

Abstract
ATP-binding cassette transporters Pdr5 and Yor1 from Saccharomyces cerevisiae control the asymmetric distribution of phospholipids across the plasma membrane as well as serving as ATP-dependent drug efflux pumps. Mutant strains lacking these transporter proteins were found to exhibit very different resistance phenotypes to two inhibitors of sphingolipid biosynthesis that act either late (aureobasidin A [AbA]) or early (myriocin [Myr]) in the pathway leading to production of these important plasma membrane lipids. These pdr5Δ yor1 strains were highly AbA resistant but extremely sensitive to Myr. We provide evidence that these phenotypic changes are likely due to modulation of the plasma membrane flippase complexes, Dnf1/Lem3 and Dnf2/Lem3. Flippases act to move phospholipids from the outer to the inner leaflet of the plasma membrane. Genetic analyses indicate that lem3Δ mutant strains are highly AbA sensitive and Myr resistant. These phenotypes are fully epistatic to those seen in pdr5Δ yor1 strains. Direct analysis of AbA-induced signaling demonstrated that loss of Pdr5 and Yor1 inhibited the AbA-triggered phosphorylation of the AGC kinase Ypk1 and its substrate Orm1. Microarray experiments found that a pdr5Δ yor1 strain induced a Pdr1-dependent induction of the entire Pdr regulon. Our data support the view that Pdr5/Yor1 negatively regulate flippase function and activity of the nuclear Pdr1 transcription factor. Together, these data argue that the interaction of the ABC transporters Pdr5 and Yor1 with the Lem3-dependent flippases regulates permeability of AbA via control of plasma membrane protein function as seen for the high-affinity tryptophan permease Tat2.
INTRODUCTION
The role of ATP-binding cassette (ABC) transporter proteins as drug efflux pumps is arguably the best appreciated activity of these membrane proteins (1). Mutant cell lines engineered to exhibit high-level chemotherapeutic resistance led to the identification of the foundational eukaryotic ABC transporter Mdr1 (2). The dramatic drug efflux effect of these proteins has led to a focus on these roles of ABC transporters and less emphasis on their endogenous activity. More recent work indicates that ABC transporters have important functions in the absence of any drug, including export/import of natural products (3). While each ABC transporter has a characteristic substrate range of transported molecules, these proteins have also been found to influence the lipid composition of the membrane in which they are embedded (4). Altering the lipid environment of a given membrane has the capability to alter the function of the large number of proteins sharing this membrane.
The yeast Saccharomyces cerevisiae expresses two plasma membrane ABC transporters that have been demonstrated to be required for development of the normal asymmetric distribution of phospholipids across this membrane (5). These two proteins, Pdr5 and Yor1, were first identified via their contributions to the drug resistance of cells (6–9). Work from two laboratories found that these ABC transporters were required to maintain the low exofacial distribution of phosphatidylethanolamine, a phospholipid normally enriched in the cytoplasmic leaflet of the plasma membrane (5, 10). Another group discovered that cells lacking Pdr5 and Yor1 were robustly resistant to the sphingolipid intermediate phytosphingosine (PHS) (11). This was attributed to the transcriptional induction of a 7-transmembrane-domain-containing protein called Rsb1 that was argued to be a PHS exporter (12). The transcriptional control of the RSB1 gene was shown to be dependent on the zinc cluster-containing transcription factor Pdr1 (11). Pdr1 is a key regulator of pleiotropic or multiple drug resistance in S. cerevisiae through its control of many target genes, including PDR5 and YOR1 (reviewed in reference 13).
We previously demonstrated that cells lacking Pdr5 and Yor1 also had a defect in the internalization of the high-affinity tryptophan permease Tat2 (14). A pdr5Δ yor1 strain exhibited elevated plasma membrane levels of Tat2 with an associated increase in tryptophan uptake. Importantly, pdr5Δ yor1 cells were better able to grow in the presence of limiting tryptophan than wild-type cells, linking the increased Tat2 levels to a growth phenotype. Yeast strains that are tryptophan auxotrophs are exquisitely sensitive to the levels of Tat2 in the plasma membrane, as this is the only high-affinity route for these cells to obtain tryptophan (15). We found that rsb1Δ cells had lower plasma membrane Tat2 levels and provided evidence that the control of Tat2 by Rsb1 was likely to explain both the PHS resistance phenotype and enhanced growth on low tryptophan.
In further studies of the physiology of the pdr5Δ yor1 strain, we found that this strain was also extremely resistant to the sphingolipid inhibitor aureobasidin A (AbA). AbA causes lethality by blocking the conversion of ceramide to inositol-phosphoryl-ceramide (IPC) by inhibiting the IPC synthase enzyme encoded by AUR1 (16) and KEI1 (17). The AbA resistance of a pdr5Δ yor1 strain was insensitive to tryptophan auxotrophy and Tat2 or Rsb1 levels, consistent with this being a different resistance mechanism. We carried out a genetic screen for mutations that would lower the elevated AbA resistance of the pdr5Δ yor1 strain and identified 6 different genes required for AbA tolerance. Analysis of AbA-induced intracellular signaling supports the view that pdr5Δ yor1 cells are likely to exclude AbA from their cytoplasm, an ability that would explain the elevated resistance to this sphingolipid biosynthetic inhibitor. Transcriptional microarray analysis indicated that the entire suite of Pdr1-regulated genes was induced in cells lacking Pdr5 and Yor1. Together, these data suggest that these ABC transporters are important determinants of plasma membrane function as well as serving as upstream inputs controlling the activity of nuclear transcription factors.
MATERIALS AND METHODS
Media, strains, and plasmids.
Cells were grown in rich YPD medium (1% yeast extract, 2% peptone, 2% glucose) or under amino acid selective conditions in complete supplemental medium (Difco yeast nitrogen extract without amino acids, amino acid powder from Sunrise Science Products, 2% glucose). Cell transformations with plasmid were performed using the lithium acetate method with single-stranded DNA (ssDNA) (18). All strains and plasmids used in this study are listed in Table 1 and Table 2, respectively. Gene disruption with a hisG-URA3-hisG cassette was performed as described previously (19). Complete gene deletion strains were generated using a kanMX4 or natMX4 deletion cassette (20, 21). A strain lacking all 16 multidrug resistance cell membrane transporters in the RYO568 background was called RYO623 (22). To generate plasmid constructs, we used PCR amplification and homologous recombination in yeast. For construction of pSR101, a 1× hemagglutinin (HA) amino-terminally tagged YPK1 gene was placed under the control of its native promoter in pRS316. To generate pBT16, the analogue-sensitive YPK1L424G allele was amplified from pPL220 (23) and introduced into pSR101, gapped with ClaI and MfeI. All positive clones were confirmed by sequencing.
TABLE 1.
Strains
| Straina | Genotype | Source or reference |
|---|---|---|
| SEY6210 | MATα leu2-3,112 ura3-52 lys2-801 trp1-Δ901 his3-Δ200 suc2-Δ9 GAL | Scott Emr |
| MRY3 | pdr1-Δ2::hisG | 71 |
| MRY4 | pdr1-Δ2::hisG pdr3-Δ1::hisG | 71 |
| MRY32 | pdr5-Δ1::hisG | 71 |
| MRY56 | yor1-1::hisG | 71 |
| MRY290 | pdr5-Δ1::hisG yor1-1::hisG | 14 |
| MRY454 | pdr3::kanMX4 | This study |
| MRY745 | lem3::kanMX4 | This study |
| MRY746 | lem3::kanMX4 pdr5-Δ1::hisG yor1-1::hisG | This study |
| RMY190 | ypk1::natMX4 | This study |
| RMY193 | ypk1::natMX4 pdr1-Δ2::hisG pdr3-Δ1::hisG | This study |
| RYO568b | MATα hoΔ::tetO2-GFP-URA3 can1Δ::GMToolkit-alpha [CMVpr-rtTA NATMX4 STE3pr-LEU2] lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 | 22 |
| RYO623b | MATα adp1Δ snq2Δ ycf1Δ pdr15Δ yor1Δ vmr1Δ pdr11Δ nft1Δ bpt1Δ ybt1Δ ynr070wΔ yol075cΔ aus1Δ pdr5Δ pdr10Δ pdr12Δ can1Δ::GMToolkit-alpha (CMVpr-rtTA NATMX4 STE3pr-LEU2) his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 | 22 |
| SKY52 | per1::kanMX4 | This study |
| SKY53 | per1::kanMX4 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SKY82 | atr1::kanMX4 | This study |
| SKY83 | atr1::kanMX4 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SKY90 | gcn4::kanMX4 | This study |
| SKY91 | gcn4::kanMX4 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY71 | gda1::kanMX4 | This study |
| SRY73 | gda1::kanMX4 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY92 | C21 MATa per1 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY187 | pdr1::kanMX4 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY188 | pdr3::kanMX4 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY222 | B19 MATa lem3 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY224 | B27 MATα cyc8 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY226 | B33 MATa gda1 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY228 | C1 MATa swa2 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY230 | C17 MATa bsh1 pdr5-Δ1::hisG yor1-1::hisG | This study |
| SRY232 | C25 MATα mss4 pdr5-Δ1::hisG yor1-1::hisG | This study |
All strains were generated in SEY6210 background unless otherwise specified.
