

Abstract
Chronic pancreatitis is a progressive inflammatory disorder of the pancreas, which often develops as a result of genetic predisposition. Some of the most frequently identified risk factors affect the serine protease inhibitor Kazal type 1 (SPINK1) gene, which encodes a trypsin inhibitor responsible for protecting the pancreas from premature trypsinogen activation. Recent genetic and functional studies indicated that promoter variants in the SPINK1 gene might contribute to disease risk in carriers. Here, we investigated the functional effects of 17 SPINK1 promoter variants using luciferase reporter gene expression assay in four different cell lines, including three pancreatic acinar cell lines (rat AR42J with or without dexamethasone-induced differentiation and mouse 266-6) and human embryonic kidney 293T cells. We found that most variants caused relatively small changes in promoter activity. Surprisingly, however, we observed significant variations in the effects of the promoter variants in the different cell lines. Only four variants exhibited consistently reduced promoter activity in all acinar cell lines, confirming previous reports that variants c.-108G>T, c.-142T>C, and c.-147A>G are risk factors for chronic pancreatitis and identifying c.-52G>T as a novel risk variant. In contrast, variant c.-215G>A, which is linked with the disease-associated splice-site mutation c.194+2T>C, caused increased promoter activity, which may mitigate the overall effect of the pathogenic haplotype. Our study lends further support to the notion that sequence evaluation of the SPINK1 promoter region in patients with chronic pancreatitis is justified as part of the etiological investigation.
Keywords: trypsin inhibitor, chronic pancreatitis, luciferase reporter, acinar cell, promoter mutation
chronic pancreatitis is a progressive inflammatory disorder of the pancreas leading to acinar cell loss, fibrosis, and impaired pancreatic function. Clinically, disease manifestations involve recurrent acute attacks, which can progress to chronic disease with maldigestion, chronic pain, diabetes mellitus, and increased risk for pancreatic cancer (13, 40). Chronic pancreatitis frequently develops on the basis of genetic susceptibility. Mutations in human cationic trypsinogen (PRSS1), serine protease inhibitor Kazal type 1 (SPINK1), chymotrypsin C (CTRC), carboxypeptidase 1 (CPA1), and cystic fibrosis transmembrane conductance regulator (CFTR) can act as predisposing factors (reviewed in Refs. 11, 28, 29, 38). Chronic pancreatitis is a complex genetic disease, and patients may carry several mutations in various susceptibility genes.
The SPINK1 gene (OMIM 167790) encodes pancreatic secretory trypsin inhibitor, a ∼6-kDa protein secreted by the pancreatic acinar cells, which inhibits autoactivation of trypsinogen within the pancreas and thereby protects against pancreatitis (reviewed in Ref. 30). SPINK1 is also expressed in extrapancreatic tissues and various malignancies (reviewed in Refs. 15, 30). Mutation p.N34S in the SPINK1 gene was first described in 2000, and its association with chronic pancreatitis was demonstrated in the same year (10, 34, 39). A large number of subsequent studies confirmed that p.N34S is a relatively strong and frequent (odds ratio ≥10; occurrence ≥10%) risk factor for familial, idiopathic, and tropical chronic pancreatitis; even in alcoholic chronic pancreatitis it plays a lesser but still significant role (Ref. 1 and references therein, also reviewed in Refs. 28 and 30). Even though p.N34S was presumed to cause a loss of function, the exact mechanism of action has never been identified, as no functional defect was demonstrated for p.N34S or any of the four intronic variants associated with this haplotype (5, 7, 19, 20, 24, 26). In contrast, an unambiguous loss-of-function phenotype was evident for variant c.194+2T>C (28, 35, 39, and references therein), the second most frequent SPINK1 mutation that affects a splice site in intron-3 and causes exon skipping with markedly reduced SPINK1 mRNA expression (19, 23). This intronic mutation is in complete linkage disequilibrium (i.e., found together) with the promoter variant c.-215G>A. Studies worldwide identified a large number of rare or private missense SPINK1 variants, which caused loss of secretion of the SPINK1 protein either attributable to misfolding or impaired function of the secretory signal peptide (5, 6, 20, 21). Other rare genetic changes resulting in loss of function were also found in patients with chronic pancreatitis, such as frame-shift mutations, splice-site mutations, deletion of the SPINK1 gene, or mutation of the translation initiation codon (see references in Ref. 4).
