

Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used to treat a number of conditions, and proton pump inhibitors (PPIs) are often used to prevent NSAID-induced gastric mucosal damage; however, the effects of NSAIDs on intestinal motility are poorly understood. The purpose of the present study is to determine the effects of a prototypical NSAID, indomethacin, either alone or in conjunction with the PPI omeprazole, on intestinal motility. Rats were randomly divided into four groups treated with vehicle, omeprazole, indomethacin, or a combination of indomethacin and omeprazole. Intestinal motility and transit were measured along with inflammatory mediators in the intestinal smooth muscle, markers of mucosal damage, and bacterial counts in the intestinal wall. Indomethacin, but not omeprazole, caused mucosal injury indicated by lower gut bleeding; however, both omeprazole and indomethacin suppressed contractile activity and frequency in the distal part of the small intestine. Cotreatment with omeprazole did not reduce indomethacin-induced intestinal bleeding. Furthermore, although indomethacin caused increased inflammation as indicated by increased edema development and inflammatory mediators, cotreatment with omeprazole did not reduce inflammation in the intestinal smooth muscle or prevent the increased bacterial count in the intestinal wall induced by indomethacin. We conclude that both NSAID and PPI treatment suppressed contractile activity in the distal regions of the small intestine. The suppression of intestinal contractility was associated with increased inflammation in both cases; however, indomethacin and omeprazole appear to affect intestinal motility by different mechanisms.
Keywords: indomethacin, omeprazole, intestinal motility, inflammation
nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used for the treatment of a number of different conditions including rheumatoid arthritis, ankylosing spondylitis, osteoarthritis, and patent ductus arteriosus. The association of NSAID use with mucosal injury in the gastrointestinal (GI) tract is well established. NSAIDs have been shown to cause mucosal damage, not only in the stomach, but also in the small and large intestine (9, 17, 18, 21). In a recent study, lower GI damage accounted for 30–40% of all serious GI events in patients taking NSAIDs (17). However, side effects related to motility disorders are also common with NSAID use, including nausea, vomiting, abdominal pain, and constipation (13). The effects of NSAID use on gastrointestinal smooth muscle function are not well understood.
Several studies have shown that indomethacin induces increased motility in the lower gut (15, 24, 33). Nylander (24) showed that indomethacin treatment (3 mg/kg) increased motility in the duodenum of rats, measured as increased luminal pressure. Satoh et al. (33) showed that indomethacin (5 mg/kg) increased both the amplitude and frequency of contractions in the ileum when administered after feeding but not in fasted cats. Korolkiewicz et al. (15) showed that indomethacin (30 mg/kg) attenuated the inhibitory effects of gut manipulation on intestinal transit. The Nylander, Satoh, and Korolkeiwicz studies all show that indomethacin increases intestinal motility; however, in contrast to these studies, constipation has long been recognized as a common side effect of NSAIDs, suggesting that NSAIDs reduce intestinal motility (13, 37, 38). Furthermore, NSAID use was found to be a risk factor for chronic constipation (6). Constipation would suggest a decrease in intestinal motility, not an increase as shown in earlier studies; this discrepancy in experimental data vs. known side effects of indomethacin warrants further study.
Proton pump inhibitors (PPIs) are often used in conjunction with NSAIDs to reduce the incidence of NSAID-induced gastric mucosal damage. Wallace et al. (44) recently showed that PPIs can exacerbate NSAID-induced mucosal injury in the small intestine. However, the effects of PPIs on small intestinal motility are unknown. The effects of PPIs on small intestinal bacterial overgrowth (SIBO) have been somewhat controversial. Several studies reporting dysbiosis with PPI use (44, 46), including a meta-analysis; in contrast, other studies, including a large clinical study, showed no association of SIBO with PPI use (20, 29). Intestinal motility changes may influence whether SIBO develops in patients taking PPIs and NSAIDs. Thus the effects of NSAIDs and PPIs on motility in the small intestine are important. The purpose of the present study is to determine the effects of a prototypical NSAID, indomethacin, either alone or in conjunction with the PPI omeprazole, on intestinal motility.
MATERIALS AND METHODS
Animal model.
All procedures were approved by the University of Texas Medical School Institutional Animal Care and Use Committee and are consistent with the NIH “Guide for the Care and Use of Laboratory Animals.” Male Sprague-Dawley rats weighing between 250–300 g were used for all experiments. Rats were randomly divided into four groups. The Control group was vehicle treated only (saline). The PPI group was given omeprazole (37.5 mg/kg subcutaneously twice a day for 4 days). This dosage of omeprazole has been shown to suppress gastric acid secretion by ∼90% in rats (36, 44). The Indo group was given a single 20 mg/kg dose of indomethacin, subcutaneously, 24 h before euthanasia (on third day of omeprazole treatment). The PPI/Indo group was given a combination of omeprazole and indomethacin as described above.
Luminal hemoglobin content.
The intestinal luminal contents were collected by flushing 8 ml of cold 0.9% saline into the proximal duodenum and collecting the perfusate at the ileocecal valve. The flushed sample was kept at 4°C, vortexed for 2 min, and centrifuged (3 min at 2,000 rpm). Hemoglobin content was measured in the supernatants as described by Crosby and Furth (8). In a subset of rats in the indomethacin and control treatments only, the small intestine was divided into 10 equal sections and flushed separately, and the hemoglobin was measured in each intestinal segment.
Measurement of intestinal contractility.
Intestinal contractile activity, in the longitudinal axis, was measured as described in Refs. 7 and 25. Full-thickness intestinal strips (∼10 mm in length) from ileal or jejunal sections of the small intestine, two from each section, were mounted in 25-ml organ baths filled with Krebs solution (in mM: 103 NaCl, 4.7 KCl, 2.5 CaCl2, 25 NaHCO3, 1.1 NaH2PO4, 15 glucose). The solution was buffered with albumin to avoid edema formation during incubation in the tissue chamber and gassed with 5% CO2-95% O2. Isometric force was monitored by an external force displacement transducer (Experimetria, Budapest, Hungary) connected to a PowerLab (AD Instruments, Colorado Springs, CO). Each strip was stretched to 0.5-g tension and allowed to equilibrate for 30 min. After equilibration, 10 min of basal contractile activity data were recorded. After recording of contractile activity, length of each strip was measured and tissue was removed, dried, and weighed. Contractile activity parameters were all calculated over 5 min of recorded data. Total contractile activity was calculated as the area under the curve (integral from minimum). Amplitude was calculated as average cycle height. All force development was normalized to tissue cross-sectional area. All measurements were performed in duplicate on two separate intestinal strips and averaged.
