

Abstract
Proper control of Ca2+ signaling is mandatory for effective cell migration, which is critical for embryonic development, wound healing, and cancer metastasis. However, how Ca2+ coordinates structural components and signaling molecules for proper cell motility had remained elusive. With the advance of fluorescent live-cell Ca2+ imaging in recent years, we gradually understand how Ca2+ is regulated spatially and temporally in migrating cells, driving polarization, protrusion, retraction, and adhesion at the right place and right time. Here we give an overview about how cells create local Ca2+ pulses near the leading edge, maintain cytosolic Ca2+ gradient from back to front, and restore Ca2+ depletion for persistent cell motility. Differential roles of Ca2+ in regulating various effectors and the interaction of roles of Ca2+ signaling with other pathways during migration are also discussed. Such information might suggest a new direction to control cancer metastasis by manipulating Ca2+ and its associating signaling molecules in a judicious manner.
1. Introduction
Calcium is one of the most important chemical elements for human beings. At the organismic level, calcium together with other materials composes bone to support our bodies [1]. At the tissue level, the compartmentalization of calcium ions (Ca2+) regulates membrane potentials for proper neuronal [2] and cardiac [3] activities. At the cellular level, increases in Ca2+ trigger a wide variety of physiological processes, including proliferation, death, and migration [4]. Aberrant Ca2+ signaling is therefore not surprising to induce a broad spectrum of diseases in metabolism [1], neuron degeneration [5], immunity [6], and malignancy [7]. However, though tremendous efforts have been exerted, we still do not fully understand how this tiny divalent cation controls our lives.
Such a puzzling situation also exists when we consider Ca2+ signaling in cell migration. As an essential cellular process, cell migration is critical for proper physiological activities, such as embryonic development [8], angiogenesis [9], and immune response [10], and pathological conditions, including immunodeficiency [11], wound healing [12], and cancer metastasis [13]. In either situation, coordination between multiple structural (such as F-actin and focal adhesion) and regulatory (such as Rac1 and Cdc42) components is required for cell migration processes (or modules), including polarization, protrusion, retraction, and adhesion [8]. Since Ca2+ signaling is meticulously controlled temporally and spatially in both local and global manners, it serves as a perfect candidate to regulate cell migration modules. However, although the significant contribution of Ca2+ to cell motility has been well recognized [14], it had remained elusive how Ca2+ was linked to the machinery of cell migration. The advances of live-cell fluorescent imaging for Ca2+ and cell migration in recent years gradually unravel the mystery, but there is still a long way to go.
In the present paper, we will give a brief overview about how Ca2+ signaling is polarized and regulated in migrating cells, its local actions on the cytoskeleton, and its global effect on cell migration and cancer metastasis. The strategies employing Ca2+ signaling to control cell migration and cancer metastasis will also be discussed.
2. History: The Journey to Visualize Ca2+ in Live Moving Cells
The attempt to unravel the roles of Ca2+ in cell migration can be traced back to the late 20th century, when fluorescent probes were invented [15] to monitor intracellular Ca2+ in live cells [16]. Using migrating eosinophils loaded with Ca2+ sensor Fura-2, Brundage et al. revealed that the cytosolic Ca2+ level was lower in the front than the back of the migrating cells. Furthermore, the decrease of regional Ca2+ levels could be used as a marker to predict the cell front before the eosinophil moved [17]. Such a Ca2+ gradient in migrating cells was also confirmed by other research groups [18], though its physiological significance had not been totally understood.
In the meantime, the importance of local Ca2+ signals in migrating cells was also noticed. The use of small molecule inhibitors and Ca2+ channel activators suggested that local Ca2+ in the back of migrating cells regulated retraction and adhesion [19]. Similar approaches were also recruited to indirectly demonstrate the Ca2+ influx in the cell front as the polarity determinant of migrating macrophages [14]. Unfortunately, direct visualization of local Ca2+ signals was not available in those reports due to the limited capabilities of imaging and Ca2+ indicators in early days.