Variants of unknown significance were also detected in the promoter region of SPINK1 by multiple studies. In 2011, Boulling et al. (4) performed resequencing of the proximal promoter region in French, German, and Indian patients and controls, together with functional analysis of 11 promoter variants. With the use of a luciferase reporter gene expression assay followed by EMSA analysis, they found that variants c.-53C>T, c.-142T>C, and c.-147A>G caused reduced promoter activity, whereas variants c.-81C>T and c.-215G>A increased activity in COLO-357 cells, a human cell line derived from the metastasis of a pancreatic adenocarcinoma. The authors proposed that variants that decrease promoter activity are likely risk factors for chronic pancreatitis, whereas gain-of-function variants may have a protective effect. More recently, the Hungarian Pancreatic Study Group published promoter-sequencing data for their chronic pancreatitis cohort and performed limited functional analysis on newly identified variants using dexamethasone-differentiated AR42J rat acinar cells (14). Statistically significant decreases in promoter activity were reported for variants c.-14G>A, c.-108G>T, and c.-246A>G, whereas variant c.-215G>A showed twofold increased activity.
The present study was undertaken to confirm and extend previous observations in an attempt to clarify which SPINK1 promoter variants might be true risk factors for chronic pancreatitis. Because reporter gene analysis inherently suffers from methodological uncertainties related to selection of cell line and the arbitrary nature of the promoter segment studied, we sought to investigate the reproducibility of published data in multiple cell lines. Furthermore, compared with the 11 variants examined by Boulling et al. (4), we studied a total of 17 variants including 6 variants published recently.
MATERIALS AND METHODS
Nomenclature.
Nucleotide numbering reflects coding DNA numbering with c.1 corresponding to the first nucleotide of the translation initiation codon. Promoter variants were numbered relative to the first nucleotide upstream of the initiation codon, which was designated c.–1. The genomic reference sequence used for SPINK1 was from NC_000005.9, the Homo sapiens chromosome 5, GRCh37.p13 primary assembly.
Construction of luciferase reporter plasmids harboring the SPINK1 promoter.
The DNA sequence between c.–541 and c.35 of the SPINK1 gene was cloned into the pGL3-Basic vector (Promega, Madison, WI) upstream of the firefly luciferase reporter gene using restriction sites KpnI and HindIII. This plasmid was designated pGL3-SPINK1. Promoter variants were generated by overlap extension PCR mutagenesis and cloned into the pGL3-SPINK1 plasmid. Note that the same construct was used recently by Hegyi et al. (14), whereas Boulling et al. (4) used a promoter sequence corresponding to the region from c.–1171 to c.–1 of the SPINK1 gene.
Cell culture and transfection.
AR42J rat pancreatic acinar cells were obtained from ATCC (cat. CRL-1492; Manassas, VA) and used with or without dexamethasone-induced differentiation. The mouse 266-6 pancreatic acinar cell line was purchased from ATCC (cat. CRL-2151) and human embryonic kidney (HEK) 293T cells were from GenHunter (cat. Q401; Nashville, TN). Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies, Grand Island, NY) supplemented with 4 mM glutamine, 1% penicillin/streptomycin, and fetal bovine serum (10% for HEK 293T and 266-6, 20% for AR42J, purchased from Life Technologies). Before transfections, cells were seeded into six-well plates at a density of 1.5 million cells per well (HEK 293T and 266-6) or 1 million cells per well (AR42J) and incubated overnight. For dexamethasone-differentiated AR42J cells, treatment was performed with 100 nM dexamethasone for 48 h before transfection. Transfection medium was prepared by mixing 10 μl Lipofectamine 2000 (Life Technologies) with 2 μg pGL3-SPINK1 plasmid and 10–40 ng pRL-SV40 Renilla luciferase plasmid (Promega) in 0.5 ml OPTI-MEM medium (Life Technologies) and incubating the mixture for 20 min at 22°C. Transfection was carried out by substituting 0.5 ml from the 2 ml of DMEM medium covering the cells with the transfection mix. After incubation overnight (18 h), the transfection medium was removed, and cells were rinsed, covered with 2 ml OPTI-MEM medium, and incubated for 30 h before luciferase expression was assayed. For dexamethasone-differentiated AR42J cells, the OPTI-MEM medium contained 100 nM dexamethasone.
Dual-luciferase reporter gene assay.
At 48 h after transfection, cells were rinsed with PBS, overlaid with 500 μl passive lysis buffer (Promega), and scraped from the culture plates. The cell suspension was then subjected to a freeze-thaw cycle in liquid nitrogen and incubated for 15 min at 22°C. Cell debris was pelleted by centrifugation (10,000 revolution/min, 30 s), and the supernatant was collected for luciferase activity assay. Luciferase expression was measured using the Dual-Glo Luciferase Assay System (Promega). Aliquots of cell extracts (20 μl) were mixed with 100 μl Luciferase Assay Reagent II, and the luminescence was measured with a Veritas luminometer (Turner Biosystems, Sunnyvale, CA). After recording the firefly luciferase activity, Renilla luciferase was measured by adding 100 μl Stop and Glo Reagent. Relative luciferase activity was determined by dividing the firefly and Renilla luciferase luminescence results and expressing this firefly/Renilla ratio as a percentage of the wild-type pGL3-SPINK1 value.
