstract
We characterized a phosphoinositide phospholipase C (PI-PLC) from the procyclic form (PCF) of Trypanosoma brucei. The protein contains a domain organization characteristic of typical PI-PLCs, such as X and Y catalytic domains, an EF-hand calcium-binding motif, and a C2 domain, but it lacks a pleckstrin homology (PH) domain. In addition, the T. brucei PI-PLC (TbPI-PLC) contains an N-terminal myristoylation consensus sequence found only in trypanosomatid PI-PLCs. A peptide containing this N-terminal domain fused to green fluorescent protein (GFP) was targeted to the plasma membrane. TbPI-PLC enzymatic activity was stimulated by Ca2+ concentrations below the cytosolic levels in the parasite, suggesting that the enzyme is constitutively active. TbPI-PLC hydrolyzes both phosphatidylinositol (PI) and phosphatidylinositol 4,5-bisphosphate (PIP2), with a higher affinity for PIP2. We found that modification of a single amino acid in the EF-hand motif greatly affected the protein's Ca2+ sensitivity and substrate preference, demonstrating the role of this motif in Ca2+ regulation of TbPI-PLC. Endogenous TbPI-PLC localizes to intracellular vesicles and might be using an intracellular source of PIP2. Knockdown of TbPI-PLC expression by RNA interference (RNAi) did not result in growth inhibition, although enzymatic activity was still present in parasites, resulting in hydrolysis of PIP2 and a contribution to the inositol 1,4,5-trisphosphate (IP3)/diacylglycerol (DAG) pathway.
INTRODUCTION
Trypanosoma brucei is a protist parasite that belongs to the order Kinetoplastida, whose members are characterized by the presence of a kinetoplast, a DNA-containing structure within the single mitochondrion of the parasite. T. brucei has a complex life cycle involving invertebrate and vertebrate hosts. The procyclic form (PCF) is the proliferative form in the tsetse fly, and the long, slender bloodstream form (BSF) is the proliferative form in the mammalian host. Trypanosoma brucei brucei (called T. brucei in this article) is a parasite of African ruminants that produces the disease nagana, while T. b. gambiense and T. b. rhodesiense are the subspecies that infect humans. Currently, approximately 50,000 people are infected annually with an agent of human African trypanosomiasis (HAT), which is endemic in 36 sub-Saharan African countries (1). Efforts to understand the biology of the parasite are essential because they provide insights into novel ways by which we can disrupt the life cycle of the parasite and/or target the parasite while leaving the host unharmed.
The inositol 1,4,5-trisphosphate/diacylglycerol (IP3/DAG) signaling pathway regulates many processes, such as cell division, secretion, and differentiation, in eukaryotic cells (2). The central enzyme in this pathway, the phosphoinositide phospholipase C (PI-PLC), catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the second messengers inositol 1,4,5-trisphospate (IP3) and diacylglycerol (DAG) (3). IP3 gates a channel in the endoplasmic reticulum, the IP3 receptor (IP3R), which releases calcium ions (Ca2+) into the cytosol. DAG activates a protein kinase stimulating downstream signaling pathways (4).
Previous work from our laboratory characterized Ca2+ stores in T. brucei and found that this parasite stores Ca2+ in an acidic compartment termed the acidocalcisome (5). Recently, a T. brucei IP3R (TbIP3R) was shown to be a functional Ca2+ release channel located in T. brucei acidocalcisomes and to be essential for growth of both procyclic (PCF) and bloodstream (BSF) forms and for the ability of BSF trypanosomes to establish an infection (6). Considering the central role that IP3 plays in Ca2+ homeostasis, we became interested in the characterization of the enzyme that generates this second messenger, the T. brucei PI-PLC (TbPI-PLC). Understanding the biochemical characteristics of this enzyme will increase the understanding of the function of IP3 signaling in T. brucei, which may be important for differentiation and adaptation of the parasite during its life cycle.
Kinetoplastid PI-PLCs show some unique features not shared with any other PI-PLCs. For example, they have a myristoylation consensus sequence at the N terminus, instead of a pleckstrin homology (PH) domain (7), and this sequence has been shown to play a role in the localization of the enzyme to the plasma membrane of T. cruzi (8). A very interesting characteristic of the trypanosomatid enzymes is that their overall domain organization is most similar to that of the mammalian zeta-type PI-PLC (PI-PLC-ζ) present in sperm (9). PI-PLC-ζ is released into the oocyte upon fertilization, where it is activated by cytosolic Ca2+ and causes Ca2+ oscillations which are responsible for the whole downstream cascade of signaling pathways leading to the development of the embryo (9, 10).
In this work, we characterize the activity of the TbPI-PLC1 and study its function and localization in PCF trypanosomes, as well as its potential regulation.
MATERIALS AND METHODS
Culture methods and in vitro growth analysis.
PCF trypanosomes (strain 29-13) were a gift from George A. M. Cross (Rockefeller University) and were grown as described before (11, 12). For RNA interference (RNAi) studies, PCF trypanosomes at a density of 1 × 106 cells were induced with 1 μg/ml of tetracycline and diluted every 2 days.
Chemicals and reagents.
Qia gel extraction and MinElute PCR purification kits were purchased from Qiagen. TRIzol reagent and Alexa 488- and 546-conjugated rat, rabbit, and mouse secondary antibodies were obtained from Invitrogen. Rabbit TbVP1 antibody (13) was a gift from Norbert Bakalara (École Nationale Superieure de Chimie de Montpellier, Montpellier, France). Rabbit TbBiP antibody (14) was a gift from James Bangs (University of Wisconsin, Madison, WI). Purified anti-hemagglutinin (anti-HA) antibody was obtained from Roche. TbPPDK antibody was a gift from Frédéric Bringaud (University of Bordeaux, Bordeaux, France). Commercial anti-PIP2 was obtained from VWR. A mouse anti-green fluorescent protein (anti-GFP) monoclonal antibody was purchased from Molecular Probes. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse IgG antibodies were purchased from Santa Cruz Biotechnology. myo-[3H]inositol (7.27 Ci/mmol) and [3H]PIP2 (5.0 Ci/mmol) were purchased from PerkinElmer Life Sciences. His-tagged Superflow cartridges were obtained from Clontech. A Gene Tailor site-directed mutagenesis kit was obtained from Invitrogen. PIP2 and phosphatidylinositol (PI) were obtained from Avanti-Polar Lipids, Inc. (Alabaster, AL). The pUB39 vector was a gift from George A. M. Cross (Rockefeller University, New York, NY). The p2T7Ti vector (15) was a gift from John Donelson (University of Iowa, Iowa City, IA). The pET32 vector and other ligation-independent cloning vectors were obtained from Novagen. The pMOTag4H vector (16) was a gift from Thomas Seebeck (University of Bern, Bern, Switzerland).
Cloning and expression of TbPI-PLC1.