Diploid strain from BY4741 × BY4742.
TABLE 2.
Plasmids
| Name | Description | Source or reference |
|---|---|---|
| pSR101 | pRS316 YPK1PRO-1×HA-YPK1 CEN6 URA3 | This study |
| pBT16 | pRS316 YPK1PRO-1×HA-YPK1L424G CEN6 URA3 | This study |
| pPL215 | p416Met25 YPK1-3HA URA3 | 23 |
| pSH14HA | ORM1PRO-HA-ORM1-ORM1TERM CEN6 HIS3 | 72 |
| pDR3.3 | Sau3A partial fragment of yeast genomic DNA containing PDR5-coding region was inserted into BamHI site of YEp24, URA3 2μ | 73 |
| MRB700 | pdr5 mutant allele derived from YJM789, YEplac195, URA3 2μ | 56 |
| p238 | Constitutively active GCN4C allele in YCp50, URA3 CEN4 | 61 |
Drug sensitivity assay.
For drug sensitivity plating assays, we used mid-log-phase cultures (optical density at 600 nm [OD600] of 1). Fifteen thousand cells were spotted in 1:10 dilutions onto plates containing aureobasidin A (Clontech catalogue number 630466), myriocin (Myr) (Sigma catalogue number M1177), phytosphingosine (PHS) (Avanti catalogue number 239083), or fluconazole (Flu) (LKT Laboratories, Inc., catalogue number F4682). Plates were incubated at 30°C for 2 days.
EMS mutagenesis.
The protocol for ethyl methanesulfonate (EMS) mutagenesis was adapted from that of Steven Hahn's laboratory, Fred Hutchinson Cancer Research Center. The pdr5Δ yor1 (MRY290) strain was treated with EMS for 40 min to induce 50% lethality. To recover mutagenized clones, 15,000 cells were plated onto YPD at 500 cells per plate density. Recovered clones were further analyzed for their sensitivity to 75 ng/ml aureobasidin A (AbA) by replica plating. EMS mutants that showed strong sensitivity to AbA were backcrossed to the parental pdr5Δ yor1 strain three times. Final strains were transformed with the YCp50 library with selection for AbA resistance. Boundaries of the complementing DNA contained in the plasmids were determined by sequencing with YCp50-specific primers 5′-GCCGCCCAGTCCTGCTCGCTTCGCT and 3′-TGATGTCGGCGATATAGGCGCCAGC. Three mutants (lem3, swa3, and per1) could not be complemented from our YCp50 library. We prepared genomic DNA from these mutant strains and the isogenic wild-type strain. These DNA samples were subjected to Illumina sequencing and analyzed for the altered genes contained in each mutant. We confirmed that the mutations detected were responsible for the AbA sensitivity phenotype in at least one of two ways. All mutants were transformed with plasmids containing the wild-type gene obtained from a genomic library (24). The presence of the appropriate wild-type gene returned AbA resistance to its high level. We also disrupted wild-type copies of LEM3 or PER1 with a kanMX4 allele and determined that these null mutations led to AbA hypersensitivity in all backgrounds tested. For these EMS mutant strains, yeast sphingolipids were extracted and measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) according to previously established protocols (25).
Western blot analysis.
Four OD600 units of mid-log-phase cells were collected per each sample. Total protein extract was isolated using an NaOH-trichloroacetic acid (TCA) method as previously described (26). Equal amounts of total protein extract were resolved on 8% SDS-polyacrylamide gels and transferred to Whatman Protran nitrocellulose membranes (GE Healthcare Life Science catalogue number Z670960). The membrane was blocked in 5% nonfat dry milk in1× TBST (50 mM Tris [pH 7.4], 300 mM NaCl, 0.05% Tween 20) and probed with 1:5,000 mouse monoclonal HA.11 (16B12) (Covance catalogue number MMS-101P), polyclonal anti-phospho-T662-Ypk1 (23), or monoclonal mouse 12G10 anti-alpha-tubulin (procured from the Developmental Studies Hybridoma Bank at the University of Iowa) primary antibodies in Odyssey blocking buffer (phosphate-buffered saline [PBS]) (Li-Cor catalogue number 927-40000) overnight. IRD dye 680RD goat anti-rabbit (Li-Cor catalogue number 926-68071) and IRD dye 800CW goat anti-mouse (Li-Cor catalogue number 926-32210) were used as secondary antibodies at a 1:15,000 dilution in Odyssey blocking buffer (PBS). For imaging and quantitation of the protein signal, we used the Li-Cor Odyssey infrared imaging system, application software version 3.0.
Microarray analyses.
Cells were grown in YPD medium prior to RNA isolation. One microgram of total RNA was reverse transcribed and labeled with Cy5 or Cy3 using the amino-allyl protocol described at transcriptome.ens.fr/sgdb/protocols/. We used custom yeast microarrays 8x15k made by Agilent, in which each gene probe is present in duplicate (GEO platform number GPL6035). These Agilent arrays were hybridized with the labeled cDNA using the Agilent hybridization protocol. After washing following the supplier's recommendations, the slides were scanned using a 2-μm Agilent microarray scanner. The images were analyzed using the feature extraction software and normalized by global lowess using Goulphar (27). For each gene, the Cy5/Cy3 ratios corresponding to the duplicated probes were averaged. The experiments were performed four times using biologically independent samples and dye swap. The averaged log2 of the mutant/wild-type ratios and the standard deviation between the two replicates were calculated for each gene. The complete microarray data are available in Table S1 in the supplemental material. The Gene Ontology category enrichment analyses were performed using FUNSPEC with default parameters (cutoff P value = 0.01 and no Bonferroni correction) (28). The enrichments in specific transcription factor-target gene regulatory interactions were measured using the “rank TF” tool of YEASTRACT with default parameters (29).
Microarray data accession number.
The complete microarray data have been deposited in the GEO database under accession number GSE65264.
RESULTS
Links between ABC transporters and sphingolipid biosynthesis.
Strains lacking the plasma membrane ABC transporter proteins Pdr5 and Yor1 have been found to exhibit striking resistance to exposure to long-chain base phytosphingosine (PHS) (11). Since PHS is an intermediate in sphingolipid biosynthesis, we tested the ability of the pdr5Δ yor1 strain to tolerate inhibitors of sphingolipid biosynthesis to probe activity of this biosynthetic pathway. We used the IPC synthase inhibitor aureobasidin A (AbA) and the serine palmitoyltransferase inhibitor myriocin (Myr) (Fig. 1A). We used the ergosterol biosynthetic inhibitor fluconazole (Flu) and PHS to provide phenotypic controls for previously characterized phenotypes of the pdr5Δ yor1 strain. Strains were grown to mid-log phase and serial dilutions plated on rich media containing drugs at various concentrations.
FIG 1.

Sphingolipid biosynthetic inhibitors and their interaction with strains lacking ABC transporters. (A) A simplified diagram depicting sphingolipid biosynthesis and the locations at which different inhibitors block this pathway. Spt, serine palmitoyltransferase; LCB, long-chain base or sphingoid base; GPI anchors, glycosylphosphatidylinositol-linked protein biosynthesis; IPC, inositol phosphorylceramide; MIPC, mannosyl-IPC; M(IP)2C, mannosyl-(di-inositol phosphoryl)-ceramide. (B) Isogenic yeast strains were grown to mid-log phase and tested for drug resistance by spotting the indicated number of cells on rich medium (YPD) or YPD supplemented with drugs. The resulting plates were incubated at 30°C. (C) Strains were transformed with low-copy-number plasmid vectors to restore uracil (pRS316:URA3) or tryptophan (pRS134:TRP1) prototrophy. Transformants were spotted on YPD or YPD plus the indicated drugs.
Although both AbA and Myr act to inhibit sphingolipid biosynthesis, the behaviors of the pdr5Δ yor1 strain in response to these drugs were very different. Resistance to AbA was strongly elevated in this genetic background, while these same strains were hypersensitive to Myr (Fig. 1B). This differential resistance behavior does not support the idea that a simple increase in sphingolipid biosynthesis is likely to explain the AbA resistance. As expected, the pdr5Δ yor1 cells were sensitive to Flu and resistant to PHS exposure.
PHS toxicity is most pronounced in tryptophan auxotrophs, as the PHS-dependent diversion of Tat2 to the vacuole removes the only high-affinity route cells possess to acquire this amino acid (30). Restoration of tryptophan prototrophy strongly elevates tolerance to PHS (31). Since resistance to AbA and PHS was elevated in the absence of Pdr5 and Yor1, we wanted to determine if AbA resistance was also sensitive to tryptophan prototrophic status. Isogenic wild-type, pdr5Δ, or pdr5Δ yor1 cells were transformed with plasmids expressing the URA3 or TRP1 gene, restoring uracil or tryptophan prototrophy, respectively. Transformants were then plated on media containing AbA or PHS (Fig. 1C).