Statistical analysis.
We used Student's t-test to evaluate the statistical significance of the differences in activity between promoter variants and the wild-type SPINK1 promoter. A P value <0.05 was considered significant.
RESULTS
SPINK1 promoter variants studied.
As shown in Table 1 and Fig. 1, in our study, we included SPINK1 variants that were found upstream of the translation initiator codon and were reported in a peer-reviewed publication. For simplicity, we refer to all variants collectively as promoter variants throughout the paper, even though some are located in the 5′ untranslated region of the mRNA. Variants listed in databases but not described in the scientific literature were excluded. All in all, 17 variants were studied; including all 11 variants investigated by the 2011 study by Boulling et al. (4) and the three new variants recently characterized by the Hungarian Pancreatic Study group (14).
Table 1.
SPINK1 promoter variants studied
| Variant | CP Carriers | Control Carriers | References |
|---|---|---|---|
| c.-2C>A | 1 | 0 | 2 |
| c.-7T>G | 5 | 8 | 3 |
| c.-14G>A | 1 | 0 | 14 |
| c.-22C>T | 1 | 4 | 4, 10, 18, 34 |
| c.-41G>A | 10 | 3 | 2, 10, 12, 18, 34 |
| c.-52G>T | 1 | 0 | 35 |
| c.-53C>T | 2 | 1 | 4, 35, 39 |
| c.-81C>T | 1 | 0 | 18 |
| c.-108G>T | 1 | 0 | 14 |
| c.-142T>C | 5 | 0 | 4, 18 |
| c.-147A>G | 7 | 1 | 4, 18 |
| c.-164G>C | 5 | 14 | 3, 4 |
| c.-170G>A | 1 | 0 | 4 |
| c.-215G>A | 45 (11 hm) | 4 | 3, 4, 14, 16, 17, 22, 23, 26, 27, 32, 35, 36 |
| c.-215G>T | 3 | 0 | 2, 4 |
| c.-246A>G | 1 | 0 | 14 |
| c.-253T>C | common variant | common variant | 3, 8–10, 12, 14, 17, 22, 33, 37, 39 |
Note that variant c.-41G>A is in linkage with variant c.174C>T (p.C58=) in exon 3, and variant c.-215G>A is linked with pathogenic variant c.194+2T>C in intron 3. The 45 reported carriers for variant c.-215G>A included 11 homozygous (hm) subjects. SPINK1, serine protease inhibitor Kazal type 1; CP, chronic pancreatitis.
Fig. 1.
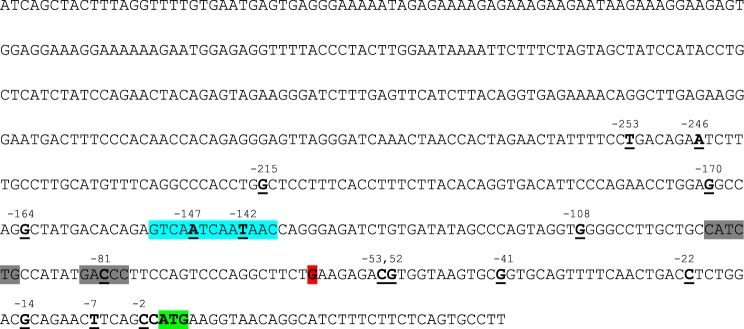
The serine protease inhibitor Kazal type 1 (SPINK1) promoter sequence used in this study as cloned into the pGL3-Basic plasmid. The sequence corresponds to the region from c.–541 to c.35 of the human SPINK1 gene. The positions of the promoter variants are underlined and emboldened. The original SPINK1 translation initiation codon is highlighted in green. Note that this codon is not in frame with the downstream luciferase start codon. The major transcriptional start site at c.–60 is shown in red. Binding sites for transcription factors hepatocyte nuclear factor 1 and pancreas-specific transcription factor 1 are indicated in blue and gray, respectively.
Assessment of SPINK1 promoter activity using luciferase reporter gene expression.