The T. brucei pUB39 expression vector was used for expression studies. The inserted fragment of VSG117 cDNA was removed by digestion with HindIII and BamHI. First, a primer pair to amplify the coding region for the first 18 amino acids, i.e., the TbPI-PLC1(1–18) primer pair (Table 1), was generated to incorporate HindIII and XhoI sites. Second, a pair of primers for the GFP tag was used to amplify the open reading frame (ORF) of Aequorea victoria GFP, incorporating an XhoI site and a BamHI site (Table 1). The linearized fragments were coligated into HindIII- and BamHI-digested pUB39 to generate pUB39/GFP and create a GFP fusion product. For the expression of the truncated version of TbPI-PLC1, the TbPI-PLC1(19–714) primer pair was used for amplification of the coding region for amino acids 19 to 714 and for incorporation of HindIII and XhoI sites (Table 1). The fragment was cloned into the pUB39/GFP vector digested with HindIII and XhoI, replacing the previously cloned myristoylation sequence with a truncated version. A forward primer (Table 1) and the reverse primer used previously were used to amplify GFP alone and to clone it into the pUB39 vector. All constructs were confirmed by sequencing (Yale University DNA Analysis Facility, New Haven, CT) and linearized by NotI digestion.
TABLE 1.
Primers used in this study
| Construct | Primer direction | Primer sequence (5′–3′)a |
|---|---|---|
| TbPI-PLC(1–18) | Forward | CAAGCTTATGGGAGGGTGTACCTCACGTGGGCTCTCA |
| Reverse | CTCGAGGGAGTAACATGCGAGTTTCT | |
| TbPI-PLC(19–714) | Forward | AAGCTTATGCACCGTACCGGCAACCTCGTT |
| Reverse | CTCGAGCTGATAAGATACTTGAACCATT | |
| GFP tag | Forward | CTCGAGGGAGTAACATGCGAGTTTCT |
| Reverse | GGGATCCTTACTTGTACAGCTCGTCCAG | |
| GFP alone | Forward | CCAAGCTTATGGTGAGCAAGGGCGAGGAG |
| Reverse | GGGATCCTTACTTGTACAGCTCGTCCAG | |
| TbPI-PLCpET32 | Forward | GACGACGACAAGATGGGAGGGTGTACGTCACGTGGGCTC |
| Reverse | GAGGAGAAGCCCGGTTACTACTGATAAGATACTTGAACCAT | |
| TbPI-PLCHA tag | Forward | TTACGTGCCCTAAAAAAAGGAATTCGCCAGGTCCCCCTTCGAGACCTCAAAGGATCTATTATACATGGCTCTTTTTTAATGGTTCAAGTATCTTATCAGGGTACCGGGCCCCCCCTCGAG |
| Reverse | AGCTACGCTGTGAAGACCATGGCACAACATGGTGCCATAAAGTCGGGTACATGCAACGCACACTTGTAACCCAATTGAAGAACACGATATTCTCAAACTCTGGCGGCCGCTCTAGAACTAGTGGAT | |
| TbPI-PLCpET32D109A | Forward | TGATATGGATAAAGTACGACGCCGACAACAGCGG |
| Reverse | GTCGTACTTTATCCATATCATCATGAACTT |
Underlining indicates restriction sites except for the HA tag sequences, where underlining for the forward refers to TbPI-PLC1 coding sequence before and excluding the stop codon and underlining for the reverse refers to the 21 bp for pMOTag4H.
Primers were designed to amplify the TbPI-PLC1 gene for cloning into the pET32 ligation-independent cloning system (Table 1). This system incorporates two histidine tags, both N and C terminally, and it creates a thioredoxin fusion tag that facilitates the production of soluble protein. The primers were used to amplify the TbPI-PLC1 gene from genomic DNA by using Finnzymes Phire HS DNA polymerase. To prepare an insert that was compatible for ligation, the PCR product was treated with T4 DNA polymerase according to the manufacturer's protocol and incubated at 22°C for 30 min, followed by inactivation of the enzyme at 75°C for 20 min. The manufacturer's protocol was used to anneal the insert into the EK/LIC vector. A ligation reaction was used to transform NovaBlue GigaSingles competent cells according to the manufacturer's protocol. A Qiagen miniprep plasmid kit was used to isolate the plasmid and to transform expression bacteria [Escherichia coli BL21(DE3)-RIPL]. The colonies were screened by digestion with NcoI and KpnI. Colony 1 was confirmed by sequencing and was used for the expression of TbPI-PLC in bacteria by use of established protocols, and Clontech columns were used for purification according to the manufacturer's protocols. The eluates were pooled and dialyzed overnight with 10 mM Tris-HCl, pH 7.0. Samples of the eluates were analyzed by SDS-PAGE for protein expression.
Cell transfections.
PCF trypanosomes were transfected according to a published protocol (17). Approximately 2.5 × 107 PCF trypanosomes in mid-log phase were used. The cuvette was electroporated with two pulses from a Bio-Rad Gene Pulser instrument set at 1,500 V and 25 μF. Stable transfectants were selected using the appropriate antibiotic.
Epitope tagging.
Forward and reverse primers were designed to contain 100 nucleotides of the TbPI-PLC1 ORF before the stop codon and the reverse complement of 100 nucleotides of the 3′-untranslated region (3′-UTR) followed by 21 nucleotides of the backbone of pMOTag4h, in frame (Table 1). The primers amplified the HA tagging cassette with a hygromycin resistance gene from pMOTag4H. Amplification was verified by electrophoresis, and DNA was purified using a Qiagen PCR purification kit, precipitated, and transfected into parasites following the previously published one-step epitope tagging protocol (16).
Western blot analysis.
Cells were harvested and washed twice with phosphate-buffered saline (PBS) and then lysed with RIPA buffer (150 mM NaCl, 20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1% SDS, and 0.1% Triton X-100) containing a protease inhibitor cocktail (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 2.5 U/ml of Benzonase). Either total cell lysates or supernatant and pellet fractions were used according to previously published protocols. Immunoblots were visualized using Pierce ECL Western blot substrate according to the manufacturer's protocol.
Fluorescence microscopy.
Live cells were imaged using direct fluorescence microscopy. For immunofluorescence assays, parasites were fixed with 4% paraformaldehyde in PBS, pH 7.4, for 1 h. Cells were washed two times with PBS and allowed to adhere to 1-mg/ml poly-l-lysine-coated coverslips. Cells were permeabilized with 0.3% Triton X-100 in PBS, pH 7.4, for 3 min. Parasites were blocked for 1 h at room temperature with a blocking solution containing 50 mM NH4Cl, 3% bovine serum albumin (BSA), 1% fish gelatin, and 5% goat serum. Cells were incubated with a Roche monoclonal anti-HA antibody (1:20) and either the polyclonal rabbit antibody against TbVP1 (1:300), the rabbit anti-TbBiP antibody (1:300), the mouse anti-TbPPDK antibody (1:30), or the mouse monoclonal anti-PIP2 antibody (1:50) for 1 h. The secondary antibodies were Alexa 488-conjugated goat anti-rat (anti-HA) and/or Alexa 546-conjugated goat anti-rabbit/mouse at 1:1,000 for 1 h. All images of parasites were captured with an Olympus 1X-71 inverted fluorescence microscope with a Photometrix Coolsnap charge-coupled device (CCD) camera, using Delta Vision software from Applied Precision. Images were processed using Softworx deconvolution software. Differential interference contrast and fluorescence images were captured under nonsaturating conditions, using the same exposure times.