AbA resistance was unaffected by tryptophan prototrophy. Both wild-type and pdr5Δ cells remained sensitive to AbA irrespective of their ability to biosynthesize tryptophan. As seen before (14, 31), Trp+ transformants were highly tolerant to PHS compared to isogenic Trp− cells. These data indicate that the mechanism of AbA resistance in cells lacking Pdr5 and Yor1 is different from that of PHS tolerance and seems unlikely to involve increased sphingolipid biosynthetic pathway activity.
Pdr pathway activity and AbA resistance.
We wondered if the finding that increased AbA resistance was seen in the absence of Pdr5 and Yor1 could be related to earlier findings that loss of particular ABC transporter-encoding genes triggered a potentially compensatory induction of the remaining genes (32). To evaluate this possibility, we employed a strain engineered to lack all genes encoding the ABC transporter proteins in S. cerevisiae (22). Matched congenic control strains were grown and placed on medium containing AbA (Fig. 2A).
FIG 2.
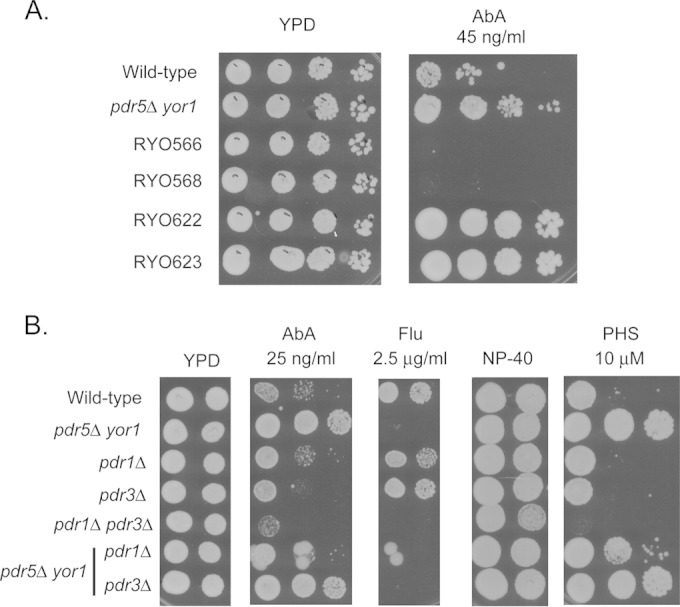
Genetic analysis of participation of other ABC transporter-encoding genes or Pdr1/3 transcription factors in pdr5Δ yor1 phenotypes. (A) Serial dilutions of congenic strains containing (RYO566/568) or lacking (RYO622/623) the genes encoding the 16 ABC transporter proteins were placed on rich medium (YPD) lacking or containing the indicated concentration of AbA. Isogenic wild-type and pdr5Δ yor1 strains used in all other panels were included as controls for the effect of AbA. (B) The indicated strains were tested for resistance by spotting different numbers of cells on drug-containing media. Plates were then incubated at 30°C to allow spots to develop.
Loss of all 16 ABC transporter-encoding genes still produced a strain exhibiting a robust AbA-resistant phenotype. We conclude from this experiment that the cause of the enhanced AbA resistance seen in the absence of Pdr5 and Yor1 is not related to changes in other ABC transporter proteins. Since both PDR5 and YOR1 are transcriptionally regulated by the homologous zinc cluster-containing transcription factors Pdr1 and Pdr3, we examined the participation of these positive regulatory proteins in the increased AbA resistance seen in a pdr5Δ yor1 strain. Isogenic strains lacking the indicated PDR genes were placed on media containing various drugs and compared for their ability to respond to these stresses (Fig. 2B).
Removal of Pdr1 but not Pdr3 dramatically lowered AbA resistance in a pdr5Δ yor1 strain. This result is consistent with Pdr1 playing a more important role in the AbA resistance phenotype induced in response to loss of Pdr5 and Yor1. A similar increased importance of Pdr1 compared to Pdr3 was seen in the elevated PHS resistance of the pdr5Δ yor1 strain (Fig. 2B) (11, 14). In strains containing Pdr5 and Yor1, loss of either Pdr1 or Pdr3 alone has a negligible effect on AbA resistance. Loss of both transcription factors increased AbA sensitivity in otherwise wild-type cells.
These findings indicate an important transcriptional input from Pdr1 that contributes to the AbA resistance in pdr5Δ yor1 cells. We examined this directly using transcriptional microarray experiments (see below).
Evidence that AbA accumulation is lowered in a pdr5Δ yor1 strain.
The toxicity of AbA requires this drug to enter the cell and bind to the Aur1 protein, an essential subunit of IPC synthase (16). AbA tolerance could be increased by multiple mechanisms, including lowered intracellular levels of the drug and/or efficacy of action. The AGC kinase Ypk1 is induced by AbA exposure and influences resistance to this drug via the phosphorylation and inhibition of the serine palmitoyltransferase (Spt) regulator Orm1 (and Orm2) (33, 34). The Orm proteins inhibit Spt activity (35). We used Ypk1 and its substrate Orm1 as monitors for the effect of loss of Pdr5 and Yor1 on AbA-induced changes in signaling.
Isogenic wild-type and pdr5Δ yor1 strains lacking the chromosomal YPK1 gene were constructed. These strains were transformed with epitope-tagged versions of the YPK1 gene corresponding to either the wild-type or analogue-sensitive (AS) allele carried on a low-copy-number vector plasmid. The AS allele was engineered to confer sensitivity to an ATP analogue (23), and we demonstrate here that this allele has reduced function compared to the wild-type version of this gene. Appropriate transformants were tested for the ability to confer AbA resistance to ypk1Δ or pdr5Δ yor1 ypk1Δ strains. The empty low-copy-number vector was included as a negative control. Transformants were grown to mid-log phase and then placed on media containing different concentrations of AbA (Fig. 3A).
FIG 3.
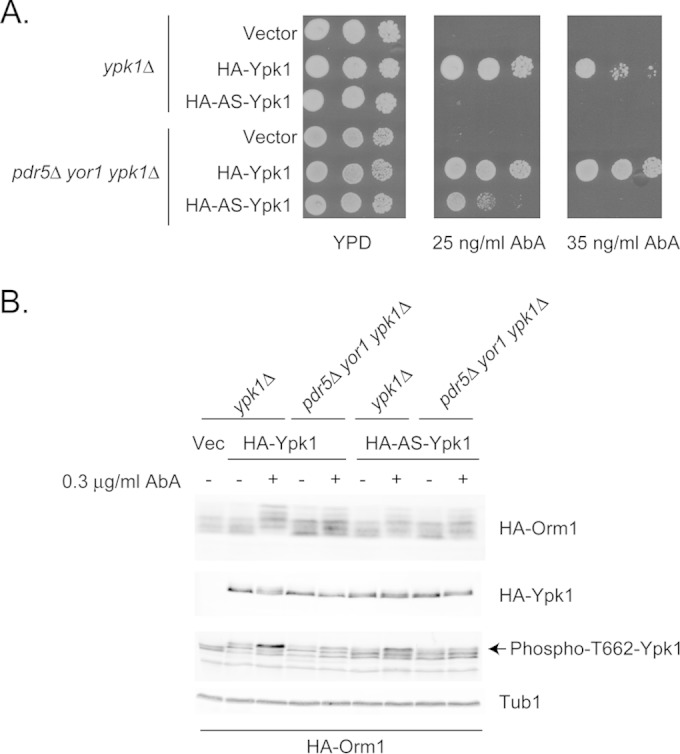
Ypk1-dependent signaling is required for AbA resistance in both wild-type and pdr5 yor1 cells. (A) Isogenic ypk1Δ and pdr5Δ yor1 ypk1Δ strains were transformed with low-copy-number plasmids expressing either wild-type Ypk1, analogue-sensitive (AS) Ypk1, or the empty vector plasmid alone. Transformants were grown to mid-log phase and then tested for AbA resistance. (B) Transformants from panel A were retransformed with a low-copy-number plasmid expressing epitope-tagged Orm1 (HA-Orm1). Transformants were grown to mid-log phase and then either left untreated (−) or challenged with 0.3 μg/ml AbA for 90 min (+). Whole-cell protein extracts were prepared and analyzed by Western blotting using the indicated antibodies. HA-tagged Orm1 and Ypk1 were well resolved by this 12% SDS-PAGE and detected simultaneously using anti-HA antiserum. TORC2-dependent phosphorylation of Ypk1 residue threonine 662 (Phospho-T662-Ypk1) was detected using a previously described antibody (23). Levels of tubulin were blotted as a control for equal protein loading (Tub1).
Loss of Ypk1 led to a dramatic increase in AbA sensitivity in both otherwise wild-type and pdr5Δ yor1 strains. The presence of the AS-Ypk1 protein was unable to restore AbA resistance to ypk1Δ cells but did support increased resistance in a pdr5Δ yor1 strain. We interpret these data to indicate that the AS-YPK1 allele encodes a hypomorphic form of Ypk1 that has reduced but still significant function compared to that of the wild-type kinase and that the ability of this protein to function is regulated by the presence of Pdr5 and Yor1.