Intestinal transit.
A Silastic catheter was introduced into the proximal duodenum of rats via vertical laparotomy incision for transit measurements. The catheter was tunneled through the musculature of the left abdominal wall and subcutaneous tissue and externalized behind the neck. The following day, a solution of 70-kDa nonabsorbable fluorescein isothiocyanate (FITC) dextran (150 μl) was injected into the duodenum via the catheter in awake animals. Forty-five minutes after administration of FITC dextran animals were euthanized. The entire small intestine was removed. The small intestine was divided into 10 equal segments and each segment was flushed with 3 ml of 10% mM Tris-buffer solution. FITC-dextran concentrations were measured in the flushed contents on a fluorescent plate reader (excitation 480, emission 520) in each sample and expressed as absorbance units. Geometric center was calculated based on a modified method published by Miller et al. (23) using the following equation: geometric center = ∑(fraction of FITC per segment × segment number).
Intestinal interstitial edema.
Wet-to-dry weight measurements were calculated as a measure of edema development. Full-thickness intestinal samples were weighed immediately after collection. After drying in a 65°C oven, samples were weighed again. Wet-to-dry weight ratio was calculated as [(wet weight) − (dry weight)]/dry weight.
NF-κB assay.
NF-κB activation was measured in the nuclear extracts by using a Transcription Factor Assay Kit (Active Motif, Carlsbad, CA) following manufacturer's directions. Briefly, nuclear extracts from each group were added to each well of a 96-well plate in which oligonucleotide containing the NF-κB consensus sequence was immobilized. After incubation with lysates, a NF-κB p65 antibody was added followed by incubation with a secondary antibody conjugated to the horseradish peroxidase (HRP) enzyme. A colorimetric HRP substrate was then added. The colorimetric reaction was stopped with oxalic acid. Wells were washed after each incubation period. The plate was read at 450-nm wavelength. A recombinant P65 standard was used to generate a standard curve. Specificity of the assay was confirmed by competition with wild-type or mutant NF-κB consensus site oligonucleotides. Each sample was assayed in duplicate and normalized to total nuclear protein.
Cytokine array.
A rat cytokine array was utilized to measure cytokine and chemokine levels in the rat intestinal smooth muscle (R&D Systems, Minneapolis, MN) following manufacturer's directions. In the cytokine array kit, capture antibodies for the following 29 cytokines and chemokines are immobilized on nitrocellulose membrane: cytokine-induced neutrophil chemoattractant (CINC)-1, CINC-2α/β, CINC-3, ciliary neurotrophic factor (CNTF), fractalkine, granulocyte macrophage colony-stimulating factor (GM-CSF), soluble intercellular adhesion molecule (sICAM)-1, interferon (IFN)-γ, interleukin (IL)-1α, IL-1β, IL-1 receptor agonist (ra), IL-2, IL-3, IL-4, IL-6, IL-10, IL-13, IL-17, interferon γ-induced protein (IP)-10, lipopolysaccharide-induced CXC chemokine (LIX), l-selectin, monokine induced by interferon-γ (MIG), macrophage inflammatory protein (MIP-1α), MIP-3α, regulated upon activation normal T-cell expressed (RANTES), thymus chemokine, tissue inhibitor of metalloproteinase (TIMP)-1, tumor necrosis factor (TNF)-α, and vascular endothelial growth factor (VEGF).
Cytoplasmic extracts from smooth muscle obtained from the distal small intestine were prepared. Cytoplasmic extracts (300 μg) were diluted to the required volume and incubated with biotinylated detection antibodies for 1 h at room temperature. After the membrane was blocked, the sample/detection antibody mixture was incubated on the membrane overnight at 4°C. The membrane was then washed, followed by incubation with streptavidin-HRP. After incubation with the chemiluminescent reagent, the membrane was exposed to X-ray film and developed. Spots were quantitated using ImageJ (34).
Bacterial counts.
Full-thickness small intestinal samples were collected in a sterile environment and flushed with sterile 0.9% saline. Intestinal tissue was chopped into 1-mm3 pieces, weighed, and placed in stomacher bags along with 3 ml sterile 0.9% saline for homogenization. Samples were blended for 2–3 min. After blending, serial dilutions were made up to 10−7 and 25 μl of each dilution was plated on brain-heart infusion agar for total aerobes. After incubation for 24 h at 37°C, bacterial counts (total aerobes) were performed. Bacterial counts were divided by total weight of tissue blended.
Statistics.
All data except for bacterial counts are shown as means ± standard error. Bacterial counts were logarithmically transformed and are shown as geometric means. Data were analyzed by one-way ANOVA.
RESULTS
Intestinal injury.
The hematocrit decreased significantly in the Indo and PPI/Indo groups compared with the Control group (Fig. 1A) (Indo, 0.31 ± 0.017 and PPI/Indo, 0.27 ± 0.022 vs. Control, 0.46 ± 0.0053), which is indicative of the development of anemia caused by NSAID-induced GI bleeding. PPI alone (0.45 ± 0.010) did not induce any significant change in hematocrit (PPI vs. Control, P = 0.59), and there were no significant differences in hematocrit between Indo and PPI/Indo groups (Indo vs. PPI/Indo, P = 0.17).
Fig. 1.