The above problems were gradually resolved in recent years with the advance of technology. First, the utilization of high-sensitive camera for live-cell imaging [20] reduced the power requirement for the light source, which eliminated phototoxicity and improved cell health. A camera with high sensitivity also improved the detection of weak fluorescent signals, which is essential to identify Ca2+ pulses of nanomolar scales [21]. In addition to the camera, the emergence of genetic-encoded Ca2+ indicators (GECIs) [22, 23], which are fluorescent proteins engineered to show differential signals based on their Ca2+-binding statuses, revolutionized Ca2+ imaging. Compared to small molecule Ca2+ indicators, GECIs' high molecular weights make them less diffusible, enabling the capture of transient local signals. Furthermore, signal peptides could be attached to GECIs so the recombinant proteins could be located to different compartments, facilitating Ca2+ measurements in different organelles. Such tools dramatically improved our knowledge regarding the dynamic and compartmentalized characteristics of Ca2+ signaling.
With the above techniques, “Ca2+ flickers” were observed in the front of migrating cells [18], and their roles in cell motility were directly investigated [24]. Moreover, with the integration of multidisciplinary approaches including fluorescent microscopy, systems biology, and bioinformatics, the spatial role of Ca2+, including the Ca2+ gradient in migrating cells, was also gradually clarified [25]. Our present understanding about Ca2+ signaling in migrating cells is briefly summarized as follows.
3. Ca2+ Transporters Regulating Cell Migration
3.1. Generators of Local Ca2+ Pulses: Inositol Triphosphate (IP3) Receptors and Transient Receptor Potential (TRP) Channels (Figure 1)
Figure 1.

Local Ca2+ pulses control retraction and adhesion around the leading edge of migrating cells. (a) Polarized receptor tyrosine kinase (RTK) signaling generates inositol triphosphate (IP3) in front of migrating cells, which sensitizes IP3 receptors (IP3R) to release Ca2+ periodically from the endoplasmic reticulum (ER). IP3R are also triggered by Ca2+-induced Ca2+ release (CICR), which originates from transient receptor potential (TRP) channels, mainly TRPM7. (b) Local Ca2+ pulses activate myosin light chain kinase (MLCK, shown as ML in the illustration), which phosphorylates myosin II for proper actin treadmilling and recycling. (c) Local Ca2+ pulse-triggered myosin contraction also enhances the formation of focal adhesion (FA) complexes, probably via force-induced positive feedback. Please notice the temporal correlation (as shown by dotted lines and arrowheads) and oscillatory dynamics between local Ca2+ pulses, front retraction, and FA. MY: myosin II; ML: myosin light chain kinase; I: integrin; ECM: extracellular matrix.
For a polarized cell to move efficiently, its front has to coordinate activities of protrusion, retraction, and adhesion [8]. The forward movement starts with protrusion, which requires actin polymerization in lamellipodia and filopodia, the foremost structure of a migrating cell [8, 13, 26]. At the end of protrusion, the cell front slightly retracts and adheres [27] to the extracellular matrix. Those actions occur in lamella, the structure located behind lamellipodia. Lamella recruits myosin to contract and dissemble F-actin in a treadmill-like manner and to form nascent focal adhesion complexes in a dynamic manner [28]. After a successful adhesion, another cycle of protrusion begins with actin polymerization from the newly established cell-matrix adhesion complexes. Such protrusion-slight retraction-adhesion cycles are repeated so the cell front would move in a caterpillar-like manner.
For the above actions to proceed and persist, the structural components, actin and myosin, are regulated in a cyclic manner. For actin regulation, activities of small GTPases, Rac, RhoA, and Cdc42 [29], and protein kinase A [30] are oscillatory in the cell front for efficient protrusion. For myosin regulation, small local Ca2+ signals are also pulsatile in the junction of lamellipodia and lamella [24]. Those pulse signals regulate the activities of myosin light chain kinase (MLCK) and myosin II, which are responsible for efficient retraction and adhesion [31, 32]. Importantly, due to the extremely high affinity between Ca2+-calmodulin complexes and MLCK [33], small local Ca2+ pulses in nanomolar scales are sufficient to trigger significant myosin activities.