The aim of our study was to evaluate whether the routinely used reporter gene expression methods are able to predict the functional and clinical effects of SPINK1 promoter variants. To this end, we constructed an expression plasmid harboring the sequence region between c.–541 and c.35 of the SPINK1 gene, placed upstream of the firefly luciferase reporter gene (pGL3-SPINK1). This expression plasmid was used to transfect various cell lines, and promoter activity was then estimated by measuring luciferase expression levels using a luminescence assay. The study by Boulling et al. (4) used the COLO-357 human pancreatic cancer cell line, which expresses pancreatic digestive enzymes. The authors observed 11-fold increased expression of luciferase in this cell line driven by the SPINK1 promoter relative to the promoterless pGL3-Basic construct, whereas no increase was detected in HEK 293 cells (4). In the present study, we used four different cell lines, the rat acinar cell line AR42J with or without dexamethasone-induced differentiation, the mouse acinar cell line 266-6, and the HEK cell line 293T. These cell lines are well characterized, widely used, and readily available from commercial sources. AR42J and 266-6 cells express digestive enzymes, and we confirmed the presence of trypsinogen and chymotrypsinogen in the conditioned medium (data not shown). Dexamethasone treatment induces upregulation of the secretory pathway in AR42J cells with expansion of the endoplasmic reticulum, appearance of zymogen granules, and increased digestive enzyme secretion (25). In contrast, HEK 293T cells do not secrete pancreatic enzymes. When the four cell lines were transfected with pGL3-SPINK1 or the promoterless pGL3-Basic plasmid and luciferase expression was compared, AR42J acinar cells grown in the absence or presence of dexamethasone expressed about 13-fold and 17-fold higher luciferase levels, respectively, driven by the SPINK1 promoter. Luciferase expression in 266-6 cells and in HEK 293T cells was increased about fourfold over control (Fig. 2).
Fig. 2.
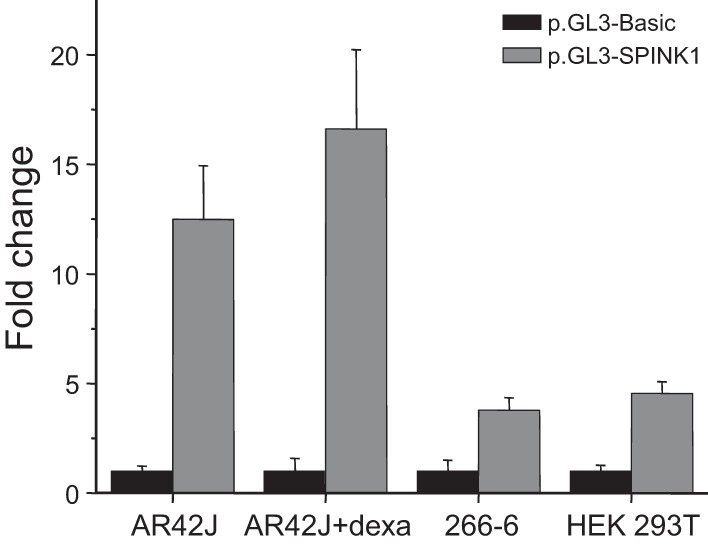
Luciferase reporter gene expression by the SPINK1 promoter. The indicated cell lines were transfected with the pGL3-SPINK1 plasmid or the promoterless pGL3-Basic plasmid, and luciferase expression was determined as described in materials and methods. The ratios of the luciferase activities (SPINK1 over control) are indicated as fold change with pGL3-Basic values arbitrarily set at unity. Means ± SD (n = 10–45 transfections) are shown. Note that measured firefly luciferase activities varied among cell lines; typical values with pGL3-Basic transfections were in the 10,000–50,000 relative light units range for AR42J, AR42J + dexamethasone (Dexa), and 266-6 cells, whereas about 2–7 million relative light units were detected with HEK 293T cells.
Effect of SPINK1 promoter variants on luciferase expression.
Results for the functional analysis of SPINK1 promoter variants in four different cell lines are shown in Table 2 and Fig. 3. Consistent with the observations by Boulling et al. (4), most variants caused minor, less than twofold changes in promoter activity, and even the largest effect was less than sixfold. Surprisingly, significant variations were detected in the effect of the promoter variants among the four cell lines tested, and variability remained apparent even among the three acinar cell lines or between AR42J cells grown in the absence or presence of dexamethasone. When the data obtained in the acinar cells were considered as relevant to the pancreas, the variants fell in four groups: 1) four variants (c.-52G>T, c.-108G>T, c.-142T>C, and c.-147A>G) exhibited some degree of loss of function in all three acinar cell lines, suggesting that these variants are risk factors for chronic pancreatitis; 2) one variant (c.-215G>A) showed a consistent gain of function in all acinar cell lines, indicating a possible protective effect against pancreatitis; 3) three variants (c.-164G>C, c.-170G>A, and c.-253T>C) had a neutral phenotype; and 4) nine variants caused variable, cell line-dependent changes in promoter activity.
Table 2.