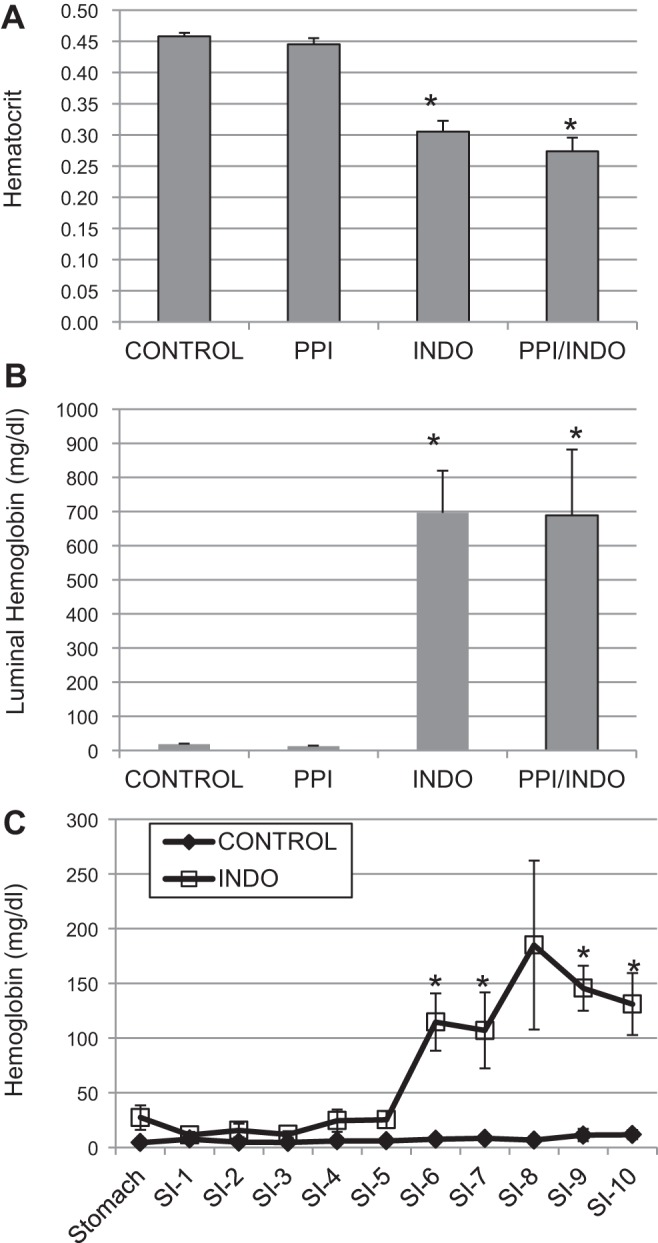
Hematocrits and luminal hemoglobin were measured as markers for mucosal injury in the small intestine. A: hematocrits measured on the day of euthanasia for Control, proton pump inhibitor (PPI), indomethacin (Indo), and PPI/Indo groups; n = 6 per group for Control and PPI groups, n = 7 for Indo and PPI/Indo groups, *P < 0.0001. B: total small intestinal lumen hemoglobin content in Control, PPI, Indo, and PPI/Indo groups. Means ± SE are shown; n = 12 per group for Control and Indo groups; n = 6 for PPI and PPI/Indo groups; *P < 0.001. C: luminal hemoglobin content in Control and Indo groups in the stomach (S) and along the length of the small intestine (SI-1 through SI-10). Means ± SE are shown; n = 4 per group; *P < 0.05.
As shown in Fig. 1B, hemoglobin in the small intestinal lumen increased significantly in both the Indo and PPI/Indo groups compared with the vehicle-treated Control group (Control, 18.08 ± 1.96 mg/dl; Indo, 696.50 ± 123.33 mg/dl; PPI/Indo, 688.74 ± 192.74 g/dl). Luminal hemoglobin was not altered compared with control values in the animals treated with omeprazole only (PPI, 12.24 ± 1.93 g/dl) (Control vs. PPI, P = 0.97). Luminal hemoglobin after a combination of omeprazole and indomethacin (PPI/Indo) treatment did not differ significantly from indomethacin treatment alone (Indo) (P = 0.96). Figure 1C shows luminal hemoglobin in the stomach and along the small intestine after indomethacin-only treatment (Indo) or vehicle treatment. Hemoglobin increased significantly in the lumen of the distal part of the length of the small intestine only (intestinal sections 6–10), suggesting that indomethacin induced mucosal injury predominantly in the distal jejunum and ileum.
Contractile activity.
Contractile activity was measured in proximal and distal sections of the small intestine. As shown in Fig. 2A, contractile activity was modestly but significantly decreased in the jejunal region of the small intestinal after omeprazole treatment (Control, 27.43 ± 2.70 g·s·cm−2 vs. PPI, 14.26 ± 2.74 g·s·cm−2) but not after indomethacin treatment or combination treatment with indomethacin and omeprazole (22.24 ± 3.31 and 19.02 ± 5.37, Indo and Indo/PPI, P = 0.32 and 0.13, respectively). Contractile activity in the distal (ileal) small intestine decreased significantly in all treatment groups compared with the Control group (Control, 49.20 ± 7.82 g·s·cm−2 vs. 15.54 ± 3.41, 32.14 ± 3.56, and 31.89 ± 2.91 g·s·cm−2 in PPI, Indo, and PPI/Indo groups, respectively) with changes in the PPI-treated group being most marked. In the jejunal region of the small intestine, the average contraction amplitude decreased significantly in the PPI group only compared with Control (Control, 0.12 ± 0.017 vs. PPI, 0.05 ± 0.008) as shown in Fig. 2B. In the distal small intestine, the average contraction amplitude decreased significantly in all three treatment groups compared with Control (Control, 0.27 ± 0.044 vs. 0.10 ± 0.025, 0.16 ± 0.054, and 0.16 ± 0.039, PPI, Indo and PPI/Indo groups, respectively). Changes in contraction frequency in the jejunal and ileal sections of the small intestine are shown in Fig. 2C. Frequency of contractions decreased significantly in the distal small intestine compared with the proximal small intestine as expected. Contraction frequency did not change significantly in the jejunum between the four treatment groups; however, frequency decreased significantly in all three treatment groups, PPI, Indo, and PPI/Indo, in the ileum compared with the Control group (Control, 0.40 ± 0.01 contractions/s vs. 0.33 ± 0.005, 0.36 ± 0.01, and 0.29 ± 0.01 contractions/s in PPI, Indo, and PPI/Indo groups, respectively).
Fig. 2.

Spontaneous contractile activity was measured in the proximal and distal small intestine in an isolated organ bath system. A: proximal and distal intestinal contractile activity, calculated as the integral from minimum, in Control, PPI, Indo, and PPI/Indo groups. B: average contraction amplitude in Control, PPI, Indo, and PPI/Indo groups. C: frequencies of spontaneous contractions in the proximal and distal sections of the small intestine are shown in Control, PPI, Indo, and PPI/Indo groups. Means ± SE are shown; n = 10 for Control and Indo groups; n = 6 for PPI and PPI/Indo groups; *P < 0.05; **P < 0.005.