PI-PLC activity assay.
The protein concentration was determined, and the recombinant enzyme was stored in 40% glycerol at −80°C. Analysis of the enzymatic activity of PI-PLC was performed by measuring the release of soluble IP3 from the hydrolysis of [3H]PIP2. The standard reaction was performed as described previously (7). Briefly, the reaction mixtures contained 30,000 cpm of radioactive PIP2 (or PI) and also contained cold PIP2 (or PI), at various concentrations and dried under a nitrogen stream. The buffer included 20 mM Tris-HCl, pH 7.2, 2.5 mM EGTA, 100 mM NaCl, and CaCl2 at various concentrations to obtain the calculated concentration of free calcium by using the MaxChelator computer program. The phospholipid mixture was sonicated for 2 s. The reactions were initiated by the addition of 3.5 μg of enzyme followed by incubation at 30°C for 20 min. The reaction was stopped by adding 500 μl of chloroform-methanol-HCl (100:100:0.6) and 150 μl of 5 mM EGTA in 1 N HCl. Samples were centrifuged to separate the organic and aqueous phases. The aqueous phase was removed and counted using a scintillation counter. Activity was analyzed in response to changes in the Ca2+ concentration, pH, time of reaction assay, and incubation temperature to establish the activity parameters. The Km, Vmax, and kcat values were calculated by nonlinear regression analysis using Prism software.
Site-directed mutagenesis.
The Invitrogen Gene Tailor site-directed mutagenesis system was used. Briefly, primers were designed to include overlapping regions and the creation of a nucleotide mutation: GAC (aspartate) to GCC (alanine). With the aim of mutating the coding sequence for aspartic acid 109 to that for alanine in TbPI-PLC1, a primer pair (Table 1) was designed to incorporate this change. The TbPI-PLC1 gene was methylated, amplified using the mutated primers, and transformed into DH5α bacteria. Colonies were screened by restriction digestion, and plasmids were confirmed by sequencing. The mutated gene was digested with BamHI and SalI and cloned into the ligation-dependent vector pET32a. The expression of TbPI-PLC1-D109A was done in BL21(DE3) bacteria, and the protein was purified as described above for TbPI-PLC1 and assayed for activity using the same established parameters.
Generation of RNAi constructs.
To knock down the expression of the TbPI-PLC1 gene by double-stranded RNA expression, the p2T7Ti vector was used. This vector utilizes dually inducible T7 promoters. A 500-bp fragment corresponding to nucleotides 1304 to 1790 (shown in bold) was amplified using the following primers: TbPI-PLC RNAi forward, 5′-CCGCTCGAGGAATTCAAGGGGTGCTGAAAAG-3′ (with an XhoI site); and TbPI-PLC RNAi reverse, 5′-CGGGATCCCCTGGGAAGACAGAAAGCT-3′ (with a BamHI site). The amplified fragment was then cloned into the digested p2T7Ti vector. The construct was verified by sequencing and purified for cell transfections.
Northern blot analysis.
RNAs were isolated by use of TRIzol reagent and quantified. RNA samples (10 μg) were separated in 1% formaldehyde-agarose gels and then transferred to a Zeta Probe nylon membrane by overnight capillary transfer. RNAs were cross-linked to the membrane by baking at 80°C. The same primer pair was used to generate probes and subsequently labeled with [α-32P]dCTP following the Prime-a-Gene labeling protocol. β-Tubulin was labeled and used as a loading control. The membranes were hybridized in 7% SDS, 0.5 M Na2HPO4, pH 7.2, overnight at 65°C in a rotating hybridizer. Membranes were washed once with 1× SSC (0.15 M NaCl plus 0.015 M sodium citrate), 0.1% SDS at 65°C and once with 0.1× SSC, 0.1% SDS at 65°C for 30 min. The blots were visualized by autoradiography.
Parasite lysates.
Exponentially growing PCF 29-13, TbPI-PLC1-OE, or TbPI-PLC1 RNAi trypanosomes were harvested and washed twice with PBS and then subjected to freeze-thawing by exposing cells to liquid nitrogen (5-min freeze and 1-min thaw at 37°C) for 3 to 5 cycles or until cells were broken. The pellet was resuspended in approximately 200 μl of 20 mM Tris-HCl, pH 7.2. The protein concentration was measured using a bicinchoninic acid (BCA) protein assay kit, and an enzymatic activity assay was performed immediately. The PI-PLC1 activity assay was carried out as previously described, using 35 μg of total lysate in each reaction mixture.
RESULTS
Molecular characterization of TbPI-PLC.
Analysis of the T. brucei genome in the TriTryp database (http://tritrypdb.org) showed the presence of one putative phosphoinositide phospholipase C gene (TbPI-PLC1; Tb927.11.5970) (accession no. XP_828672) that is an ortholog of the previously described T. cruzi PI-PLC (TcPI-PLC) gene (7, 18) and another gene previously identified as encoding a phospholipase C-like protein (19) (TbPI-PLC2; Tb927.6.2090) (accession no. AAZ11771). There is only 29% identity between TbPI-PLC1 and TbPI-PLC2, and TbPI-PLC2 lacks some of the features of TbPI-PLC1 that are characteristic of PI-PLCs, such as a Y domain, and also possesses an additional PDZ (PSD-95-Dlg-ZO1) domain. The ORF of TbPI-PLC1 consists of 2,145 bp predicting an encoded protein of 714 amino acids with a molecular mass of 80.4 kDa. In addition to the highly conserved catalytic X (amino acids 246 to 392) and Y (amino acids 453 to 571) domains, TbPI-PLC1 has an EF-hand domain (amino acids 95 to 130) in the N-terminal region and a C2 domain (amino acids 580 to 713) (Fig. 1A). The TbPI-PLC1 EF-hand motif consists of approximately 36 amino acids in a helix-loop-helix arrangement (20). Within this motif, there is a 12-residue loop region (residues 108 to 119) that has residues (aspartate and glutamate) in locations that may play a role in Ca2+ binding (21). The overall domain organization of TbPI-PLC1 is most similar to that of the mammalian sperm-specific PLC-ζ protein (accession no. AF435950) (Fig. 1A). Sequence alignment shows an overall 27% identity and 40% similarity between the TbPI-PLC1 and mouse PLC-ζ proteins. However, one prominent difference between TbPI-PLC1 and PLC-ζ is the presence of an N-myristoylation consensus sequence predicted by the NMT program (http://mendel.imp.univie.ac.at/myristate/) (Fig. 1A), which is highly similar to the N-myristoylation sequence of T. cruzi (7).