Effect of SPINK1 promoter variants on luciferase expression
| AR42J | AR42J + Dexa | 266-6 | HEK 293T | COLO-357 | |
|---|---|---|---|---|---|
| Wild type | 100 | 100 | 100 | 100 | 100 |
| c.-2C>A | 128 | 54 | 126 | 115 | ND |
| c.-7T>G | 151 | 118 | 88 | 76 | 97 |
| c.-14G>A | 147 | 80 | 118 | 108 | ND |
| c.-22C>T | 107 | 46 | 133 | 127 | 103 |
| c.-41G>A | 240 | 57 | 104 | 109 | 98 |
| c.-52G>T | 85 | 20 | 68 | 81 | ND |
| c.-53C>T | 145 | 31 | 72 | 64 | 75 |
| c.-81C>T | 177 | 45 | 85 | 104 | 130 |
| c.-108G>T | 72 | 31 | 74 | 136 | ND |
| c.-142T>C | 25 | 13 | 66 | 213 | 67 |
| c.-147A>G | 17 | 11 | 35 | 77 | 56 |
| c.-164G>C | 119 | 95 | 127 | 224 | 101 |
| c.-170G>A | 122 | 118 | 106 | 87 | 93 |
| c.-215G>A | 136 | 201 | 162 | 561 | 123 |
| c.-215G>T | 94 | 50 | 114 | 256 | 104 |
| c.-246A>G | 109 | 53 | 119 | 96 | ND |
| c.-253T>C | 122 | 92 | 107 | 110 | ND |
Relative luciferase activities were expressed as percentage of the wild-type value. Average values from 3–6 transfections are shown; experimental variability (SD) is indicated in Figure 3. Data for the COLO-357 cell line were taken from Boulling et al. (4). Dexa, dexamethasone; ND, not determined.
Fig. 3.

Effect of SPINK1 promoter variants on luciferase expression. The indicated cell lines were transfected with SPINK1 promoter constructs, and luciferase expression was determined as described in materials and methods. Relative luciferase activities were expressed as percentages of the wild-type value. Means ± SD (n = 3–6 transfections) are shown. Data for 4 variants in the dexamethasone-differentiated AR42J cells were taken from Hegyi et al. (14). *P < 0.05, using Student's t-test. Dexa, dexamethasone.
In the nonacinar HEK 293T cell line, none of the variants caused a substantial (i.e., more than 50%) loss of promoter function. In contrast, three variants (c.-142T>C, c.-164G>C, and c.-215G>T) exhibited more than twofold increased activity, whereas variant c.-215G>A elicited a more than fivefold increase in luciferase expression.
DISCUSSION
In the present study, we surveyed all published proximal promoter variants in the SPINK1 gene for possible functional defects using a luciferase reporter assay. Because SPINK1 serves as an essential defense mechanism against intrapancreatic trypsinogen activation, changes in SPINK1 expression are expected to alter risk for chronic pancreatitis. Identification of variants causing loss or gain of promoter function, therefore, is important for their clinical classification as potentially pathogenic or protective. Many of these variants are rare, and genetic analysis alone cannot conclusively determine disease association. Previously, Boulling et al. (4) investigated 11 variants using the human COLO-357 cell line. The authors found relatively small changes in promoter activity (see Table 2) and designated variants c.-53C>T, c.-142T>C, and c.-147A>G as likely pathogenic (loss of function) and variants c.-81C>T and c.-215G>A as protective (gain of function). More recently, limited characterization using dexamethasone-differentiated AR42J cells was described for five variants; reduced promoter activity was reported for variants c.-14G>A, c.-108G>T, and c.-246A>G and increased activity for variant c.-215G>A (14). Here we extended these studies and investigated a total of 17 variants and utilized four different cell lines.
In agreement with the earlier study by Boulling et al. (4), we found that SPINK1 promoter variants typically have relatively small effects on promoter activity. Unexpectedly, however, we observed significant cell line-dependent variability in the effects of these variants, which makes interpretation difficult. Therefore, we took a conservative approach and designated only those variants functionally altered that consistently showed the same or similar phenotype in all three acinar cell lines tested. Four loss-of-function variants were identified, c.-52G>T, c.-108G>T, c.-142T>C, and c.-147A>G; two of these (c.-142T>C and c.-147A>G) were also studied by Boulling et al. (4) and described as functionally impaired. Variants c.-52G>T and c.-108G>T are private mutations identified in a German and a Hungarian patient with chronic pancreatitis, respectively (Table 1). In published reports, variants c.-142T>C and c.-147A>G were detected more frequently in patients (5 and 7 occurrences, respectively), and only variant c.-147A>G was found in a single control subject (Table 1). Taken together, the available genetic information for these four loss-of-function variants is not conclusive but consistent with a pathogenic phenotype. From a mechanistic aspect, variants c.-142T>C and c.-147A>G map to a positive regulatory region (see Fig. 1) of the SPINK1 promoter and seem to alter binding of the transcription factor hepatocyte nuclear factor 1 (4). Variant c.-52G>T maps in the vicinity of the transcription start site in a region that was found protected in a DNAse I foot-printing analysis, and it may affect binding of RNAse polymerase II (41). Variant c.-108G>T maps upstream of a bipartite binding site for pancreas-specific transcription factor 1, and it may alter binding of this transcription factor (4). Finally, our data did not confirm unambiguously the previously published finding that variant c.-53C>T caused a loss of function and was consequently pathogenic (4). Genetic studies are also inconclusive, as the variant was described in two patients and a healthy control (Table 1). However, given the proximity of this variant to the transcription start site (see Fig. 1) and the loss of function detected in two of the acinar cell lines tested, we conclude that variant c.-53C>T remains a potential candidate for a risk factor, but additional studies are required to clarify its significance in chronic pancreatitis. We were also unable to confirm the loss-of-function phenotype for variants c.-14G>A and c.-246A>G described recently by Hegyi et al. (14), as reduced promoter activity was only detected in dexamethasone-differentiated AR42J cells but not in the other cell lines tested.