Intestinal transit was measured in awake, unanesthetized animals. Figure 3A shows the average % dye in each intestinal segment (proximal to distal, 1–10). The Control and PPI groups exhibited a distinct dye peak corresponding to segments 6–7 in the middistal jejunum. In contrast, the Indo and PPI/Indo groups exhibited a broad, poorly defined dye peak. Quantification of the dye peak is shown in Fig. 3B. The percent dye at the peak was significantly lower in the Indo and PPI/Indo groups compared with the Control and PPI groups (Control and PPI, 49.70 ± 3.97 and 65.76 ± 8.59 vs. Indo and PPI/Indo, 23.87 ± 1.59 and 29.84 ± 1.70). As shown in Fig. 3C, transit was significantly slower (lower geometric center) in both the indomethacin-treated group (Indo) and the indomethacin and omeprazole-treated group (PPI/Indo) compared with the Control group (Control, 6.38 ± 0.43 vs. Indo, 4.9 ± 0.23 and PPI/Indo, 4.09 ± 0.42). Omeprazole treatment alone did not significantly alter intestinal transit (PPI, 6.36 ± 0.18, P = 0.97 vs. Control). Transit in the indomethacin-treated group (Indo) was not significantly different from the Indo/PPI group (P = 0.10).
Fig. 3.

Intestinal transit measured by propagation of nonabsorbable FITC-dextran dye through the small intestine is shown. A: average dye per intestinal segment for each treatment group. B: percent dye at the peak segment for each treatment group. C: calculated geometric center reflecting the speed of intestinal transit for each treatment group. Means ± SE are shown; n = 6 per group; *P < 0.05; **P < 0.005.
Inflammatory markers.
Wet-to-dry weight ratios, indicating interstitial edema development, were measured in whole thickness distal small intestine in each group, as shown in Fig. 4A. Wet-to-dry weight ratios increase significantly in all three drug treatment groups compared with Control in the distal small intestine (3.44 ± 0.06 in Control vs. 3.85 ± 0.11, 3.88 ± 0.10, and 3.76 ± 0.17 in PPI, Indo, and PPI/Indo groups, respectively). There were no significant differences in wet-to-dry weight ratios between the PPI, Indo, and PPI/Indo groups.
Fig. 4.
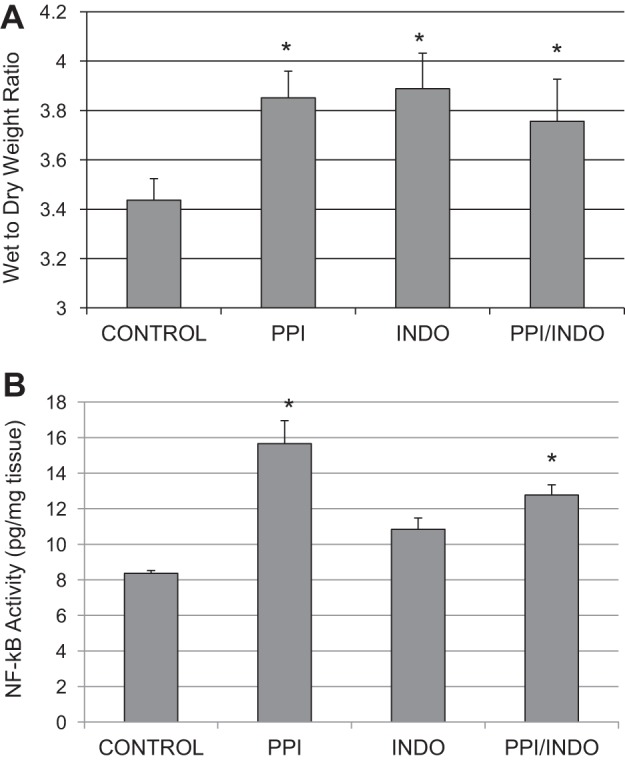
Interstitial edema development and activation of NF-κB in the intestinal smooth muscle were measured as indicators of inflammation in the distal small intestine. A: wet-to-dry weight ratios, indicating edema development, in the distal small intestine in Control, PPI, Indo, and PPI/Indo groups. Means ± SE are shown; n = 8 in Control and Indo groups; n = 4 in PPI and PPI/Indo groups; *P < 0.005. B: nuclear NF-κB DNA binding activity, indicating NF-κB activation, in smooth muscle of the distal small intestine from Control, PPI, Indo, and PPI/Indo groups. Means ± SE are shown; n = 9 in Control and Indo groups; n = 5 in PPI and PPI/Indo groups; *P < 0.05.
Nuclear NF-κB DNA binding activity was measured in distal intestinal smooth muscle as a marker of NF-κB activation as shown in Fig. 4B. NF-κB activation increased significantly in the PPI and PPI/Indo groups but not in the Indo group compared with the Control group (Control, 8.36 ± 0.81 pg/mg nuclear protein vs. 15.65 ± 3.65, 10.84 ± 0.83, and 12.77 ± 1.06 pg/mg nuclear protein in PPI, Indo, and PPI/Indo groups, respectively).
Inflammatory cytokines were measured by using an antibody-based microarray in distal intestinal smooth muscle. As shown in Fig. 5, A–C, inflammatory markers including IL-1α, IL-1β, and CINC-1 were significantly increased in the intestinal smooth muscle in the Indo and PPI/Indo groups compared with Control. However, these inflammatory markers were not increased significantly in the PPI group. TIMP-1 and LIX-1 also increased significantly in both the Indo and PPI/Indo groups but not the PPI group (data not shown). MIP-1 and TNF-α increased significantly in the PPI/Indo group only but not the other groups (data not shown). On the other hand, IFN-γ increased significantly in the PPI group compared with Control as shown in Fig. 5D. IFN-γ did not increase significantly in the Indo or Indo/PPI groups (P = 0.25 and 0.30 in Indo and Indo/PPI groups, respectively).
Fig. 5.

An antibody-based inflammatory mediator array was used to measure changes in inflammatory mediators in the distal small intestinal smooth muscle. Changes in IL-1α (A), IL-1β (B), CINC-1 (C), and IFN-γ (D) in intestinal smooth muscle from Control, PPI, Indo, and PPI/Indo groups are shown. Means ± SE are shown; n = 4, 5, 5, and 6 in Control, PPI, Indo, and PPI/Indo groups, respectively; *P < 0.05; **P < 0.001.