FIG 1.

The N terminus of TbPI-PLC1 is sufficient for plasma membrane localization of a fusion protein. (A) Overall domain organization of TbPI-PLC1 and mammalian PLC-ζ. EF, EF-hand domain; M, myristoylation domain. (B) Constructs for overexpression of the first 18 amino acids and a truncated form of TbPI-PLC1 bound to GFP. (C) Western blot analysis of GFP, N-terminal TbPI-PLC(1–18)::GFP (NtGFP), and C-terminal TbPI-PLC1(19–714)::GFP (CtGFP) expressed in PCF trypanosomes. Lysates containing 30 μg protein were subjected to SDS-PAGE on 10% polyacrylamide gels and transferred to a nitrocellulose membrane. The blot was probed with antibodies against GFP as described in Materials and Methods. Bands of 28, 30, and 110 kDa were detected for PCF homogenates. Migration of molecular mass standards is shown to the left and right of the gels. (D and E) TbPI-PLC1(1–18)::GFP localized to the plasma membranes and flagella of PCF trypanosomes as detected by direct fluorescence microscopy (D), whereas truncated TbPI-PLC(19–714)::GFP localized throughout the cell (E). Bars = 10 μm.
Role of the N-myristoylation sequence.
Previous studies with TcPI-PLC (8) revealed that the N-terminal 20 amino acids of the protein have the necessary and sufficient information to target GFP to the surface of cells. We therefore investigated whether this was also the case with TbPI-PLC1. GFP fusion constructs were generated and cloned into the pUB39 vector for overexpression in PCF trypanosomes (Fig. 1B). The first 18 amino acids of the TbPI-PLC1 sequence, TbPI-PLC1(1–18), fused to GFP, and the truncated PI-PLC1 protein, TbPI-PLC1(19–714), were transfected into PCF (strain 29-13) trypanosomes. Cells were induced by the addition of tetracycline (1 μg/ml), and lysates were analyzed by Western blotting for expression of a protein of the expected size with anti-GFP (Fig. 1C). Direct fluorescence of live parasites demonstrated localization of TbPI-PLC1(1–18) to the plasma membrane (Fig. 1D), whereas localization of TbPI-PLC1(19–714) was cytosolic (Fig. 1E). This demonstrated that the N-myristoylation consensus sequence is sufficient for localization to the plasma membrane.
Endogenous localization.
Attempts to obtain a specific antibody against TbPI-PLC failed. Therefore, to determine the localization of endogenous TbPI-PLC1, in situ tagging was used to incorporate a tagging cassette at the C-terminal end of one of the chromosomal copies of the TbPI-PLC1 gene in PCF trypanosomes. This strategy incorporates a 3×HA tag and a hygromycin resistance gene. Localization was analyzed by immunofluorescence microscopy with anti-HA. TbPI-PLC1 localized to intracellular vesicles, and further colocalization studies with intracellular markers of acidocalcisomes (vacuolar proton pyrophosphatase [TbVP1]) (13), the endoplasmic reticulum (TbBiP) (14), and glycosomes (pyruvate phosphate dikinase [TbPPDK]) (22) showed that TbPI-PLC1 did not colocalize with any of these markers (Fig. 2A). Western blot analysis using anti-HA confirmed the expression of the protein at the expected size (Fig. 2B). The localization of TbPI-PLC1 to vesicles instead of the plasma membrane was unexpected, considering our data from the overexpression of the myristoylation sequence and the truncated version. However, it has been demonstrated that PLCs can utilize alternate sources of PIP2. For example, the sperm-specific PLC-ζ protein localizes to distinct vesicles inside the egg (23). Likewise, PIP2 was shown to localize to distinct vesicular structures inside the egg cortex, where PLC-ζ hydrolyzed this intracellular source of PIP2 (23). We used anti-PIP2 to study the distribution of PIP2 with respect to the observed distribution of HA-tagged TbPI-PLC1 (Fig. 2C). Our results demonstrated PIP2 localization to vesicles throughout the cells, with a distribution similar to that of TbPI-PLC1.
FIG 2.

Localization of TbPI-PLC in PCF trypanosomes. TbPI-PLC1 has a punctate cytosolic localization and does not colocalize with either TbBiP, PPDK, or VP1 (A) but partially colocalizes with antibodies against PIP2 (C). Bars in panel C, 5 μm. DIC, differential interference contrast microscopy. (B) Western blot analysis of TbPI-PLC1 expressed in PCF trypanosomes (two clones, i.e., HA1 and HA2). Lysates containing 30 μg protein were subjected to SDS-PAGE on 10% polyacrylamide gels and transferred to a nitrocellulose membrane. The blot was probed with antibodies against HA as described in Materials and Methods. A band of 80 kDa was detected for PCF homogenates. Migration of molecular mass standards is shown to the left of the gel.
Expression, purification, and activity of recombinant TbPI-PLC1.
TbPI-PLC1 was expressed in bacteria, and the purified protein was analyzed for its enzymatic properties. We investigated the enzyme activity when cells were subjected to free calcium (Ca2+) concentrations ranging from 1 nM to 10 mM (Fig. 3A). We also optimized the time of incubation and the incubation temperature (data not shown), as well as the pH (Fig. 3B). We observed a peak of activity at a Ca2+ concentration of 10 nM for PIP2 and a peak of activity with PI when Ca2+ was present at 100 nM (Fig. 3A). In addition, the enzyme was most active at pH 7.2 (Fig. 3B). Enzyme activities at different concentrations of PIP2 (Fig. 3C) and PI (Fig. 3D) were investigated, and the Vmax and Km values were calculated for both substrates. The lower Km for PIP2 indicates a preference of the enzyme for PIP2 compared to PI as the substrate (Table 2).
FIG 3.

TbPI-PLC1 activity as a function of Ca2+ concentration (A), pH (B), or substrate concentration (C and D). Experimental conditions were as described in Materials and Methods, using the same buffer with different Ca2+ concentrations and 5 μM PIP2 (blue line) or PI (red line) (A) or with 10 μM Ca2+ and different pHs (B) or different concentrations of PIP2 (C) or PI (D). Values are means ± standard errors of the means (SEM) (n = 3).
TABLE 2.
Kinetic parameters for purified recombinant TbPI-PLC with PIP2 and PI as substrates
| Substrate | Km (μM) | Vmax (nmol min−1 mg−1) | kcat (min−1) | kcat/Km (M−1 min−1) |
|---|---|---|---|---|
| PIP2 | 4.1 ± 0.7 | 34.4 ± 1.0 | 0.27 ± 0.008 | 6.6 × 104 |
| PI | 22.6 ± 4.6 | 13.5 ± 0.8 | 0.11 ± 0.007 | 1.2 × 104 |
Ca2+ binding.