With respect to gain-of-function variants, only variant c.-215G>A exhibited consistently elevated activity in all cell lines tested. The same phenotype was reported for this variant by Boulling et al. (4) and Hegyi et al. (14). Although the higher promoter activity suggests a protective function, this variant is always found in complete linkage disequilibrium with the intron 3 splice-site mutation c.194+2T>C, which causes exon skipping, markedly reduced SPINK1 expression, and strongly increased risk for chronic pancreatitis (19, 23, 28). It is intriguing to find that the negative effect of this splice-site mutation is mitigated by a promoter mutation that increases expression. One can speculate that, in the absence of this promoter mutation, the splice mutation may not be compatible with life, at least not in the homozygous state as seen with SPINK3 knockout mice (31). It is noteworthy that variant c.-215G>T, which affects the same nucleotide as c.-215G>A, exhibited variable promoter activity in our acinar cell lines and was described as neutral by Boulling et al. (4). This variant was found in three subjects of Indian origin with chronic pancreatitis, who also carried the p.N34S variant (Table 1), which likely was responsible for their disease risk (9). Finally, variant c.-81C>T yielded conflicting results in the different acinar cell lines, and thus we were unable to replicate the gain-of-function phenotype described by Boulling et al. (4). Similarly, variable results were obtained for a handful of other variants (c.-2C>A, c.-7T>G, c.-14G>A, c.-22C>T, c.-41G>A, and c.-246A>G); hence their pathological significance remains indeterminate pending further studies.
Data obtained in the HEK 293T cell line are unlikely to be pertinent to the pancreas and chronic pancreatitis but may be relevant to extrapancreatic tissues including tumors, where SPINK1 expression seems to contribute to pathology. Interestingly, none of the variants exhibited significantly impaired activity (all were ≥64% of wild-type), whereas four variants exhibited more than twofold increased activity. As discussed above, despite its impressive 5.6-fold increased activity, variant c.-215G>A is unlikely to cause elevated SPINK1 expression because of its linkage with c.194+2T>C. On the other hand, variants c.-142T>C, c.-164G>C, and c.-215G>T may act as risk factors for certain cancers.
In summary, we studied the functional effect of 17 SPINK1 promoter variants and identified four loss-of-function variants as likely predisposing factors for chronic pancreatitis and a gain-of-function variant, which modifies the deleterious effect of a linked splice-site mutation. Our data extend and refine prior studies by Boulling et al. (4) and Hegyi et al. (14) and confirm the notion that sequence evaluation of the SPINK1 promoter region in patients with chronic pancreatitis is warranted as part of an etiological workup.
GRANTS
This work was supported by NIH Grants R01 DK058088, R01 DK082412, and R01 DK095753 (to M. Sahin-Tóth) and by a scholarship from the Rosztoczy Foundation (to E. Kereszturi).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.H.D., A.G., E.K., and M.S.-T. conception and design of research; M.H.D., A.G., and E.K. performed experiments; M.H.D., A.G., and E.K. analyzed data; M.H.D., A.G., E.K., and M.S.-T. interpreted results of experiments; M.H.D., A.G., and M.S.-T. prepared figures; M.H.D., A.G., and M.S.-T. drafted manuscript; M.H.D., A.G., and M.S.-T. edited and revised manuscript; M.H.D., A.G., E.K., and M.S.-T. approved final version of manuscript.
ACKNOWLEDGMENTS
M. Derikx thankfully acknowledges the support of Prof. Joost P. H. Drenth (Department of Gastroenterology and Hepatology, Radboud UMC, Nijmegen, The Netherlands).