To determine the effect of the pdr5Δ yor1 background on Ypk1-dependent signaling, we used two different reporters for the activity status of Ypk1. Experiments from several labs demonstrated that Orm1 is a Ypk1 substrate that is negatively regulated by phosphorylation (33, 36, 37). Activation of Ypk1 requires phosphorylation on threonine 662 via the TORC2 kinase complex (36, 38). Relevant transformants from the experiments described above carrying either the wild-type or AS form of Ypk1 were grown to mid-log phase and then either left untreated or challenged with AbA for 90 min. Whole-cell protein extracts were prepared and then analyzed by Western blotting with several different antibodies (Fig. 3B).
Ypk1 phosphorylation of Orm1 was increased upon AbA challenge, as reported previously (33, 34). However, AbA-induced phosphorylation was lower in pdr5Δ yor1 strains. No significant increase in Orm1 phosphorylation could be detected in the presence of the AS-Ypk1 kinase in either genetic background. Direct evaluation of the TORC2-dependent activation of Ypk1 was accomplished using an anti-phosphothreonine 662 (p662T) antibody described previously (23). Levels of p662T Ypk1 were strongly increased upon AbA challenge in ypk1Δ cells but clearly reduced in the presence of the pdr5Δ yor1 mutations. This same behavior was seen for the AS-Ypk1 kinase. Expression of Ypk1 was similar under all conditions.
These data argue that the degree of AbA-induced stress is reduced in the absence of Pdr5 and Yor1 and support the view that less AbA is able to access its intracellular target (Aur1).
Genetic search for second-site suppressors of AbA resistance of pdr5Δ yor1 strains.
The data discussed above indicate that the increased AbA resistance seen in a strain lacking Pdr5 and Yor1 is unlikely to be caused by increased expression of other ABC transporters or enhanced Ypk1 signaling. To identify genes involved in the elevated AbA resistance seen in a pdr5Δ yor1 mutant background, we mutagenized a strain lacking these two ABC transporter-encoding genes with a chemical mutagen. Survivors were recovered on rich medium and then screened for loss of AbA resistance. AbA-sensitive mutants were backcrossed to a pdr5Δ yor1 strain of opposite mating type and the diploids evaluated for AbA resistance. These diploids were sporulated and segregants retested for AbA sensitivity. Sensitive mutants were backcrossed to an unmutagenized pdr5Δ yor1 strain at least two more times. The genes altered in these AbA-sensitive strains are referred to as maintenance of aureobasidin resistance (MAR) genes.
We have identified 6 different complementation groups of MAR loci that were all represented by single isolates in our original screen. All of the mutant alleles were recessive. These genes were recovered in one of two manners. First, we used a YCp50-based library to clone three genes by selecting for restored AbA resistance in a marX pdr5Δ yor1 background. These genes were GDA1, CYC8, and MSS4. Second, three genes were identified by sequencing the genome of a marX pdr5Δ yor1 strain of interest as well as the wild-type strain. Candidate mutations were selected by comparing the DNA sequences and the causative allele confirmed by complementation. Genes identified in this fashion were SWA2, PER1, and LEM3.
Two of the MAR loci encode enzymes involved in biosynthetic pathways. GDA1 produces a guanosine diphosphatase that acts to stimulate mannose import into the Golgi apparatus (39). This sugar is used for glycosylation of both proteins and sphingolipids, and loss of Gda1 causes defects in both of these processes (40). PER1 is required for biosynthesis of glycosylphosphatidylinositol (GPI)-anchored proteins (41). Work from others established that loss of GPI-anchored protein biosynthesis led to a profound increase in AbA tolerance (42). Interestingly, the recessive allele of PER1 identified here corresponded to an amino acid substitution mutation in the conserved catalytic region of this enzyme (G103E) (41).
CYC8 is a transcriptional coregulator, involved in both negative (43) and positive (44) control of gene expression. While the pleiotropic nature of CYC8-dependent target genes complicated our understanding of this mutation, the change responsible for the phenotype in this mutant was localized by DNA sequencing to a missense mutation changing glycine 337 to aspartate, located in tetratricopeptide repeat 9 (TPR9). These TPRs are important in protein-protein interaction, and TPR9 has been implicated in promoter-specific gene regulation (45).
The remaining three genes are involved in events at the plasma membrane. SWA2 encodes another TPR-containing protein that is involved in disassembling clathrin-coated vesicles (46). The SWA2 mutation is predicted to be a glycine-to arginine change at position 449. Swa2 is important in endocytosis and has defects in cortical endoplasmic reticulum inheritance (47). MSS4 encodes the only phosphatidylinositol 4′,5′-phosphate (PI-4,5-P2) kinase in the cell and is an essential gene (48, 49). As seen with other MSS4 alleles (48), the mutation isolated here is temperature sensitive and consists of two changes in the coding sequence (T271I T724M). Finally, LEM3 encodes an essential subunit of the plasma membrane phospholipid flippase (50, 51). The lesion affecting the LEM3 gene was the introduction of stop codon at position 308 in place of a tryptophan codon (TGG to TGA).
To compare the behaviors of these different mutations, we grew isolates that had been outcrossed twice to an unmutagenized version of the starting pdr5Δ yor1 strain. These strains were tested for the ability to grow in the presence of AbA or PHS by spot test assay (Fig. 4A).
FIG 4.

Isolation of second-site suppressors of the AbA resistance of a pdr5Δ yor1 strain. (A) The indicated EMS-generated mutant strains generated in the pdr5Δ yor1 background were tested for resistance to AbA and PHS as before. (B) The strains in panel A were grown to mid-log phase and total lipids extracted as described previously (74). Levels of the two sphingoid bases in S. cerevisiae (DHS and PHS) were determined by mass spectrometry.
All of these mutant strains were highly sensitive to AbA, although some differences were seen. Strains lacking wild-type versions of MSS4, LEM3, PER1, and SWA2 were the most sensitive to AbA while loss of GDA1 and CYC8 caused a less severe phenotype. Interestingly, none of these mutants exhibited significantly increased sensitivity to PHS. As described above for the tryptophan sensitivity of PHS resistance, these phenotypes support the view that the mechanisms of AbA and PHS tolerance are distinct.
We also analyzed all these strains for their levels of sphingoid bases (PHS and dihydrosphingosine [DHS]) via mass spectrometry (Fig. 4B). Only loss of PER1 caused a large increase in sphingoid base levels, with the relative amounts of DHS and PHS being twice those in the other strains. Mutants lacking PER1 have previously been shown to normally synthesize complex sphingolipids (containing inositol) (41). These data provide the first demonstration that Per1 is required for normal metabolism of sphingoid bases, at least in the context of the pdr5Δ yor1 strain.
Finally, we confirmed for PER1, GDA1, and LEM3 that the mutations we isolated were loss-of-function alleles by constructing isogenic deletion derivatives. We amplified the respective kanMX4 allele from the S. cerevisiae disruption collection (52) and introduced these mutations into isogenic wild-type and pdr5Δ yor1 strains. Single disruption mutations of each of these genes caused AbA sensitivity in both backgrounds (see Fig. S1 in the supplemental material), consistent with the EMS-generated alleles corresponding to loss-of-function forms of the genes.
Interaction of Pdr5 and Lem3/flippase activity.
Work from two other laboratories provided evidence that Pdr5 and Yor1 can act as phospholipid floppase proteins, moving phospholipids from the inner to the outer leaflet of the plasma membrane (5, 10). The action of floppase proteins is balanced by the inward movement of phospholipids catalyzed by flippase proteins. In S. cerevisiae, Lem3 is an essential contributor to the plasma membrane flippase activity. Lem3 acts as an auxiliary subunit that is required for exit of the flippase complex from the endoplasmic reticulum (53). Lem3 associates with the catalytic subunits encoded by DNF1 and DNF2 to produce the plasma membrane flippase. Identification of a lem3 mutation as a suppressor of the AbA resistance induced in a pdr5Δ yor1 strain prompted us to examine the possible interaction between Pdr5 and Lem3-dependent flippase activity.
We constructed isogenic lem3Δ derivatives of our wild-type and pdr5Δ yor1 strains to evaluate the contribution of flippase to the AbA and Myr phenotypes caused by loss of Pdr5 and Yor1. As can be seen from Fig. 5A, loss of Lem3 had dramatically different effects on the resistance to these sphingolipid biosynthesis inhibitors. Introduction of the lem3Δ allele into either wild-type or pdr5Δ yor1 strains led to a large increase in tolerance to Myr while causing a dramatic sensitization to AbA exposure. The elevated resistance to Myr caused by loss of Lem3 has been noted by two other groups (36, 54). The epistatic relationship between the LEM3 and PDR5/YOR1 genes is most consistent with Lem3 acting downstream from the ABC transporters.
FIG 5.

Interaction of Lem3-dependent flippase with ABC transporters determines resistance to sphingolipid biosynthesis. (A) The indicated strains were tested for resistance to either myriocin (Myr) or AbA. The LEM3 alleles represent lem3Δ::kanMX4 deletions that were recovered from the collection of genomic disruption mutations (75). (B) Loss of Fpk1/2 kinases phenocopies lem3Δ. Disruption alleles of FPK1, FPK2, or both were introduced into wild-type and pdr5Δ yor1strains. These mutants were tested for resistance to Myr and AbA. (C) Separation of resistance phenotypes influenced by Pdr5.