Bacterial growth.
Changes in total aerobic bacterial counts measured in whole-thickness intestinal tissue were measured in Control, PPI, Indo, and PPI/Indo groups as shown in Fig. 6. Total aerobes increased significantly in the indomethacin-treated groups (Indo and PPI/Indo) compared with both the Control and the PPI group. PPI treatment had no significant effect on total aerobic bacterial growth (P = 0.33, Control vs. PPI). Treatment with both omeprazole and indomethacin (PPI/Indo) did not significantly increase bacterial growth in the small intestine compared treatment with indomethacin alone (Indo).
Fig. 6.
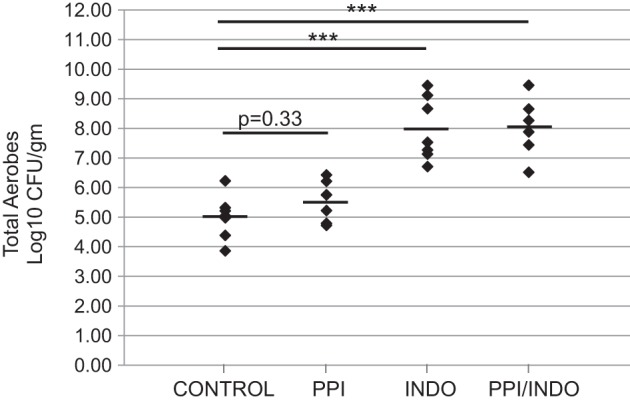
Number of total aerobes in the full-thickness small intestinal wall for Control, PPI, Indo, and PPI/Indo groups; n = 6–8 per group; mean is indicated for each group; ***P < 0.00001. Rel, relative.
DISCUSSION
Nonsteroidal anti-inflammatory drugs (NSAIDS) including indomethacin are used extensively for inflammatory conditions such as rheumatoid arthritis and osteoarthritis. Both experimental and clinical studies have shown that NSAID use causes mucosal injury in both the stomach and small intestine (2, 4, 11). Proton pump inhibitors such as omeprazole are often used in conjunction with NSAIDs to prevent gastroduodenal mucosal injury (30, 31). However, the ability of PPIs to attenuate NSAID-induced lower gut injury has been questioned in several recent studies (44). In particular, recent studies show that PPIs protect against gastric mucosal damage but do little to protect the small intestine, a major site for NSAID-induced injury (19, 22). Although the effects of NSAIDs and PPIs on intestinal mucosa have been widely studied, the effects of these drugs on intestinal smooth muscle are unclear. We show, in the present study, that indomethacin, but not omeprazole, causes mucosal injury leading to lower gut bleeding (Fig. 1); however, both omeprazole and indomethacin suppressed contractile activity and frequency in the distal part of the small intestine (Fig. 2). Cotreatment with omeprazole did not reduce indomethacin-induced intestinal bleeding (Fig. 1). Furthermore, cotreatment with omeprazole did not reduce inflammation in the intestinal smooth muscle or bacterial translocation induced by indomethacin (Figs. 4 and 5). In addition to PPIs, histamine H2 receptor antagonists, another class of antisecretory drugs, have also been shown to exacerbate NSAID-induced injury to the duodenum; however, the effects of these drugs on distal intestinal motility and injury should also be tested (5, 33).
The distribution of luminal hemoglobin (Fig. 1C) suggests that mucosal injury/bleeding was predominantly limited to the distal part of the small intestine. Although PPIs have been shown to protect the stomach from NSAID-induced damage (46), the protective effects of PPIs in the small intestine are unclear. Whereas several studies have shown that PPIs can protect the small intestine from NSAID-induced mucosal injury, other studies have shown that PPIs exacerbate or fail to protect against NSAID-induced mucosal injury in the small intestine (19, 44). For example, lansoprazole was shown to prevent indomethacin-induced intestinal ulceration (46). On the other hand, Wallace et al. (44) showed that omeprazole treatment significantly exacerbated NSAID-induced mucosal injury in the small intestine. We found no significant effect of PPI administration on indomethacin-induced small intestinal bleeding in our study. Differences in the studies may be a result of differences in the severity of injury due to treatment regime or type of NSAID used in the studies.
Spontaneous contractile activity in the proximal small intestine was unaffected by indomethacin treatment; however, omeprazole treatment inhibited contractile activity in the proximal small intestine alone or in combination with indomethacin (Fig. 2, A and C). Frequency of contractions were unaffected by either indomethacin or omeprazole treatment in the proximal part of the small intestine; however, the average contraction amplitude was suppressed by omeprazole treatment in the proximal small intestine (Fig. 2, B and C). The effects of PPIs on jejunal contractile activity may be related to PPI effects on gastric emptying (32). A large number of studies have shown that omeprazole delays gastric emptying (3, 26–28, 40). We speculate that delayed gastric emptying may lead to decreased volume in the proximal small intestine and therefore decreased contractile activity in the jejunum as shown in Fig. 2.
Contractile activity in the ileal region of the small intestine was decreased in both the omeprazole and indomethacin-treated groups. The combination of omeprazole and indomethacin did not significantly worsen contractile function compared with either treatment alone. Both frequency and average contraction amplitude were decreased in the treated groups compared with untreated and are likely responsible for the decreased contractile activity (Fig. 2). Several investigators showed that indomethacin increased motility in the small intestine (24, 39). These studies were performed within 2–3 h after administration of a lower dose of indomethacin. Furthermore, motility was measured via luminal pressure in the duodenum. Intestinal contractile activity in our study was measured 24 h after administration of indomethacin and differences in contractility were detected predominantly in the ileum. Thus discrepancies may reflect when and where contractile activity was measured. Furthermore, measurements in the organ bath system in our study may reflect smooth muscle dysfunction rather than neurally mediated changes in motility. Kurt et al. (16) showed that omeprazole decreased spontaneous intestinal contractile activity when added to the organ bath. This study supports our data showing the inhibitory effects of omeprazole on intestinal smooth muscle contractility.