REFERENCES
- 1.Aoun E, Chang CC, Greer JB, Papachristou GI, Barmada MM, Whitcomb DC. Pathways to injury in chronic pancreatitis: decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS One 3: e2003, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Audrézet MP, Chen JM, Le Maréchal C, Ruszniewski P, Robaszkiewicz M, Raguénès O, Quéré I, Scotet V, Férec C. Determination of the relative contribution of three genes—the cystic fibrosis transmembrane conductance regulator gene, the cationic trypsinogen gene, and the pancreatic secretory trypsin inhibitor gene—to the etiology of idiopathic chronic pancreatitis. Eur J Hum Genet 10: 100–106, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bernardino AL, Guarita DR, Mott CB, Pedroso MR, Machado MC, Laudanna AA, Tani CM, Almeida FL, Zatz M. CFTR, PRSS1 and SPINK1 mutations in the development of pancreatitis in Brazilian patients. JOP 4: 169–177, 2003. [PubMed] [Google Scholar]
- 4.Boulling A, Witt H, Chandak GR, Masson E, Paliwal S, Bhaskar S, Reddy DN, Cooper DN, Chen JM, Férec C. Assessing the pathological relevance of SPINK1 promoter variants. Eur J Hum Genet 19: 1066–1073, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boulling A, Le Maréchal C, Trouvé P, Raguénès O, Chen JM, Férec C. Functional analysis of pancreatitis-associated missense mutations in the pancreatic secretory trypsin inhibitor (SPINK1) gene. Eur J Hum Genet 15: 936–942, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Boulling A, Keiles S, Masson E, Chen JM, Férec C. Functional analysis of eight missense mutations in the SPINK1 gene. Pancreas 41: 329–330, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Boulling A, Chen JM, Callebaut I, Férec C. Is the SPINK1 p.Asn34Ser missense mutation per se the true culprit within its associated haplotype? WebmedCentral Genetics 3: WMC003084, 2012. [Google Scholar]
- 8.Chandak GR, Idris MM, Reddy DN, Mani KR, Bhaskar S, Rao GV, Singh L. Absence of PRSS1 mutations and association of SPINK1 trypsin inhibitor mutations in hereditary and non-hereditary chronic pancreatitis. Gut 53: 723–728, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandak GR, Idris MM, Reddy DN, Bhaskar S, Sriram PV, Singh L. Mutations in the pancreatic secretory trypsin inhibitor gene (PSTI/SPINK1) rather than the cationic trypsinogen gene (PRSS1) are significantly associated with tropical calcific pancreatitis. J Med Genet 39: 347–351, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JM, Mercier B, Audrezet MP, Ferec C. Mutational analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene in hereditary and sporadic chronic pancreatitis. J Med Genet 37: 67–69, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JM, Férec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet 10: 63–87, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Lira M, Bonamini D, Castellani C, Unis L, Cavallini G, Assael BM, Pignatti PF. Mutations in the SPINK1 gene in idiopathic pancreatitis Italian patients. Eur J Hum Genet 11: 543–546, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Gupte AR, Forsmark CE. Chronic pancreatitis. Curr Opin Gastroenterol 30: 500–505, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Hegyi E, Geisz A, Sahin-Tóth M, Derikx M, Németh BC, Balázs A, Hritz I, Izbéki F, Halász A, Párniczky A, Takács T, Kelemen D, Sarlós P, Hegyi P, Czakó L. SPINK1 promoter variants in chronic pancreatitis. Pancreas. In press 2015. [DOI] [PubMed] [Google Scholar]
- 15.Itkonen O, Stenman UH. TATI as a biomarker. Clin Chim Acta 431: 260–269, 2014. [DOI] [PubMed] [Google Scholar]
- 16.Kalinin VN, Kaifi JT, Schwarzenbach H, Sergeyev AS, Link BC, Bogoevski D, Vashist Y, Izbicki JR, Yekebas EF. Association of rare SPINK1 gene mutation with another base substitution in chronic pancreatitis patients. World J Gastroenterol 12: 5352–5356, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaneko K, Nagasaki Y, Furukawa T, Mizutamari H, Sato A, Masamune A, Shimosegawa T, Horii A. Analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene mutations in Japanese patients with chronic pancreatitis. J Hum Genet 46: 293–297, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Keiles S, Kammesheidt A. Identification of CFTR, PRSS1, and SPINK1 mutations in 381 patients with pancreatitis. Pancreas 33: 221–227, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Kereszturi E, Király O, Sahin-Tóth M. Minigene analysis of intronic variants in common SPINK1 haplotypes associated with chronic pancreatitis. Gut 58: 545–549, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Király O, Wartmann T, Sahin-Tóth M. Missense mutations in pancreatic secretory trypsin inhibitor (SPINK1) cause intracellular retention and degradation. Gut 56: 1433–1438, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Király O, Boulling A, Witt H, Le Maréchal C, Chen