To further probe the genetic relationship between the floppase activity produced by Pdr5 and Yor1 and the flippase activity dependent on Lem3, we constructed a series of strains lacking positive regulators of the flippase (Fig. 5B). These two related genes, FPK1 and FPK2, encode protein kinases that stimulate flip activity (55). Loss of Fpk1 is sufficient to cause Myr resistance and a marked decrease in flip (54, 55). Loss of both Fpk1 and Fpk2 caused elevated Myr resistance and AbA hypersensitivity. This relationship was retained in pdr5Δ yor1 cells. Together, these genetic interactions suggest that Pdr5 and Yor1 act as negative regulators of AbA resistance but positively modulate Myr resistance. The effect of Pdr5 and Yor1 is overridden by disruption of LEM3, consistent with these ABC transporters acting upstream of the Lem3-dependent flippase.
Since these data suggest that Pdr5 might be acting by modulating flippase function, we wondered if the activity of Pdr5 as a drug efflux transporter could be separated from its influence on flippase function. To examine this possibility, we used a recently described allele of PDR5 that lacked the ability to provide fluconazole resistance but produced a protein that was normally expressed and localized (56). This mutant form of Pdr5 was obtained from a clinical isolate of S. cerevisiae and is referred to as PDR5* here. PDR5* has 17% amino acid alterations between its second transmembrane domain region and that of the standard S288C PDR5 gene (56). Plasmid vectors expressing either the wild-type (PDR5) or this clinical (PDR5*) allele were introduced into a pdr5Δ yor1 strain and transformants tested for several different resistance phenotypes (Fig. 5C).
Expression of either the wild-type or PDR5* allele in pdr5Δ yor1 cells strongly elevated Myr resistance, as expected for a positive regulator of tolerance to this drug. As expected, expression of the PDR5* allele did not complement the fluconazole sensitivity of the pdr5Δ yor1 strain but did partially reverse both the AbA and PHS resistance, suggesting that this form of Pdr5 retained low but significant activity. Wild-type PDR5 was able to fully restore fluconazole and Myr resistance and normal AbA sensitivity. These data are consistent with the view that drug efflux activity (represented by fluconazole resistance) can be separated from Myr resistance (potentially mediated by flippase).
Loss of Pdr5 and Yor1 induces a Pdr1-dependent transcriptional response and represses the Gcn4 regulon.
As mentioned above, Pdr1 was required for the increased AbA resistance in the absence of Pdr5 and Yor1 (Fig. 2B). We used transcriptional microarrays to profile the genomic changes in gene expression induced in a pdr5Δ yor1 strain compared with its isogenic wild-type strain, We analyzed the enrichments in gene ontology categories and specific transcription factor regulatory interactions in the lists of the 50 most induced and 50 most repressed genes (see Table S1 in the supplemental material). These analyses pointed out two striking patterns of regulation that characterized the pdr5Δ yor1 strain (Fig. 6A).
FIG 6.

Loss of Pdr5 and Yor1 induces expression of the Pdr regulon. (A) The pdr5Δ yor1 strain induces the PDR regulon and represses a subset of Gcn4 targets. MA (Bland-Altman) plot of the pdr5Δ yor1 comparison is shown. The x axis is the log2 of fluorescence intensity. The y axis is the log2 of the expression ratios (mutant/wild-type). Red squares are genes from the PDR regulon. Green triangles are Gcn4 gene targets. (B) The PDR response of the pdr5Δ yor1 strain is Pdr1 dependent. The x axis is the log2 of pdr5Δ yor1 strain/pdr5Δ yor1 pdr1Δ strain expression ratios. The y axis is the log2 of the pdr5Δ yor1 strain/wild-type strain expression ratios. Red squares are genes from the PDR regulon. Green triangles are Gcn4 gene targets. (C) Gcn4 antagonizes phenotypes caused by loss of Pdr5 and Yor1. A low-copy-number plasmid containing a constitutively expressed allele of GCN4 (GCN4c) (61) or the empty vector (pRS316) was introduced into wild-type and pdr5Δ yor1 strains. Transformants were then tested for resistance to AbA and Myr by plate assay.
First, removal of the PDR5 and YOR1 genes induced the Pdr response. Indeed, the upregulated genes were mostly involved in drug response (P value = 3.3 × 10−5) and stress response (P value = 1.5 × 10−7). Moreover, according to the Yeastract database, the transcription factor which is the most strongly associated with the 50 most upregulated genes in our experiments is Pdr1, with 74% of these genes being documented as Pdr1 targets (P value = 10−15). More precisely, 17 out of the 25 genes being previously defined as constituting the “Pdr1 regulon” (57–60) were found among the 50 most upregulated genes (Fig. 6A). Microarray comparisons of the pdr5Δ yor1 and pdr5Δ yor1 pdr1Δ transcriptomes confirmed that this PDR response was dependent on the presence of the PDR1 gene, with the notable exceptions of PDR3 and PDR15, which remained overexpressed in the triple mutant (Fig. 6B). This result is consistent with our previous observation that the loss of the transcription factor Pdr1 prevents the normal development of pdr5Δ yor1-induced AbA resistance (Fig. 2B).
Second, the list of the 50 most downregulated genes in the pdr5Δ yor1 strain is enriched in loci encoding enzymes involved in amino acid biosynthesis (P value = 1.3 × 10−10), particularly those required for arginine (P value = 5.5 × 10−9) and leucine (P value = 4.3 × 10−6) production. Strikingly, almost all of these 50 genes are referred as Gcn4 targets in the Yeastract database (92%; P value = 3 × 10−15) (Fig. 6A). To determine if the decreased expression of Gcn4-responsive genes influenced the resistance phenotypes of the pdr5Δ yor1 strain, we introduced a plasmid that drove constitutive expression of Gcn4. This plasmid allows the expression of a GCN4 allele lacking the 4 upstream AUG codons in the 5′ untranslated region (UTR) of the GCN4 mRNA that normally repress translation of the coding sequence (61). This plasmid drives elevated and unregulated levels of Gcn4. This Gcn4-overproducing plasmid was introduced into wild-type and pdr5Δ yor1 cells. Transformants were tested for resistance to AbA and Myr (Fig. 6C). The presence of the Gcn4-overexpressing plasmid caused a dramatic reduction in AbA resistance in both wild-type and pdr5Δ yor1 cells. Gcn4 overproduction was also able to restore Myr resistance to pdr5Δ yor1 cells but had minor effects on this phenotype in wild-type cells. These data provide evidence that the reduction in Gcn4-dependent gene expression is a factor contributing to the AbA-resistant, Myr-sensitive phenotype seen in pdr5Δ yor1 cells.
In conclusion, pdr5Δ yor1 cells induce the Pdr1 regulon and repress Gcn4 targets. Both effects are important for the response of pdr5Δ yor1 strain to sphingolipid biosynthesis inhibitors, since the inactivation of Pdr1 or the overproduction of Gcn4 reversed the mutant phenotypes.
DISCUSSION
The vast majority of phenotypes caused by loss of the ABC transporters Pdr5 and Yor1 involve increased sensitivity to many chemically dissimilar drugs (62). Our finding here of the large increase in resistance to the IPC synthase inhibitor AbA was unexpected and striking when considered along with earlier observations that the pdr5Δ yor1 strain was also highly resistant to the ceramide precursor PHS (11). These observations, along with findings that genes involved in sphingolipid biosynthesis were members of the Pdr regulon (63, 64), suggested the presence of a regulatory link between the plasma membrane ABC transporters and sphingolipid biosynthesis that could be mediated by Pdr1. Our microarray experiments supported this view and provide evidence for the existence of a presently unidentified signal that elevates Pdr1 activity in a pdr5Δ yor1 background. This signal is Pdr1 specific, as disruption of the related PDR3 gene failed to reduce AbA (or PHS) resistance (11, 65). The finding that Pdr1-regulated transcription is induced in the absence of Pdr5 and Yor1 extends a previous observation that compensatory activation of genes encoding other ABC transporter proteins can be detected in a similar genetic background (32). The data reported here demonstrate that the full Pdr regulon is induced in a pdr5Δ yor1 strain and is not restricted to ABC transporter-encoding genes. Taken together, these observations suggest that some Pdr1-specific signal is produced in the pdr5Δ yor1 background that leads to the increased expression of target genes.
While Pdr1 activity is induced upon loss of Pdr5 and Yor1, we do not believe that the increased AbA resistance of a pdr5Δ yor1 strain can be explained by an increase in sphingolipid biosynthesis, since resistance to other pathway inhibitors such as Myr or rustmicin decreased when Pdr5 was lost (66). Additionally, our direct measurements of long-chain bases demonstrated that no consistent increase in the levels of these key sphingolipid biosynthetic intermediates could be seen (Fig. 4B). These nonequivalent phenotypes point out the inherent complexity of analysis of a multifunctional protein like Pdr5 (or Yor1). These ABC transporters have a direct action on drug resistance through their activity as drug exporters but also can influence membrane function via their control of phospholipid asymmetry. The phenotype of strains lacking these membrane transporter proteins will be a summation of the direct loss of their drug-pumping activities combined with the secondary phenotypes triggered by loss of their lipid-transporting functions. It is this second role in regulation of membrane composition that we believe is responsible for AbA resistance via export of this drug from the cell.