In agreement with the contractile activity, indomethacin treatment, either alone or in combination with omeprazole, slowed intestinal transit compared with the untreated group (Fig. 3). The dye peak in the indomethacin-treated groups was broad and the percentage of dye at the peak in the Indo group was significantly lower compared with the Control group (Fig. 3, A and B), indicating a less organized peristaltic contraction. These data along with the contractile data (Fig. 2) suggest that both smooth muscle dysfunction and lack of coordination of peristaltic contractions occurred in the indomethacin-treated groups, contributing to the slowed intestinal transit. Interestingly, although contractile activity, measured in the organ bath, decreased in the omeprazole-treated group, intestinal transit was not significantly suppressed. Despite the decreased contraction amplitude, peristaltic contractions appeared to be well coordinated as demonstrated by the well-defined dye peak and the significantly higher percentage of dye at the peak in the omeprazole treatment group compared with the untreated group (Fig. 3, A and B). Intestinal transit was measured in fasted animals and duodenal pH has a significant effect on interdigestive migrating myoelectric complex (35, 45). Thus the pH in the duodenum may have stimulated strong peristaltic contractions in the intact animals despite the dysfunctional smooth muscle. In the indomethacin-treated groups, the dye peak was much more diffuse (Fig. 3A) and the percent dye at the peak was significantly lower than in untreated control animals (Fig. 3B).
Since most of the decreased contractile activity occurred in the distal part of the small intestine along with the mucosal injury as reflected by luminal hemoglobin, we measured inflammatory markers in the ileal smooth muscle. Intestinal edema developed after both omeprazole and indomethacin, indicating the development of inflammation (Fig. 4). These data indicate that smooth muscle dysfunction was likely due to inflammation in both omeprazole and indomethacin treatment; however, the type of inflammation appears to be different for omeprazole compared with indomethacin treatment as indicated by the differing pattern of inflammatory mediators (Fig. 5). IFN-γ increased in the omeprazole-treated group, but not in the indomethacin-treated groups. Since no increases in luminal hemoglobin was detected in the omeprazole treatment group, the mechanism by which IFN-γ increased after omeprazole is unknown at this time and warrants further investigation. NF-κB activation was also increased in the intestinal smooth muscle after omeprazole treatment in the absence of any measurable mucosal damage (indicated by absence of hematocrit and luminal hemoglobin changes). IFN-γ is likely responsible for the increased NF-κB activation in the intestinal smooth muscle of the omeprazole-treated group. NF-κB has been shown to suppress intestinal smooth muscle contractility by us and other investigators (10, 12, 43). For example, we showed that NF-κB activation inhibited intestinal contractile activity in a hydrostatic intestinal edema model (43). Increased NF-κB activation may be related to increased interferon-γ levels in the intestinal smooth muscle in the PPI group; however, the mechanism by which PPI increased interferon-γ is unclear. We did not detect increased bacterial growth in the omeprazole-treated group; however, there may have been changes in a particular group of bacteria due to PPI-induced pH changes in the stomach and small intestine that could have caused the NF-κB activation and increased interferon-γ levels. Wallace et al. (44) demonstrated that omeprazole induced intestinal dysbiosis.
Contractile changes in the indomethacin-treated groups (Indo and PPI/Indo) are likely due to mucosal damage leading to increased inflammation in the intestinal smooth muscle in this group. Inflammatory mediators including IL-1α, IL-1β, and CINC-1 were significantly increased in the indomethacin-treated groups due to indomethacin-induced mucosal injury. Inflammation has been reported to both increase and decrease intestinal motility; thus the effects of inflammation on motility are unclear and likely to depend on which inflammatory mediators are involved (1, 12, 14, 41, 42). IL-1β, in particular, is associated with suppression of both intestinal and colonic motility (12, 14, 41, 42). Murthy and coworkers (12) showed that IL-1β decreases colonic motility via downregulation of CPI-17, an endogenous inhibitor of myosin light chain phosphatase. Bauer and colleagues (42) showed that IL-1β increased and intestinal motility decreased in a peripheral traumatic injury model. Thus increased IL-1β likely inhibits intestinal motility; however, the effects of IL-1α and CINC-1 on intestinal motility are less clear. In general, the effects of inflammatory mediators on smooth muscle function are poorly understood and warrant further investigations.
Bacterial counts measured in the intestinal wall from both indomethacin-treated groups were significantly higher than the control group, indicating bacterial translocation in these groups (Fig. 6). Both intestinal contractility and intestinal transit were compromised in these groups; our data suggest that the mucosal damage combined with decreased intestinal transit caused the increased bacterial translocation in these groups, leading eventually to increased inflammation in the intestinal smooth muscle. The increased inflammation, in turn, likely contributed to the decreased intestinal motility and consequently the decreased intestinal transit through a feedback loop. Figure 7 shows the hypothetical model of intestinal smooth muscle injury induced by indomethacin.
Fig. 7.
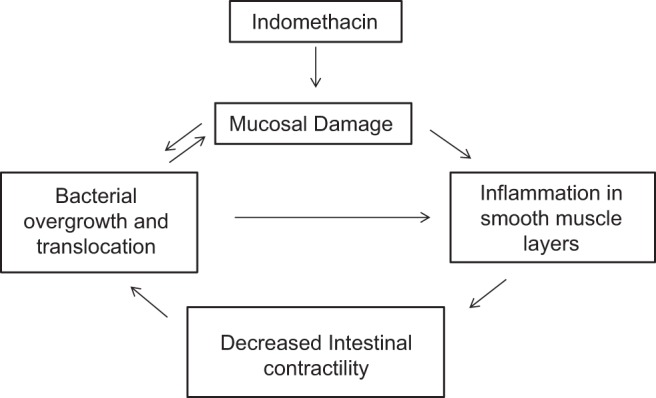
Model for indomethacin-induced inhibition of small intestinal smooth muscle contractility. Indomethacin causes mucosal damage, which initiates a feedback loop involving inflammatory damage to smooth muscle layers, which decreases intestinal smooth muscle contractility. The decreased intestinal contractility enhances the bacterial overgrowth and translocation initiated by the damaged mucosa.
One drawback of the study was that intestinal transit was measured in fasted animals, thus interdigestive contractile activity was measured. Decreased contractile activity in the organ bath occurred after both omeprazole and indomethacin treatment, indicating intestinal smooth muscle dysfunction in both treatments. Further study is needed to determine whether this smooth muscle dysfunction may have a more pronounced effect on intestinal transit of solid food compared with the liquid dye movement tracked in the present study.