The critical roles of local Ca2+ pulses in migrating cells raise the question where those Ca2+ signals come from. In a classical signaling model, most intracellular Ca2+ signals originate from endoplasmic reticulum (ER) through inositol triphosphate (IP3) receptors [34, 35], which are activated by IP3 generated via receptor-tyrosine kinase- (RTK-) phospholipase C (PLC) signaling cascades. It is therefore reasonable to assume that local Ca2+ pulses are also generated from internal Ca2+ storage, that is, the ER. In an in vitro experiment, when Ca2+ chelator EGTA was added to the extracellular space, local Ca2+ pulses were not immediately eliminated from the migrating cells [24], supporting the above hypothesis. Moreover, pan-RTK inhibitors that quenched the activities of RTK-PLC-IP3 signaling cascades reduced local Ca2+ pulses efficiently in moving cells [25]. The observation of enriched RTK and PLC activities at the leading edge of migrating cells was also compatible with the accumulation of local Ca2+ pulses in the cell front [25]. Therefore, polarized RTK-PLC-IP3 signaling enhances the ER in the cell front to release local Ca2+ pulses, which are responsible for cyclic moving activities in the cell front.
In addition to RTK, the readers may wonder about the potential roles of G protein-coupled receptors (GPCRs) on local Ca2+ pulses during cell migration. As the major pathway to activate PLC, GPCRs coupled to the Gαq/11 subunit [36] trigger the cleavage of phosphatidylinositol (4,5)-bisphosphate (PIP2) by PLC to generate diacylglycerol (DAG) and IP3, which subsequently releases Ca2+ from the ER as Ca2+ pulses and spikes [37]. Indeed, Ca2+ oscillations induced by GPCR pathways have been observed in various cell types [38, 39]. However, the GPCRs coupled to Gαq/11 and PLC include serotonergic, adrenergic, muscarinic, glutamatergic, and histamine receptors [37], most of which do not directly affect cell migration. In contrast, growth factors contributing to cell migration, such as fibroblast growth factor (FGF) [40], epidermal growth factor (EGF) [41], and vascular endothelial growth factor (VEGF) [42], activate RTK signaling pathways. Therefore, it is more likely for the RTK rather than GPCR pathway to be responsible for local Ca2+ pulses in migrating cells. Nonetheless, more studies are required to clarify this important question.
The readers may also be curious how nonpulsatile RTK-PLC signaling generates oscillatory local Ca2+ pulses. In fact, IP3-induced Ca2+ oscillation has been reported repeatedly in various physiological circumstances [34, 38, 43, 44]. One possibility is that RTK signaling sensitizes IP3 receptors in the front ER but does not directly open those Ca2+ channels. Alternatively, the cyclic Ca2+ channel opening could be triggered by Ca2+-induced Ca2+ release (CICR), which is the activation of IP3 receptors by small changes of local Ca2+ levels [45, 46]. In the protruding cell front, the change of membrane tension may open stretch-activated transient receptor potential (TRP) channels [47], offering the required CICR. Indeed, TRP channels have been extensively reported as major contributors for cell migration and cancer metastasis [48]. Specifically, TRPM7 has been revealed to enhance cancer cell metastasis [49, 50], by mediating Ca2+ influx [51], altering Ca2+ flickers [18, 52], and regulating cell-matrix adhesion [53]. Therefore, oscillatory small Ca2+ pulses in the migrating cell front are probably the integrated results of polarized RTK signaling interacting with pulsatile membrane stretch and TRP channel opening, to release Ca2+ periodically from the front ER.
3.2. Maintainers of Basal Ca2+ Levels and Gradient: Sarcoplasmic/Endoplasmic Reticulum Ca2+-ATPase (SERCA) and Plasma Membrane Ca2+-ATPase (PMCA) (Figure 2)
Figure 2.
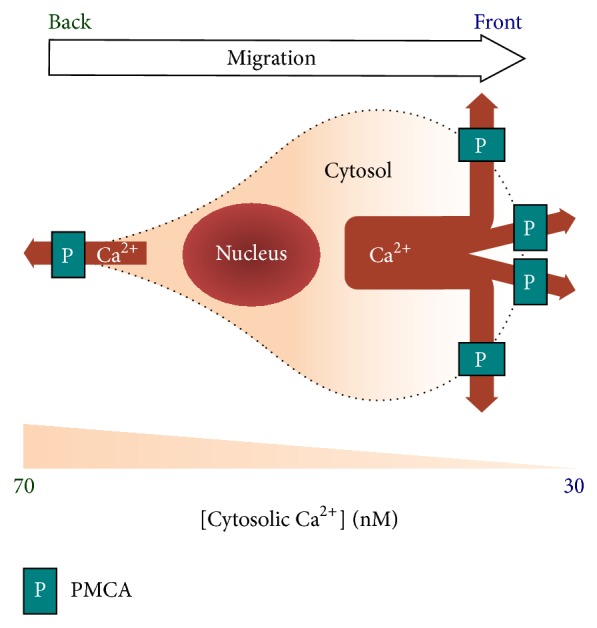
Cytosolic Ca2+ levels are low in the front and high in the back of the migrating cell. The Ca2+ gradient is created by the differential distribution of plasma membrane Ca2+-ATPase (PMCA, shown as P in the illustration), resulting in higher pump activity to move cytosolic Ca2+ out of the cell in the front than the back. Low Ca2+ in the front “starves” myosin light chain kinase (MLCK), which is essential for its reactivity to local Ca2+ pulses. High Ca2+ in the back facilitates the turnover of stable focal adhesion complexes. (See Figure 4 and the text for more details.)