The Ca2+ sensitivity of recombinant TbPI-PLC1 was surprisingly high compared to the Ca2+ sensitivity of other PI-PLCs. For example, TcPI-PLC has a peak of activity at approximately 10 μM Ca2+ (7), while PLC-ζ activity peaks at approximately 1 μM Ca2+ (24). We also observed that TbPI-PLC1 had low activity (Vmax of 34.4 nmol min−1 mg−1) compared to those of other PLC enzymes, such as TcPI-PLC, with a Vmax of 930 nmol min−1 mg−1 (7). EF-hand motifs are the best-known Ca2+-binding motifs (25–28). Twelve residues of the EF-hand structure typically form a pattern, Dx[DN]xDGx[ILV][DSTN]xxE, that is involved in Ca2+ binding (29). The binding of Ca2+ can have an effect on structural stability and can regulate activity (29). We compared the binding loop of TbPI-PLC1 (Tb) with the binding loops of TcPI-PLC (accession no. AF093565) (Tc) and human PLC-ζ (accession no. NP_149114) (ζ) (Fig. 4A, inset). TbPI-PLC1 has a negatively charged aspartic acid instead of an alanine in the same position of TcPI-PLC, creating a series of three negatively charged aspartates, which has not been observed in other loops. We hypothesized that changing this residue to alanine, as found in the TcPI-PLC calcium-binding loop, would alter the activity and Ca2+ sensitivity of the enzyme. Site-directed mutagenesis was used to change the aspartate at the 109th position (corresponding to the second residue in the binding loop) to alanine. The mutated protein was expressed and purified under the same conditions as those used for TbPI-PLC1 and then assayed for the release of [3H]IP3 from [3H]PIP2. Interestingly, the enzyme exhibited an increase in activity with PIP2 as the substrate (Fig. 4A) and a substantial decrease of activity toward PI (Fig. 4B). However, there was no change in the peak of activity in the presence of Ca2+. These results demonstrate that Ca2+ binding modulates the substrate preference and activity of the enzyme.
FIG 4.
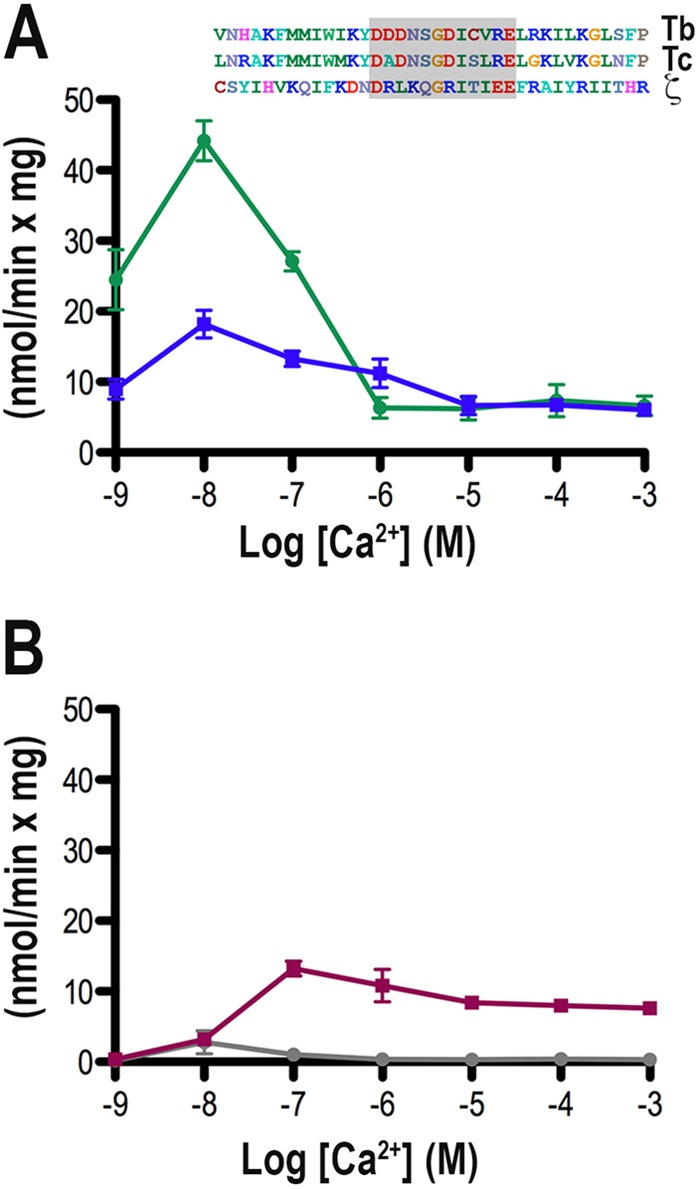
PI-PLC activity of TbPI-PLC1-D109A. Experimental conditions were as described in Materials and Methods. Incubations were done using the same buffer with 5 μM PIP2 (A) or PI (B) and different Ca2+ concentrations. All values are means ± SEM (n = 3). The inset shows a sequence alignment of the TbPI-PLC1 EF-hand domain (Tb) compared to those of T. cruzi PI-PLC (AF093565) (Tc) and human PI-PLC-ζ (NP_149114.2). The loop containing Asp109 that was mutated to Ala is shaded.
RNAi knockdown and overexpression.
Knockdown of TbPI-PLC1 expression by use of inducible double-stranded RNA did not result in growth defects in PCF trypanosomes (Fig. 5A). Northern blot analyses showed that the mRNA was downregulated after 1 to 10 days of tetracycline addition (Fig. 5B). To investigate whether downregulation and upregulation of TbPI-PLC1 expression lead to changes in cellular activity, we prepared lysates from PCF trypanosomes overexpressing TbPI-PLC1 or from TbPI-PLC1 RNAi transgenic mutants and compared them with lysates from the parental line. Endogenous levels of PI-PLC activity in the parental line lysates (strain 29-13), using PIP2 as the substrate, were tested at different Ca2+ concentrations. A peak of activity was observed at 100 nM (Fig. 5C), a Ca2+ concentration in the range of the estimated intracellular Ca2+ concentration of PCF trypanosomes (30). Lysates from the PCF parental line (strain 29-13), uninduced TbPI-PLC1-overexpressing parasites, and uninduced RNAi transgenic parasites showed similar activities. However, induction of overexpression resulted in 2-fold higher activity, while induction of RNAi significantly reduced PIP2 hydrolysis, although it did not eliminate it completely (Fig. 5D). Collectively, these results show that PI-PLC activity in parasite lysates is able to hydrolyze PIP2 and to produce the second messenger IP3 and that this activity can be altered by genetic manipulations. The results also suggest that RNAi was not totally effective at suppressing PI-PLC activity in the lysates.
FIG 5.