In summary, both NSAID and PPI treatment suppressed contractile activity in the distal regions of the small intestine. The suppression of intestinal contractility was associated with increased inflammation in both cases; however, the mechanism appears to be different. Decreased contractility due to PPI appears to be due to increased IFN-γ and NF-κB activity whereas decreased motility after NSAID treatment is associated with increased IL-1β protein expression. In the long term, decreased intestinal motility may contribute to mucosal injury/bleeding and potentially bacterial overgrowth and translocation leading to increased septic complications. Our model of indomethacin-induced decrease in intestinal contractility is shown in Fig. 7. Omeprazole decreased intestinal contractile activity but did not appear to exacerbate and/or attenuate mucosal injury under our experimental conditions. The use of PPIs has been shown to have beneficial effects in the upper GI tract; however, the protective effects of PPIs on small intestinal injury and transit in the fed state require further scrutiny.
GRANTS
This study was funded in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK56338 (L. M. Lichtenberger, E. J. Dial, K. S. Uray).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
L.M.L., E.J.D., and K.S.U. conception and design of research; L.M.L., E.J.D., and K.S.U. interpreted results of experiments; L.M.L., D.B., T.M.P., E.J.D., and K.S.U. edited and revised manuscript; L.M.L., D.B., T.M.P., E.J.D., and K.S.U. approved final version of manuscript; D.B., T.M.P., and K.S.U. performed experiments; D.B., T.M.P., and K.S.U. analyzed data; K.S.U. prepared figures; K.S.U. drafted manuscript.
REFERENCES
- 1.Akiho H, Ihara E, Nakamura K. Low-grade inflammation plays a pivotal role in gastrointestinal dysfunction in irritable bowel syndrome. World J Gastrointest Pathophysiol 1: 97–105, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med 327: 749–754, 1992. [DOI] [PubMed] [Google Scholar]
- 3.Benini L, Castellani G, Bardelli E, Sembenini C, Brentegani MT, Caliari S, Vantini I. Omeprazole causes delay in gastric emptying of digestible meals. Dig Dis Sci 41: 469–474, 1996. [DOI] [PubMed] [Google Scholar]
- 4.Bjarnason I, Price AB, Zanelli G, Smethurst P, Burke M, Gumpel JM, Levi AJ. Clinicopathological features of nonsteroidal antiinflammatory drug-induced small intestinal strictures. Gastroenterology 94: 1070–1074, 1988. [DOI] [PubMed] [Google Scholar]
- 5.Blackler RW, Gemici B, Manko A, Wallace JL. NSAID-gastroenteropathy: new aspects of pathogenesis and prevention. Curr Opin Pharmacol 19: 11–16, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Chang JY, Locke GR, Schleck CD, Zinsmeister AR, Talley NJ. Risk factors for chronic constipation and a possible role of analgesics. Neurogastroenterol Motil 19: 905–911, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Chu J, Miller CT, Kislitsyna K, Laine GA, Stewart RH, Cox CS, Uray KS. Decreased myosin phosphatase target subunit 1(MYPT1) phosphorylation via attenuated rho kinase and zipper-interacting kinase activities in edematous intestinal smooth muscle. Neurogastroenterol Motil 24: 257–266, e109, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crosby WH, Furth FW. A modification of the benzidine method for measurement of hemoglobin in plasma and urine. Blood 11: 380–383, 1956. [PubMed] [Google Scholar]
- 9.Fortun PJ, Hawkey CJ. Nonsteroidal antiinflammatory drugs and the small intestine. Curr Opin Gastroenterol 23: 134–141, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Hernandez LV, Gonzalo S, Castro M, Arruebo MP, Plaza MA, Murillo MD, Grasa L. Nuclear factor kappaB is a key transcription factor in the duodenal contractility alterations induced by lipopolysaccharide. Exp Physiol 96: 1151–1162, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi K, Umegaki E, Watanabe T, Yoda Y, Morita E, Murano M, Tokioka S, Arakawa T. Present status and strategy of NSAIDs-induced small bowel injury. J Gastroenterol 44: 879–888, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Hu W, Mahavadi S, Li F, Murthy KS. Upregulation of RGS4 and downregulation of CPI-17 mediate inhibition of colonic muscle contraction by interleukin-1β. Am J Physiol Cell Physiol 293: C1991–C2000, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones RH, Tait CL. Gastrointestinal side-effects of NSAIDs in the community. Br J Clin Pract 49: 67–70, 1995. [PubMed] [Google Scholar]
- 14.Kinoshita K, Sato K, Hori M, Ozaki H, Karaki H. Decrease in activity of smooth muscle L-type Ca2+ channels and its reversal by NF-κB inhibitors in Crohn's colitis model. Am J Physiol Gastrointest Liver Physiol 285: G483–G493, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Korolkiewicz RP, Ujda M, Dabkowski J, Ruczynski J, Rekowski P, Petrusewicz J. Differential salutary effects of nonselective and selective COX-2 inhibitors in postoperative ileus in rats. J Surg Res 109: 161–169, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Kurt A, Altun A, Bagcivan I, Koyuncu A, Topcu O, Aydin C, Kaya T. Effects of proton pump inhibitors and H2 receptor antagonists on the ileum motility. Gastroenterol Res Pract 2011: 218342, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laine L, Connors LG, Reicin A, Hawkey CJ, Burgos-Vargas R, Schnitzer TJ, Yu Q, Bombardier C. Serious lower gastrointestinal clinical events with nonselective NSAID or coxib use. Gastroenterology 124: 288–292, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Laine L, Curtis SP, Langman M, Jensen DM, Cryer B, Kaur A, Cannon CP. Lower gastrointestinal events in a double-blind trial of the cyclo-oxygenase-2 selective inhibitor etoricoxib and the traditional nonsteroidal anti-inflammatory drug diclofenac. Gastroenterology 135: 1517–1525, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Lim YJ, Phan TM, Dial EJ, Graham DY, Lichtenberger LM. In vitro and in vivo protection against indomethacin-induced small intestinal injury by proton pump inhibitors, acid pump antagonists, or indomethacin-phosphatidylcholine. Digestion 86: 171–177, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo WK, Chan WW. Proton pump inhibitor use and the risk of small intestinal bacterial overgrowth: a meta-analysis. Clin Gastroenterol Hepatol 11: 483–490, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Maiden L, Thjodleifsson B, Theodors A, Gonzalez J, Bjarnason I. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology 128: 1172–1178, 2005. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy DM. GI bleeding: problems that persist. Gastrointest Endosc 70: 225–228, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Miller MS, Galligan JJ, Burks TF. Accurate measurement of intestinal transit in the rat. J Pharmacol Methods 6: 211–217, 1981. [DOI] [PubMed] [Google Scholar]