The elevated Pdr1 activity in pdr5Δ yor1 strains is accompanied by a corresponding reduction in Gcn4-mediated transactivation. While the precise cause of this response is not understood, we speculate that reduced Gcn4 function may be related to the likely defect in protein delivery to the vacuole that we have previously characterized (14). Extensive genetic analyses have clearly established a link between subcellular protein trafficking and Gcn4 transactivation (67). Loss of various Vps proteins involved in traffic to the vacuole depressed the ability of Gcn4 to activate transcription of target genes. This earlier study also demonstrated that mutants with abnormal endosomal trafficking did not uniformly inhibit activator function, consistent with our finding here that Pdr1-mediated gene expression is stimulated when Gcn4 activation is lowered. Together, these data suggest that alterations in endosomal trafficking caused by pdr5Δ yor1 alter nuclear gene expression.
We were struck by the identification of a LEM3 allele as a strong suppressor of AbA resistance in the pdr5Δ yor1 strain. Lem3 is required for normal development of flippase activity at the plasma membrane in association with the catalytic subunits Dnf1 and Dnf2 (53). Lem3-dependent flippase activity is required for wild-type PHS resistance (11), likely through its role in the normal delivery of Tat2 to the plasma membrane (68). Mutants lacking Lem3 are highly sensitive to AbA but also extremely resistant to Myr (54, 69). These same phenotypes are seen when a lem3Δ mutation is introduced into a pdr5Δ yor1 strain. This strongly suggests that the influence of Lem3 is not via modulation of Pdr5 function but rather through an influence on another protein.
Since both Lem3 and Pdr5/Yor1 are known to impact function of Tat2, we suggest that at least part of the phenotypes influenced by loss of these phospholipid asymmetry regulating proteins is through regulation of other membrane transporters. Tat2 serves as an illustration of this target protein, as we have demonstrated that the tryptophan phenotype is not important in AbA resistance (making Tat2 an unlikely participant in this phenotype). Based on work from two other labs, Myr appears to enter the cell via the action of flippase (54, 69). The role of Lem3-dependent flippase as the importer of Myr would explain virtually all the genetic effects we have seen for this inhibitor. Recently, Lem3 was shown to be required for Myr uptake by direct measurement of intracellular Myr levels (54). Finally, overproduction of Pdr5 lowered AbA resistance but strongly increased Myr tolerance.
From these data, we suggest the model shown in Fig. 7. If Tat2 is used as a representative membrane transporter, then Pdr5 and Yor1 are positive regulators of its internalization, and the Lem3-dependent flippase is a negative regulator as shown (68). A membrane transporter that could efflux AbA would be an ideal candidate for such a target of Pdr5/Yor1- and Lem3-dependent regulation. We are currently carrying out genetic screens searching for a relevant target protein.
FIG 7.
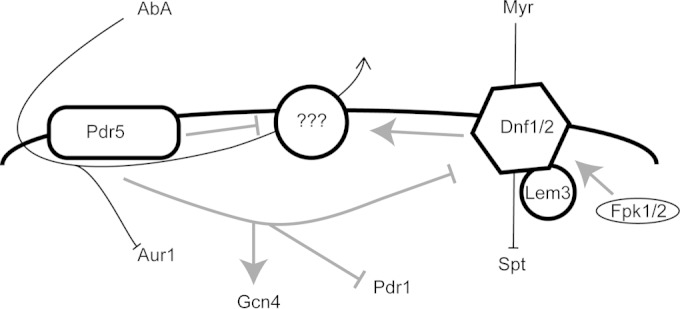
Interaction model for Pdr5 and Lem3-dependent flippase. A diagram of the plasma membrane with important proteins indicated is shown. Movement of drugs is represented by black lines, while the genetic interactions suggested by our data are shown as gray lines. Positive effects are indicated by arrows, while inhibitory effects are denoted as a T bar. The question marks indicate a currently unknown AbA exporter that is suggested by these data.
The conclusion that loss of Pdr5 and Yor1 inhibits uptake of AbA is based on the finding that pdr5Δ yor1 strains treated with this inhibitor do not exhibit the normal induction of sphingolipid biosynthesis that is seen in wild-type cells (33, 34). AbA is a cyclic peptide (70), and there is no known means to directly assay its uptake. A further argument against pdr5Δ yor1 cells exhibiting increased AbA resistance due to an increase in sphingolipid biosynthetic pathway function comes from earlier work using a different drug that also inhibits IPC synthase. This drug, called rustmicin, was found to have increased efficacy against cells lacking Pdr5, although both AbA and rustmicin act to inhibit IPC synthase activity (66). Coupled with our failure to detect any change in steady-state production of sphingolipids by either mass spectrometry (Fig. 4B) or pulse-labeling of cultures (data not shown), this can be most simply explained by a reduction in AbA entry to the cells. Furthermore, these findings also indicate that TORC2-dependent signaling is unlikely to be responsive to loss of Pdr5 and Yor1.
The epistatic relationship of Pdr5/Yor1 and flippase indicates that the ABC transporters are most likely to be acting upstream of the flippase proteins. The phenotypes seen upon deletion of PDR5 and YOR1 were reversed by loss of flippase activity via removal of LEM3. The increased Myr resistance seen when PDR5 was present in a high-copy-number plasmid can also be explained by inhibition of flippase activity (and Myr uptake) via Pdr5. The predominant role of the flippase in determining both resistance to AbA and Myr suggests that these phenotypes are routed through this critical modulator of phospholipid asymmetry.
These data provide a new illustration of the importance of ABC transporters in directly exporting drugs from cells but also in controlling the composition of membranes in which they are resident. The genetic interaction detected here between Pdr5/Yor1 and Lem3-dependent flippases suggests that these two opposing influences on phospholipid distribution across the plasma membrane are in communication. This link would help explain how cells ensure appropriate asymmetry and provide a clarifying discovery in terms of the multidrug resistance phenomenon. In the case of Pdr5 specifically, evidence has been provided that changes in expression of this single membrane protein can influence resistance to hundreds of different compounds (62). We suggest that a significant fraction of these resistance effects might emerge from the influence of Pdr5 on the content of phospholipids in the plasma membrane, which in turn can regulate the function of proteins localized there (such as Tat2). This notion of Pdr5 in particular and ABC transporters in general as direct drug exporters but also regulators of other membrane transporters could help explain why these proteins are able to influence such a broad range of resistance phenotypes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ted Powers, Amy Chang, Wenjun Guan, and Jeremy Thorner for generous sharing of antibodies, strains, and plasmids as well as important discussions. We thank Bernice Thommandru for technical assistance during the course of these experiments. Yeast sphingolipid analysis was performed at the Lipidomics Core Facility at the Medical University of South Carolina.
This work was supported in part by NIH grant GM49825 to W.S.M.-R. Analysis of yeast genomic sequences was made possible through the NIH P41 GM103533 grant funding the Yeast Resource Center. M.J.D. is a Rita Allen Scholar and Fellow in the Genetic Networks program at the Canadian Institute for Advanced Research. A.B.S. was supported by National Cancer Institute grant F30 CA165440 and National Institute on Aging grant T32 AG000057 and is an ARCS Scholar alumnus.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00021-15.