Effect of inhibition of TbPI-PLC1 expression by tetracycline-inducible RNAi on cell growth. (A) Growth of PCF trypanosomes in the absence (squares) or presence (circles) of 1 μg/ml tetracycline for the indicated numbers of days. Values are means of triplicate determinations for a representative experiment. (B) Northern blot analysis of TbPI-PLC1 RNAi of PCF trypanosomes grown in the absence (−) or presence (+) of tetracycline (Tet). (Top) Total RNA was subjected to gel electrophoresis before transfer to a nylon membrane and then hybridized with a 32P-labeled probe corresponding to the TbPI-PLC1 coding sequence. (Bottom) Tubulin is shown as a loading control. (C) PIP2 hydrolysis was measured in PCF trypanosome lysates as described in Materials and Methods, using 5 μM PIP2 and different Ca2+ concentrations. (D) Activity in lysates from the parent line (WT), trypanosomes overexpressing TbPI-PLC1 (OE), or RNAi transgenic trypanosomes (RNAi) in the absence (−) or presence (+) of tetracycline (Tet). Data are means and SEM (n = 3). ***, P < 0.05 compared to wild-type lysates (Student's t test).
DISCUSSION
Our laboratory previously described the presence of a functional IP3/diacylglycerol pathway in T. brucei (30) and showed that the IP3 receptor of the parasite is localized to acidocalcisomes and is essential for growth and infectivity (6). In this work, we demonstrate that the TbPI-PLC1 gene encodes a functional PI-PLC that has an intracellular localization in PCF trypanosomes despite its possession of an N-myristoylation consensus sequence that can target a GFP fusion protein to the plasma membrane. The enzyme prefers PIP2 over PI as a substrate and is active at Ca2+ concentrations found physiologically in the parasite under steady-state conditions (30). The EF-hand domain of TbPI-PLC is involved in its Ca2+ sensitivity and determination of its substrate preference, as demonstrated by experiments in which mutation of this domain affected the activity of the enzyme in the presence of Ca2+ and enhanced its preference for PIP2 as the substrate. Knockdown of TbPI-PLC did not affect the growth of PCF trypanosomes but was unable to completely eliminate the activity of the enzyme in lysates, while overexpression of the enzyme increased the activity of lysates, demonstrating that the enzyme is functional in the parasite.
We previously reported that the T. cruzi orthologue of TbPI-PLC1 has an N-myristoylation consensus sequence that is necessary and sufficient to target this enzyme to the plasma membrane of the parasite (8, 31). Other proteins of trypanosomatids, such as the flagellar Ca2+-binding protein of T. cruzi (32), the calflagins of T. brucei (33), and SMP-1 of Leishmania major (34), need a similar N-myristoylation consensus sequence for flagellar membrane localization. In contrast to these proteins, but in analogy with the TbPI-PLC1 protein reported here, another L. major protein, the protein phosphatase LmPPEF, contains an N-myristoylation consensus sequence that is sufficient for flagellar localization of a fusion protein, but the endogenous protein localizes to intracellular membranes (35). The Arf-like (ARF6) protein of T. brucei also has a myristoylation consensus sequence, but the endogenous protein has a vesicular localization (36). It is possible that other regions and/or signals within these proteins might be important for their subcellular localization (35). Although we cannot rule out that the C-terminal HA tag may affect the protein's localization, it is possible that a combination of N-terminal acylation and PIP2 binding is required for localization. In this regard, TbPI-PLC has an overall domain organization very similar to that of the mammalian zeta-type PI-PLC (PI-PLC-ζ) present in sperm (9). Like PI-PLC-ζ, TbPI-PLC1 lacks a pleckstrin homology (PH) domain that in mammalian PLC-δ binds with a high affinity and specificity to PIP2 (37–40), but it has a linker region proximal to the Y domain that contains a cluster of basic residues not found in the homologous region of PLC-δ (41). Figure 6 compares these sequences of the X-Y linker regions of mouse PLC-ζ and TbPI-PLC1.
FIG 6.

Comparison of the sequences of the X-Y linker regions of mouse PLC-ζ and TbPI-PLC1. Basic residues are underlined.
It has been shown that the linker of PLC-ζ has a high affinity for PIP2 (41). Interestingly, PLC-ζ causes Ca2+ oscillations in mouse eggs by targeting intracellular and not plasma membrane PIP2 (23), although the nature of the vesicles to which PLC-ζ and TbPI-PLC are localized is unknown.
While the majority of PIP2 resides in the plasma membrane of mammalian cells (42, 43), specific antibodies against PIP2 localized this lipid predominantly to intracellular membranes of PCF trypanosomes, in agreement with the intracellular localization of TbPI-PLC1. Taken together, these results suggest that TbPI-PLC1 binds to PIP2 in intracellular membranes via the X-Y linker region and that this prevents plasma membrane localization.
The enzymatic activity of TbPI-PLC1 is extremely sensitive to low Ca2+ concentrations. We observed peaks of PIP2 hydrolysis at 10 and 100 nM Ca2+ with the recombinant enzyme and trypanosome lysates, respectively. Both values are in the range of the estimated steady-state Ca2+ concentration of PCF trypanosomes (90 to 100 nM) (30). These findings suggest that the enzyme is constitutively active and that it constitutively generates IP3, which then stimulates the IP3 receptor to release Ca2+. These findings are also in agreement with the demonstration of a measurable steady-state level of IP3 in PCF trypanosome extracts (30) and with the finding that constitutively low-level IP3 receptor-mediated Ca2+ release in different vertebrate tissue culture cell lines is essential for maintenance of optimal cellular bioenergetics under normal basal conditions (44). Cells become metabolically compromised in the absence of this ongoing Ca2+ release activity as a result of diminished Ca2+ uptake by mitochondria (44). Ca2+ transfer from the endoplasmic reticulum to the mitochondria supports oxidative phosphorylation (44). In this regard, we previously demonstrated that the mitochondrial Ca2+ uniporter, which is responsible for mitochondrial Ca2+ uptake, is essential for the regulation of cell bioenergetics in T. brucei (17). The requirement for a low basal level of IP3 to constitutively stimulate the TbIP3R and mitochondrial Ca2+ uptake by PCF trypanosomes may explain the low specific activity of TbPI-PLC1 compared with that of previously described PLCs, such as TcPI-PLC (7).
TbPI-PLC1 has an EF-hand domain, a domain found in many Ca2+-modulated proteins, at its N terminus. The 12 residues of the calcium-binding loop, with the pattern Dx[DN]xDGx[ILV][DSTN]xxE, have been shown to regulate maximal enzyme activity (45). The TbPI-PLC loop has an aspartate in the second position of the loop that is not present in other loops. Site-directed mutagenesis of this aspartate to alanine increased the activity for PIP2 and decreased the activity for PI as the substrate, without changes in the peak of maximal Ca2+ stimulation of PIP2 hydrolysis, suggesting that the enzyme activity and substrate preference are regulated by the amino acid composition of the loop. In addition, this demonstrates that the EF-hand domain is responsible for the sensitivity to Ca2+, rather than the C2 domain, which is also able to bind Ca2+ (46).