JM, Rosendahl J, Battaggia C, Wartmann T, Sahin-Tóth M, Férec C. Signal peptide variants that impair secretion of pancreatic secretory trypsin inhibitor (SPINK1) cause autosomal dominant hereditary pancreatitis. Hum Mutat 28: 469–476, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kume K, Masamune A, Mizutamari H, Kaneko K, Kikuta K, Satoh M, Satoh K, Kimura K, Suzuki N, Nagasaki Y, Horii A, Shimosegawa T. Mutations in the serine protease inhibitor Kazal Type 1 (SPINK1) gene in Japanese patients with pancreatitis. Pancreatology 5: 354–360, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Kume K, Masamune A, Kikuta K, Shimosegawa T. (-215G>A; IVS3+2T>C) mutation in the SPINK1 gene causes exon 3 skipping and loss of the trypsin binding site. Gut 55: 1214, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwata K, Hirota M, Shimizu H, Nakae M, Nishihara S, Takimoto A, Mitsushima K, Kikuchi N, Endo K, Inoue M, Ogawa M. Functional analysis of recombinant pancreatic secretory trypsin inhibitor protein with amino-acid substitution. J Gastroenterol 37: 928–934, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Logsdon CD, Moessner J, Williams JA, Goldfine ID. Glucocorticoids increase amylase mRNA levels, secretory organelles, and secretion in pancreatic acinar AR42J cells. J Cell Biol 100: 1200–1208, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masamune A, Kume K, Takagi Y, Kikuta K, Satoh K, Satoh A, Shimosegawa T. N34S mutation in the SPINK1 gene is not associated with alternative splicing. Pancreas 34: 423–428, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Masamune A, Kume K, Shimosegawa T. Differential roles of the SPINK1 gene mutations in alcoholic and nonalcoholic chronic pancreatitis. J Gastroenterol 42, Suppl 17: 135–140, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Masamune A. Genetics of pancreatitis: the 2014 update. Tohoku J Exp Med 232: 69–77, 2014. [DOI] [PubMed] [Google Scholar]
- 29.Németh BC, Sahin-Tóth M. Human cationic trypsinogen (PRSS1) variants and chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol 306: G466–G473, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohmuraya M, Yamamura K. Roles of serine protease inhibitor Kazal type 1 (SPINK1) in pancreatic diseases. Exp Anim 60: 433–444, 2011. [DOI] [PubMed] [Google Scholar]
- 31.Ohmuraya M, Hirota M, Araki M, Mizushima N, Matsui M, Mizumoto T, Haruna K, Kume S, Takeya M, Ogawa M, Araki K, Yamamura K. Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology 129: 696–705, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Ota Y, Masamune A, Inui K, Kume K, Shimosegawa T, Kikuyama M. Phenotypic variability of the homozygous IVS3+2T>C mutation in the serine protease inhibitor Kazal type 1 (SPINK1) gene in patients with chronic pancreatitis. Tohoku J Exp Med 221: 197–201, 2010. [DOI] [PubMed] [Google Scholar]
- 33.Patuzzo C, Castellani C, Sagramoso C, Gomez-Lira M, Bonamini D, Belpinati F, Dechecchi MC, Assael BM, Pignatti PF. Cationic trypsinogen and pancreatic secretory trypsin inhibitor gene mutations in neonatal hypertrypsinaemia. Eur J Hum Genet 11: 93–96, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Pfützer RH, Barmada MM, Brunskill AP, Finch R, Hart PS, Neoptolemos J, Furey WF, Whitcomb DC. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 119: 615–623, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Rosendahl J, Landt O, Bernadova J, Kovacs P, Teich N, Bödeker H, Keim V, Ruffert C, Mössner J, Kage A, Stumvoll M, Groneberg D, Krüger R, Luck W, Treiber M, Becker M, Witt H. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut 62: 582–592, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Shimosegawa T, Kume K, Masamune A. SPINK1 gene mutations and pancreatitis in Japan. J Gastroenterol Hepatol 21, Suppl 3: S47–S51, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Tzetis M, Kaliakatsos M, Fotoulaki M, Papatheodorou A, Doudounakis S, Tsezou A, Makrythanasis P, Kanavakis E, Nousia-Arvanitakis S. Contribution of the CFTR gene, the pancreatic secretory trypsin inhibitor gene (SPINK1) and the cationic trypsinogen gene (PRSS1) to the etiology of recurrent pancreatitis. Clin Genet 71: 451–457, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Whitcomb DC. Genetic aspects of pancreatitis. Annu Rev Med 61: 413–24, 2010. [DOI] [PubMed] [Google Scholar]
- 39.Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet 25: 213–216, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 132: 1557–1573, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Yasuda T, Yasuda T, Ohmachi Y, Katsuki M, Yokoyama M, Murata A, Monden M, Matsubara K. Identification of novel pancreas-specific regulatory sequences in the promoter region of human pancreatic secretory trypsin inhibitor gene. J Biol Chem 273: 34413–34421, 1998. [DOI] [PubMed] [Google Scholar]