REFERENCES
- 1.Higgins CF. 1992. ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 2.Ueda K, Cardarelli C, Gottesman MM, Pastan I. 1987. Expression of a full-length cDNA for the human MDR1 gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci U S A 84:3004–3008. doi: 10.1073/pnas.84.9.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nigam SK. 2015. What do drug transporters really do? Nat Rev Drug Discov 14:29–44. doi: 10.1038/nrd4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Marques RL, Poulsen LR, Bailly A, Geisler M, Pomorski TG, Palmgren MG. 2014. Structure and mechanism of ATP-dependent phospholipid transporters. Biochim Biophys Acta doi: 10.1016/j.bbagen.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Decottignies A, Grant AM, Nichols JW, de Wet H, McIntosh DB, Goffeau A. 1998. ATPase and multidrug transport activities of the overexpressed yeast ABC protein Yor1p. J Biol Chem 273:12612–12622. doi: 10.1074/jbc.273.20.12612. [DOI] [PubMed] [Google Scholar]
- 6.Balzi E, Wang M, Leterme S, Van Dyck L, Goffeau A. 1994. PDR5: a novel yeast multidrug resistance transporter controlled by the transcription regulator PDR1. J Biol Chem 269:2206–2214. [PubMed] [Google Scholar]
- 7.Hirata D, Yano K, Miyahara K, Miyakawa T. 1994. Saccharomyces cerevisiae YDR1, which encodes a member of the ATP-binding cassette (ABC) superfamily, is required for multidrug resistance. Curr Genet 26:285–294. doi: 10.1007/BF00310491. [DOI] [PubMed] [Google Scholar]
- 8.Bissinger PH, Kuchler K. 1994. Molecular cloning and expression of the S. cerevisiae STS1 gene product. J Biol Chem 269:4180–4186. [PubMed] [Google Scholar]
- 9.Katzmann DJ, Hallstrom TC, Voet M, Wysock W, Golin J, Volckaert G, Moye-Rowley WS. 1995. Expression of an ATP-binding cassette transporter encoding gene (YOR1) is required for oligomycin resistance in Saccharomyces cerevisiae. Mol Cell Biol 15:6875–6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pomorski T, Lombardi R, Riezman H, Devaux PF, van Meer G, Holthuis JC. 2003. Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell 14:1240–1254. doi: 10.1091/mbc.E02-08-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kihara A, Igarashi Y. 2004. Cross talk between sphingolipids and glycerophospholipids in the establishment of plasma membrane asymmetry. Mol Biol Cell 15:4949–4959. doi: 10.1091/mbc.E04-06-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kihara A, Igarashi Y. 2002. Identification and characterization of a Saccharomyces cerevisiae gene, RSB1, involved in sphingoid long-chain base release. J Biol Chem 277:30048–30054. doi: 10.1074/jbc.M203385200. [DOI] [PubMed] [Google Scholar]
- 13.Paul S, Moye-Rowley WS. 2014. Multidrug resistance in fungi: regulation of transporter-encoding gene expression. Front Physiol 5:143. doi: 10.3389/fphys.2014.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson SS, Hanson PK, Manoharlal R, Brice SE, Cowart LA, Moye-Rowley WS. 2010. Regulation of yeast nutrient permease endocytosis by ATP-binding cassette transporters and a seven-transmembrane protein, RSB1. J Biol Chem 285:35792–35802. doi: 10.1074/jbc.M110.162883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe F, Iida H. 2003. Pressure-induced differential regulation of the two tryptophan permeases Tat1 and Tat2 by ubiquitin ligase Rsp5 and its binding proteins, Bul1 and Bul2. Mol Cell Biol 23:7566–7584. doi: 10.1128/MCB.23.21.7566-7584.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagiec MM, Nagiec EE, Baltisberger JA, Wells GB, Lester RL, Dickson RC. 1997. Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphoryolceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. J Biol Chem 272:9809–9817. [DOI] [PubMed] [Google Scholar]
- 17.Sato K, Noda Y, Yoda K. 2009. Kei1: a novel subunit of inositolphosphorylceramide synthase, essential for its enzyme activity and Golgi localization. Mol Biol Cell 20:4444–4457. doi: 10.1091/mbc.E09-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gietz RD, Schiestl RH. 1995. Transforming yeast with DNA. Methods Mol Cell Biol 5:255–269. [Google Scholar]
- 19.Alani E, Cao L, Kleckner N. 1987. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics 116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. [DOI] [PubMed] [Google Scholar]
- 21.Goldstein AL, McCusker JH. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15:1541–1553. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki Y, St Onge RP, Mani R, King OD, Heilbut A, Labunskyy VM, Chen W, Pham L, Zhang LV, Tong AH, Nislow C, Giaever G, Gladyshev VN, Vidal M, Schow P, Lehar J, Roth FP. 2011. Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat Methods 8:159–164. doi: 10.1038/nmeth.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niles BJ, Mogri H, Hill A, Vlahakis A, Powers T. 2012. Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by target of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc Natl Acad Sci U S A 109:1536–1541. doi: 10.1073/pnas.1117563109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones GM, Stalker J, Humphray S, West A, Cox T, Rogers J, Dunham I, Prelich G. 2008. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat Methods 5:239–241. doi: 10.1038/nmeth.1181. [DOI] [PubMed] [Google Scholar]
- 25.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. 2006. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Shahi P, Gulshan K, Naar AM, Moye-Rowley WS. 2010. Differential roles of transcriptional mediator subunits in regulation of multidrug resistance gene expression in Saccharomyces cerevisiae. Mol Biol Cell 21:2469–2482. doi: 10.1091/mbc.E09-10-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemoine S, Combes F, Servant N, Le Crom S. 2006. Goulphar: rapid access and expertise for standard two-color microarray normalization methods. BMC Bioinformatics 7:467. doi: 10.1186/1471-2105-7-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson MD, Grigull J, Mohammad N, Hughes TR. 2002. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinformatics 3:35. doi: 10.1186/1471-2105-3-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teixeira MC, Monteiro PT, Guerreiro JF, Goncalves JP, Mira NP, dos Santos SC, Cabrito TR, Palma M, Costa C, Francisco AP, Madeira SC, Oliveira AL, Freitas AT, Sa-Correia I. 2014. The YEASTRACT database: an upgraded information system for the analysis of gene and genomic transcription regulation in Saccharomyces cerevisiae. Nucleic Acids Res 42:D161–D166. doi: 10.1093/nar/gkt1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung N, Mao C, Heitman J, Hannun YA, Obeid LM. 2001. Phytosphingosine as a specific inhibitor of growth and nutrient import in Saccharomyces cerevisiae. J Biol Chem 276:35614–35621. doi: 10.1074/jbc.M105653200. [DOI] [PubMed] [Google Scholar]
- 31.Skrzypek MS, Nagiec MM, Lester RL, Dickson RC. 1998. Inhibition of amino acid transport by sphingoid long chain bases in Saccharomyces cerevisiae. J Biol Chem 273:2829–2824. doi: 10.1074/jbc.273.5.2829. [DOI] [PubMed] [Google Scholar]
- 32.Kolaczkowska A, Kolaczkowski M, Goffeau A, Moye-Rowley WS. 2008. Compensatory activation of the mutidrug transporters Pdr5p, Snq2p and Yor1p by Pdr1p in Saccharomyces cerevisiae. FEBS Lett 582:977–983. doi: 10.1016/j.febslet.2008.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, Walther TC, Loewith R. 2012. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol 14:542–547. doi: 10.1038/ncb2480. [DOI] [PubMed] [Google Scholar]
- 34.Gururaj C, Federman R, Chang A. 2013. Orm proteins integrate multiple signals to maintain sphingolipid homeostasis. J Biol Chem 288:20453–20463. doi: 10.1074/jbc.M113.472860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS. 2010. Orm family proteins mediate sphingolipid homeostasis. Nature 463:1048–1053. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roelants FM, Breslow DK, Muir A, Weissman JS, Thorner J. 2011. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 108:19222–19227. doi: 10.1073/pnas.1116948108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun Y, Miao Y, Yamane Y, Zhang C, Shokat KM, Takematsu H, Kozutsumi Y, Drubin DG. 2012. Orm protein phosphoregulation mediates transient sphingolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55-PP2A pathways. Mol Biol Cell 23:2388–2398. doi: 10.1091/mbc.E12-03-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roelants FM, Torrance PD, Thorner J. 2004. Differential roles of PDK1- and PDK2-phosphorylation sites in the yeast AGC kinases Ypk1, Pkc1 and Sch9. Microbiology 150:3289–3304. doi: 10.1099/mic.0.27286-0. [DOI] [PubMed] [Google Scholar]
- 39.Berninsone P, Miret JJ, Hirschberg CB. 1994. The Golgi guanosine diphosphatase is required for transport of GDP-mannose into the lumen of Saccharomyces cerevisiae Golgi vesicles. J Biol Chem 269:207–211. [PubMed] [Google Scholar]