The lack of an effect of RNAi of TbPI-PLC1 on growth of PCF trypanosomes may have been due to either insufficient depletion of the enzyme to levels that would affect cellular bioenergetics or the presence of alternative pathways for generating IP3, besides TbPI-PLC1. In this regard, some residual PI-PLC activity was present in the knockdown parasites, and it is possible that TbPI-PLC2 is responsible for such activity. On the other hand, Dictyostelium discoideum is known to possess an alternative pathway for the synthesis of IP3 (47), and we cannot rule out such a mechanism. However, changes in the PI-PLC activity of lysates from PCF trypanosomes in which TbPI-PLC1 was downregulated or upregulated established the functional role of this enzyme in IP3 generation.
In conclusion, our results demonstrate an important role of TbPI-PLC1 in the generation of IP3, which is important for Ca2+ release from acidocalcisomes (6) and for regulation of cellular bioenergetics (17) of trypanosomes. TbPI-PLC1, in contrast to other PI-PLCs, has a high degree of sensitivity to stimulation by Ca2+ and appears to induce Ca2+ mobilization by hydrolyzing internal PIP2 stores.
ACKNOWLEDGMENTS
We thank George A. M. Cross for providing strains 29-13 (PCF) and single marker (BSF), John Donelson for the p2T7Ti vector, Norbert Bakalara for the anti-TbVP1 antibody, Thomas Seebeck for the pMOTag4H vector, James Bangs for the antibody against T. brucei BiP, Frédéric Bringaud for the antibody against PPDK, and Jianmin Fang for his help during the initial phases of this project and for Fig. 5B.
This work was supported by Public Health Service grants AI-077538 and AI-108222 from the National Institute of Allergy and Infectious Diseases. S.K.-K. was also partially supported by NIH training grant AI060546 to the Center for Tropical and Emerging Global Diseases and by a Supplement to Promote Diversity in Health-Related Research (grant AI077538).
REFERENCES
- 1.World Health Organization. 2014. Trypanosomiasis, human African (sleeping sickness). World Health Organization fact sheet 259. WHO, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs259/en/. [Google Scholar]
- 2.Berridge MJ. 2009. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta 1793:933–940. doi: 10.1016/j.bbamcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Rhee SG. 2001. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishizuka Y. 1986. Studies and perspectives of protein kinase C. Science 233:305–312. doi: 10.1126/science.3014651. [DOI] [PubMed] [Google Scholar]
- 5.Docampo R, Scott DA, Vercesi AE, Moreno SN. 1995. Intracellular Ca2+ storage in acidocalcisomes of Trypanosoma cruzi. Biochem J 310:1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang G, Bartlett PJ, Thomas AP, Moreno SN, Docampo R. 2013. Acidocalcisomes of Trypanosoma brucei have an inositol 1,4,5-trisphosphate receptor that is required for growth and infectivity. Proc Natl Acad Sci U S A 110:1887–1892. doi: 10.1073/pnas.1216955110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furuya T, Kashuba C, Docampo R, Moreno SN. 2000. A novel phosphatidylinositol-phospholipase C of Trypanosoma cruzi that is lipid modified and activated during trypomastigote to amastigote differentiation. J Biol Chem 275:6428–6438. doi: 10.1074/jbc.275.9.6428. [DOI] [PubMed] [Google Scholar]
- 8.De Paulo Martins V, Okura M, Maric D, Engman DM, Vieira M, Docampo R, Moreno SN. 2010. Acylation-dependent export of Trypanosoma cruzi phosphoinositide-specific phospholipase C to the outer surface of amastigotes. J Biol Chem 285:30906–30917. doi: 10.1074/jbc.M110.142190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, Blayney LM, Swan K, Lai FA. 2002. PLC zeta: a sperm-specific trigger of Ca2+ oscillations in eggs and embryo development. Development 129:3533–3544. [DOI] [PubMed] [Google Scholar]
- 10.Swann K, Saunders CM, Rogers NT, Lai FA. 2006. PLCzeta (zeta): a sperm protein that triggers Ca2+ oscillations and egg activation in mammals. Semin Cell Dev Biol 17:264–273. doi: 10.1016/j.semcdb.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Lander N, Ulrich PN, Docampo R. 2013. Trypanosoma brucei vacuolar transporter chaperone 4 (TbVtc4) is an acidocalcisome polyphosphate kinase required for in vivo infection. J Biol Chem 288:34205–34216. doi: 10.1074/jbc.M113.518993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ulrich PN, Lander N, Kurup SP, Reiss L, Brewer J, Soares-Medeiros LC, Miranda K, Docampo R. 2014. The acidocalcisome vacuolar transporter chaperone 4 catalyzes the synthesis of polyphosphate in insect-stages of Trypanosoma brucei and T. cruzi. J Eukaryot Microbiol 61:155–165. doi: 10.1111/jeu.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemercier G, Dutoya S, Luo S, Ruiz FA, Rodrigues CO, Baltz T, Docampo R, Bakalara N. 2002. A vacuolar-type H+-pyrophosphatase governs maintenance of functional acidocalcisomes and growth of the insect and mammalian forms of Trypanosoma brucei. J Biol Chem 277:37369–37376. doi: 10.1074/jbc.M204744200. [DOI] [PubMed] [Google Scholar]
- 14.Bangs JD, Uyetake L, Brickman MJ, Balber AE, Boothroyd JC. 1993. Molecular cloning and cellular localization of a BiP homologue in Trypanosoma brucei. Divergent ER retention signals in a lower eukaryote. J Cell Sci 105:1101–1113. [DOI] [PubMed] [Google Scholar]
- 15.Lacount DJ, Barrett B, Donelson JE. 2002. Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J Biol Chem 277:17580–17588. doi: 10.1074/jbc.M200873200. [DOI] [PubMed] [Google Scholar]
- 16.Oberholzer M, Morand S, Kunz S, Seebeck T. 2006. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol 145:117–120. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Huang G, Vercesi AE, Docampo R. 2013. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun 4:2865. doi: 10.1038/ncomms3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nozaki T, Toh Fujii-E AM, Yagisawa H, Nakazawa M, Takeuchi T. 1999. Cloning and characterization of a gene encoding phosphatidyl inositol-specific phospholipase C from Trypanosoma cruzi. Mol Biochem Parasitol 102:283–295. doi: 10.1016/S0166-6851(99)00108-5. [DOI] [PubMed] [Google Scholar]
- 19.Emmer BT, Nakayasu ES, Souther C, Choi H, Sobreira TJ, Epting CL, Nesvizhskii AI, Almeida IC, Engman DM. 2011. Global analysis of protein palmitoylation in African trypanosomes. Eukaryot Cell 10:455–463. doi: 10.1128/EC.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kretsinger RH, Nockolds CE. 1973. Carp muscle calcium-binding protein. II. Structure determination and general description. J Biol Chem 248:3313–3326. [PubMed] [Google Scholar]