- 24.Nylander O. The impact of cyclooxygenase inhibition on duodenal motility and mucosal alkaline secretion in anaesthetized rats. Acta Physiol (Oxf) 201: 179–192, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Olsen AB, Hetz RA, Xue H, Aroom KR, Bhattarai D, Johnson E, Bedi S, Cox CS Jr, Uray K. Effects of traumatic brain injury on intestinal contractility. Neurogastroenterol Motil 25: 593–e463, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parkman HP, Urbain JL, Knight LC, Brown KL, Trate DM, Miller MA, Maurer AH, Fisher RS. Effect of gastric acid suppressants on human gastric motility. Gut 42: 243–250, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasmussen L, Oster-Jorgensen E, Qvist N, Kraglund K, Hovendal C, Pedersen SA. Short report: a double-blind placebo-controlled trial of omeprazole on characteristics of gastric emptying in healthy subjects. Aliment Pharmacol Ther 5: 85–89, 1991. [DOI] [PubMed] [Google Scholar]
- 28.Rasmussen L, Oster-Jorgensen E, Qvist N, Pedersen SA. The effects of omeprazole on intragastric pH, intestinal motility, and gastric emptying rate. Scand J Gastroenterol 34: 671–675, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Ratuapli SK, Ellington TG, O'Neill MT, Umar SB, Harris LA, Foxx-Orenstein AE, Burdick GE, Dibaise JK, Lacy BE, Crowell MD. Proton pump inhibitor therapy use does not predispose to small intestinal bacterial overgrowth. Am J Gastroenterol 107: 730–735, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Rostom A, Dube C, Wells G, Tugwell P, Welch V, Jolicoeur E, McGowan J. Prevention of NSAID-induced gastroduodenal ulcers. Cochrane Database Syst Rev 4: CD002296, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Rostom A, Moayyedi P, Hunt R; Canadian Association of Gastroenterology Consensus Group. Canadian consensus guidelines on long-term nonsteroidal anti-inflammatory drug therapy and the need for gastroprotection: benefits versus risks. Aliment Pharmacol Ther 29: 481–496, 2009. [DOI] [PubMed] [Google Scholar]
- 32.Sanaka M, Yamamoto T, Kuyama Y. Effects of proton pump inhibitors on gastric emptying: a systematic review. Dig Dis Sci 55: 2431–2440, 2010. [DOI] [PubMed] [Google Scholar]
- 33.Satoh H, Shiotani S, Otsuka N, Hatao K, Nishimura S. Role of dietary fibres, intestinal hypermotility and leukotrienes in the pathogenesis of NSAID-induced small intestinal ulcers in cats. Gut 58: 1590–1596, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schurizek BA, Kraglund K, Andreasen F, Jensen LV, Juhl B. Gastrointestinal motility and gastric pH and emptying following ingestion of diazepam. Br J Anaesth 61: 712–719, 1988. [DOI] [PubMed] [Google Scholar]
- 36.Shin JM, Sachs G. Restoration of acid secretion following treatment with proton pump inhibitors. Gastroenterology 123: 1588–1597, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Talley NJ, Fleming KC, Evans JM, O'Keefe EA, Weaver AL, Zinsmeister AR, Melton LJ 3rd. Constipation in an elderly community: a study of prevalence and potential risk factors. Am J Gastroenterol 91: 19–25, 1996. [PubMed] [Google Scholar]
- 38.Talley NJ, Weaver AL, Zinsmeister AR, Melton LJ 3rd. Functional constipation and outlet delay: a population-based study. Gastroenterology 105: 781–790, 1993. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka A, Matsumoto M, Hayashi Y, Takeuchi K. Functional mechanism underlying cyclooxygenase-2 expression in rat small intestine following administration of indomethacin: relation to intestinal hypermotility. J Gastroenterol Hepatol 20: 38–45, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Tougas G, Earnest DL, Chen Y, Vanderkoy C, Rojavin M. Omeprazole delays gastric emptying in healthy volunteers: an effect prevented by tegaserod. Aliment Pharmacol Ther 22: 59–65, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Tsuchida Y, Hatao F, Fujisawa M, Murata T, Kaminishi M, Seto Y, Hori M, Ozaki H. Neuronal stimulation with 5-hydroxytryptamine 4 receptor induces anti-inflammatory actions via alpha7nACh receptors on muscularis macrophages associated with postoperative ileus. Gut 60: 638–647, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsukamoto T, Antonic V, El Hajj II, Stojadinovic A, Binion DG, Izadjoo MJ, Yokota H, Pape HC, Bauer AJ. Novel model of peripheral tissue trauma-induced inflammation and gastrointestinal dysmotility. Neurogastroenterol Motil 23: 379–386, e164, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uray KS, Wright Z, Kislitsyna K, Xue H, Cox CS Jr. Nuclear factor-kappaB activation by edema inhibits intestinal contractile activity. Crit Care Med 38: 861–870, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Wallace JL, Syer S, Denou E, de Palma G, Vong L, McKnight W, Jury J, Bolla M, Bercik P, Collins SM, Verdu E, Ongini E. Proton pump inhibitors exacerbate NSAID-induced small intestinal injury by inducing dysbiosis. Gastroenterology 141: 1314–1322, 1322.e1–e5, 2011. [DOI] [PubMed] [Google Scholar]
- 45.Woodtli W, Owyang C. Duodenal pH governs interdigestive motility in humans. Am J Physiol Gastrointest Liver Physiol 268: G146–G152, 1995. [DOI] [PubMed] [Google Scholar]
- 46.Yoda Y, Amagase K, Kato S, Tokioka S, Murano M, Kakimoto K, Nishio H, Umegaki E, Takeuchi K, Higuchi K. Prevention by lansoprazole, a proton pump inhibitor, of indomethacin -induced small intestinal ulceration in rats through induction of heme oxygenase-1. J Physiol Pharmacol 61: 287–294, 2010. [PubMed] [Google Scholar]