- 40.Abeijon C, Yanagisawa K, Mandon EC, Hausler A, Moremen K, Hirschberg CB, Robbins PW. 1993. Guanosine diphosphatase is required for protein and sphingolipid glycosylation in the Golgi lumen of Saccharomyces cerevisiae. J Cell Biol 122:307–323. doi: 10.1083/jcb.122.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita M, Umemura M, Yoko-o T, Jigami Y. 2006. PER1 is required for GPI-phospholipase A2 activity and involved in lipid remodeling of GPI-anchored proteins. Mol Biol Cell 17:5253–5264. doi: 10.1091/mbc.E06-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kajiwara K, Watanabe R, Pichler H, Ihara K, Murakami S, Riezman H, Funato K. 2008. Yeast ARV1 is required for efficient delivery of an early GPI intermediate to the first mannosyltransferase during GPI assembly and controls lipid flow from the endoplasmic reticulum. Mol Biol Cell 19:2069–2082. doi: 10.1091/mbc.E07-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong KH, Struhl K. 2011. The Cyc8-Tup1 complex inhibits transcription primarily by masking the activation domain of the recruiting protein. Genes Dev 25:2525–2539. doi: 10.1101/gad.179275.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Proft M, Struhl K. 2002. Hog1 kinase converts the Sko1-Cyc8-Tup1 repressor complex into an activator that recruits SAGA and SWI/SNF in response to osmotic stress. Mol Cell 9:1307–1317. doi: 10.1016/S1097-2765(02)00557-9. [DOI] [PubMed] [Google Scholar]
- 45.Tzamarias D, Struhl K. 1995. Distinct TPR motifs of Cyc8 are involved in recruiting the Cyc8-Tup1 corepressor complex to differentially regulated promoters. Genes Dev 9:821–831. doi: 10.1101/gad.9.7.821. [DOI] [PubMed] [Google Scholar]
- 46.Gall WE, Higginbotham MA, Chen C, Ingram MF, Cyr DM, Graham TR. 2000. The auxilin-like phosphoprotein Swa2p is required for clathrin function in yeast. Curr Biol 10:1349–1358. doi: 10.1016/S0960-9822(00)00771-5. [DOI] [PubMed] [Google Scholar]
- 47.Du Y, Pypaert M, Novick P, Ferro-Novick S. 2001. Aux1p/Swa2p is required for cortical endoplasmic reticulum inheritance in Saccharomyces cerevisiae. Mol Biol Cell 12:2614–2628. doi: 10.1091/mbc.12.9.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desrivieres S, Cooke FT, Parker PJ, Hall MN. 1998. MSS4, a phosphatidylinositol-4-phosphate 5-kinase required for organization of the actin cytoskeleton in Saccharomyces cerevisiae. J Biol Chem 273:15787–15793. doi: 10.1074/jbc.273.25.15787. [DOI] [PubMed] [Google Scholar]
- 49.Homma K, Terui S, Minemura M, Qadota H, Anraku Y, Kanaho Y, Ohya Y. 1998. Phosphatidylinositol-4-phosphate 5-kinase localized on the plasma membrane is essential for yeast cell morphogenesis. J Biol Chem 273:15779–15786. doi: 10.1074/jbc.273.25.15779. [DOI] [PubMed] [Google Scholar]
- 50.Kato U, Emoto K, Fredriksson C, Nakamura H, Ohta A, Kobayashi T, Murakami-Murofushi K, Kobayashi T, Umeda M. 2002. A novel membrane protein, Ros3p, is required for phospholipid translocation across the plasma membrane in Saccharomyces cerevisiae. J Biol Chem 277:37855–37862. doi: 10.1074/jbc.M205564200. [DOI] [PubMed] [Google Scholar]
- 51.Hanson PK, Malone L, Birchmore JL, Nichols JW. 2003. Lem3p is essential for the uptake and potency of alkylphosphocholine drugs, edelfosine and miltefosine. J Biol Chem 278:36041–36050. doi: 10.1074/jbc.M305263200. [DOI] [PubMed] [Google Scholar]
- 52.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, et al. . 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 53.Saito K, Fujimura-Kamada K, Furuta N, Kato U, Umeda M, Tanaka K. 2004. Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol Biol Cell 15:3418–3432. doi: 10.1091/mbc.E03-11-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamane-Sando Y, Shimobayashi E, Shimobayashi M, Kozutsumi Y, Oka S, Takematsu H. 2014. Fpk1/2 kinases regulate cellular sphingoid long-chain base abundance and alter cellular resistance to LCB elevation or depletion. Microbiologyopen 3:196–212. doi: 10.1002/mbo3.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakano K, Yamamoto T, Kishimoto T, Noji T, Tanaka K. 2008. Protein kinases Fpk1p and Fpk2p are novel regulators of phospholipid asymmetry. Mol Biol Cell 19:1783–1797. doi: 10.1091/mbc.E07-07-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guan W, Jiang H, Guo X, Mancera E, Xu L, Li Y, Steinmetz L, Li Y, Gu Z. 2010. Antagonistic changes in sensitivity to antifungal drugs by mutations of an important ABC transporter gene in a fungal pathogen. PLoS One 5:e11309. doi: 10.1371/journal.pone.0011309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeRisi J, van den Hazel B, Marc P, Balzi E, Brown P, Jacq C, Goffeau A. 2000. Genome microarray analysis of transcriptional activation in multidrug resistance yeast mutants. FEBS Lett 470:156–160. doi: 10.1016/S0014-5793(00)01294-1. [DOI] [PubMed] [Google Scholar]
- 58.Devaux F, Carvajal E, Moye-Rowley S, Jacq C. 2002. Genome-wide studies on the nuclear PDR3-controlled response to mitochondrial dysfunction in yeast. FEBS Lett 515:25–28. doi: 10.1016/S0014-5793(02)02387-6. [DOI] [PubMed] [Google Scholar]
- 59.Fardeau V, Lelandais G, Oldfield A, Salin H, Lemoine S, Garcia M, Tanty V, Le Crom S, Jacq C, Devaux F. 2007. The central role of PDR1 in the foundation of yeast drug resistance. J Biol Chem 282:5063–5074. doi: 10.1074/jbc.M610197200. [DOI] [PubMed] [Google Scholar]
- 60.Banerjee D, Lelandais G, Shukla S, Mukhopadhyay G, Jacq C, Devaux F, Prasad R. 2008. Responses of pathogenic and nonpathogenic yeast species to steroids reveal the functioning and evolution of multidrug resistance transcriptional networks. Eukaryot Cell 7:68–77. doi: 10.1128/EC.00256-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mueller PP, Hinnebusch AG. 1986. Multiple upstream AUG codons mediate translational control of GCN4. Cell 45:201–207. doi: 10.1016/0092-8674(86)90384-3. [DOI] [PubMed] [Google Scholar]
- 62.Kolaczkowski M, van der Rest M, Cybularz-Kolaczkowski A, Soumillion J-P, Konings WN, Goffeau A. 1996. Anticancer drugs, ionophoric peptides and steroids as substrates of the yeast multidrug transporter Pdr5p. J Biol Chem 271:31543–31548. doi: 10.1074/jbc.271.49.31543. [DOI] [PubMed] [Google Scholar]
- 63.Hallstrom TC, Lambert L, Schorling S, Balzi E, Goffeau A, Moye-Rowley WS. 2001. Coordinate control of sphingolipid biosynthesis and multidrug resistance in Saccharomyces cerevisiae. J Biol Chem 276:23674–23680. doi: 10.1074/jbc.M101568200. [DOI] [PubMed] [Google Scholar]
- 64.Kolaczkowski M, Kolaczkowska A, Gaigg B, Schneiter R, Moye-Rowley WS. 2004. Differential regulation of ceramide synthase components LAC1 and LAG1 in Saccharomyces cerevisiae. Eukaryot Cell 3:880–892. doi: 10.1128/EC.3.4.880-892.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Panwar SL, Moye-Rowley WS. 2006. Long chain base tolerance in Saccharomyces cerevisiae is induced by retrograde signals from the mitochondria. J Biol Chem 281:6376–6384. doi: 10.1074/jbc.M512115200. [DOI] [PubMed] [Google Scholar]
- 66.Mandala SM, Thornton RA, Milligan J, Rosenbach M, Garcia-Calvo M, Bull HG, Harris G, Abruzzo GK, Flattery AM, Gill CJ, Bartizal K, Dreikorn S, Kurtz MB. 1998. Rustmicin, a potent antifungal agent, inhibits sphingolipid synthesis at inositol phosphoceramide synthase. J Biol Chem 273:14942–14949. doi: 10.1074/jbc.273.24.14942. [DOI] [PubMed] [Google Scholar]
- 67.Zhang F, Gaur NA, Hasek J, Kim SJ, Qiu H, Swanson MJ, Hinnebusch AG. 2008. Disrupting vesicular trafficking at the endosome attenuates transcriptional activation by Gcn4. Mol Cell Biol 28:6796–6818. doi: 10.1128/MCB.00800-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hachiro T, Yamamoto T, Nakano K, Tanaka K. 2013. Phospholipid flippases Lem3p-Dnf1p and Lem3p-Dnf2p are involved in the sorting of the tryptophan permease Tat2p in yeast. J Biol Chem 288:3594–3608. doi: 10.1074/jbc.M112.416263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roelants FM, Baltz AG, Trott AE, Fereres S, Thorner J. 2010. A protein kinase network regulates the function of aminophospholipid flippases. Proc Natl Acad Sci U S A 107:34–39. doi: 10.1073/pnas.0912497106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Endo M, Takesako K, Kato I, Yamaguchi H. 1997. Fungicidal action of aureobasidin A, a cyclic depsipeptide antifungal antibiotic, against Saccharomyces cerevisiae. Antimicrob Agents Chemother 41:672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katzmann DJ, Burnett PE, Golin J, Mahe Y, Moye-Rowley WS. 1994. Transcriptional control of the yeast PDR5 gene by the PDR3 gene product. Mol Cell Biol 14:4653–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu M, Huang C, Polu SR, Schneiter R, Chang A. 2012. Regulation of sphingolipid synthesis through Orm1 and Orm2 in yeast. J Cell Sci 125:2428–2435. doi: 10.1242/jcs.100578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leppert G, McDevitt R, Falco SC, Van Dyk TK, Ficke MB, Golin J. 1990. Cloning by gene amplification of two loci conferring multiple drug resistance in Saccharomyces. Genetics 125:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brice SE, Alford CW, Cowart LA. 2009. Modulation of sphingolipid metabolism by the phosphatidylinositol-4-phosphate phosphatase Sac1p through regulation of phosphatidylinositol in Saccharomyces cerevisiae. J Biol Chem 284:7588–7596. doi: 10.1074/jbc.M808325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, et al. . 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.