- 21.Moncrief ND, Kretsinger RH, Goodman M. 1990. Evolution of EF-hand calcium-modulated proteins. I. Relationships based on amino acid sequences. J Mol Evol 30:522–562. [DOI] [PubMed] [Google Scholar]
- 22.Bringaud F, Baltz D, Baltz T. 1998. Functional and molecular characterization of a glycosomal PPi-dependent enzyme in trypanosomatids: pyruvate, phosphate dikinase. Proc Natl Acad Sci U S A 95:7963–7968. doi: 10.1073/pnas.95.14.7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y, Nomikos M, Theodoridou M, Nounesis G, Lai FA, Swann K. 2012. PLCzeta causes Ca2+ oscillations in mouse eggs by targeting intracellular and not plasma membrane PI(4,5)P2. Mol Biol Cell 23:371–380. doi: 10.1091/mbc.E11-08-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouchi Z, Fukami K, Shikano T, Oda S, Nakamura Y, Takenawa T, Miyasaki S. 2004. Recombinant phospholipase Czeta has high Ca2+ sensitivity and induces Ca2+ oscillations in mouse eggs. J Biol Chem 279:10408–10412. doi: 10.1074/jbc.M313801200. [DOI] [PubMed] [Google Scholar]
- 25.Gifford JL, Walsh MP, Vogel HJ. 2007. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J 405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- 26.Grabarek Z. 2006. Structural basis for diversity of the EF-hand calcium-binding proteins. J Mol Biol 359:509–525. doi: 10.1016/j.jmb.2006.03.066. [DOI] [PubMed] [Google Scholar]
- 27.Kretsinger RH. 1976. Evolution and function of calcium-binding proteins. Int Rev Cytol 46:323–393. doi: 10.1016/S0074-7696(08)60994-8. [DOI] [PubMed] [Google Scholar]
- 28.Strynadka NC, James MN. 1989. Crystal structures of the helix-loop-helix calcium-binding proteins. Annu Rev Biochem 58:951–998. doi: 10.1146/annurev.bi.58.070189.004511. [DOI] [PubMed] [Google Scholar]
- 29.Rigden DJ, Woodhead DD, Wong PW, Galperin MY. 2011. New structural and functional contexts of the Dx[DN]xDG linear motif: insights into evolution of calcium-binding proteins. PLoS One 6:e21507. doi: 10.1371/journal.pone.0021507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreno SN, Docampo R, Vercesi AE. 1992. Calcium homeostasis in procyclic and bloodstream forms of Trypanosoma brucei. Lack of inositol 1,4,5-trisphosphate-sensitive Ca2+ release. J Biol Chem 267:6020–6026. [PubMed] [Google Scholar]
- 31.Okura M, Fang J, Salto ML, Singer RS, Docampo R, Moreno SN. 2005. A lipid-modified phosphoinositide-specific phospholipase C (TcPI-PLC) is involved in differentiation of trypomastigotes to amastigotes of Trypanosoma cruzi. J Biol Chem 280:16235–16243. doi: 10.1074/jbc.M414535200. [DOI] [PubMed] [Google Scholar]
- 32.Godsel LM, Engman DM. 1999. Flagellar protein localization mediated by a calcium-myristoyl/palmitoyl switch mechanism. EMBO J 18:2057–2065. doi: 10.1093/emboj/18.8.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emmer BT, Souther C, Toriello KM, Olson CL, Epting CL, Engman DM. 2009. Identification of a palmitoyl acyltransferase required for protein sorting to the flagellar membrane. J Cell Sci 122:867–874. doi: 10.1242/jcs.041764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tull D, Vince JE, Callaghan JM, Naderer T, Spurck T, McFadden GI, Currie G, Ferguson K, Basic A, McConville MJ. 2004. SMP-1, a member of a new family of small myristoylated proteins in kinetoplastid parasites, is targeted to the flagellum membrane in Leishmania. Mol Biol Cell 15:4775–4786. doi: 10.1091/mbc.E04-06-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mills E, Price HP, Johner A, Emerson JE, Smith DF. 2007. Kinetoplastid PPEF phosphatases: dual acylated proteins expressed in the endomembrane system of Leishmania. Mol Biochem Parasitol 152:22–34. doi: 10.1016/j.molbiopara.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price HP, Hodgkinson MR, Wright MH, Tate EW, Smith BA, Carrington M, Stark M, Smith DF. 2012. A role for the vesicle-associated tubulin binding protein ARL6 (BBS3) in flagellum extension in Trypanosoma brucei. Biochim Biophys Acta 1823:1178–1191. doi: 10.1016/j.bbamcr.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB. 1995. Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83:1037–1046. doi: 10.1016/0092-8674(95)90219-8. [DOI] [PubMed] [Google Scholar]
- 38.Ferguson KM, Lemmon MA, Sigler PB, Schlessinger J. 1995. Scratching the surface with the PH domain. Nat Struct Biol 2:715–718. doi: 10.1038/nsb0995-715. [DOI] [PubMed] [Google Scholar]
- 39.Lemmon MA. 2003. Phosphoinositide recognition domains. Traffic 4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 40.Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J. 1995. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci U S A 92:10472–10476. doi: 10.1073/pnas.92.23.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nomikos M, Mulgrew-Nesbitt A, Pallavi P, Mihalyne G, Zaitseva I, Swann K, Lai FA, Murray D, McLaughlin S. 2007. Binding of phosphoinositide-specific phospholipase C-zeta (PLC-zeta) to phospholipid membranes: potential role of an unstructured cluster of basic residues. J Biol Chem 282:16644–16653. doi: 10.1074/jbc.M701072200. [DOI] [PubMed] [Google Scholar]
- 42.Gamper N, Shapiro MS. 2007. Target-specific PIP2 signalling: how might it work? J Physiol 582:967–975. doi: 10.1113/jphysiol.2007.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM. 2002. Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem J 363:657–666. doi: 10.1042/0264-6021:3630657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Müller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaim MJ, Hallows KR, Foskett JK. 2010. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drayer AL, Meima ME, Derks MW, Tuik R, Van Haastert PJ. 1995. Mutation of an EF-hand Ca2+-binding motif in phospholipase C of Dictyostelium discoideum: inhibition of activity but no effect on Ca2+-dependence. Biochem J 311:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizo J, Sudhof TC. 1998. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem 273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 47.Stephens LR, Irvine RF. 1990. Stepwise phosphorylation of myo-inositol leading to myo-inositol hexakisphosphate in Dictyostelium. Nature 346:580–583. doi: 10.1038/346580a0. [DOI] [PubMed] [Google Scholar]