The fact that tiny cyclic Ca2+ signals induce significant changes of cell motility implies that the basal cytosolic Ca2+ level, especially that at the front of migrating cells, has to be extremely low, so the cell migration machinery, specifically myosin and focal adhesion complexes, can promptly respond to small Ca2+ changes. To achieve the above goal, the migrating cells meticulously utilize two types of Ca2+-ATPase pumps, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) and plasma membrane Ca2+-ATPase (PMCA).
3.2.1. SERCA Pumps Cytosolic Ca2+ into the ER
SERCA is transmembranously located at the ER, continuously pumping cytosolic Ca2+ into the ER lumen with fast speed and high affinity [4]. Although their activities are slightly regulated by phospholamban and protein kinase A (PKA) [54], these pumps maintain the internal Ca2+ storage with high fidelity. Once SERCA is inactivated, the ER luminal Ca2+ leaks out to the cytoplasm in no time [55]. The resulting high cytosolic Ca2+ will saturate MLCK and induce persistent contraction of myosin [25], rendering front protrusion not possible. Furthermore, SERCA dysfunction dramatically reduces the ER luminal Ca2+, disabling further Ca2+ signaling through IP3 receptors. Hence, SERCA is essential for physiological and pathological cell migration. It is therefore not surprising to see aberrant SERCA expressions in cancer progression, invasion, and metastasis [56, 57].
3.2.2. SERCA Is Not Responsible for the Cytosolic Ca2+ Gradient in Migrating Cells
Since SERCA continuously and efficiently removes Ca2+ out of the cytoplasm into the ER, it is convenient to hypothesize that cytosolic Ca2+ gradient in migrating cells results from differential SERCA activities. If the SERCA activity was higher in the front than in the back, more Ca2+ in the front cytosol would be pumped into the front ER, resulting in the low-in-front, high-in-back Ca2+ gradient in the cytoplasm and a reverse (high-in-front, low-in-back) gradient in the ER. However, blocking SERCA activities with small molecule inhibitors caused a paradoxical increase of Ca2+ gradient, in addition to the global increase of cytosolic Ca2+ [25]. Monitoring intra-ER Ca2+ with the T1ER FRET probe [58] also revealed a low-in-front, high-in-back Ca2+ gradient when the cell moved [25]. Therefore, though a pivotal molecule keeps the cytosol Ca2+ free at the basal status, SERCA does not contribute to the Ca2+ gradient in migrating cells.
3.2.3. Differential PMCA Activities Keep [Ca2+] in the Front Lower Than the Back during Cell Migration
Based on the above data, Ca2+ pumps at the plasma membrane might be better candidates for the Ca2+ gradient during cell migration. Similar to SERCA, PMCA also continuously removes cytosolic Ca2+, by pumping it to the extracellular space [4, 59]. Unlike SERCA, recent evidence revealed that PMCA inhibitors and siRNA reduced Ca2+ gradient and cell motility during cell migration [25]. Direct measurement of Ca2+ efflux through plasma membrane also demonstrated an enhancement of PMCA activity by 30–50% in the front of migrating cells [25]. Hence, differential PMCA activities might account for the Ca2+ gradient during cell migration.
It is still not totally understood how cells adjust local PMCA activities to make them high in the front and low in the back. Several modulators have been demonstrated to regulate PMCA, including calmodulin [60], PKA [61], and calpain [62]. Whether those proteins could be spatially regulated inside the cells remains elusive. In addition, PMCA was enriched in the front plasmalemma of moving cells [25], suggesting that its differential distribution might account for the well-recognized front-low, back-high Ca2+ gradient during cell migration. Still, how PMCA is accumulated in the cell front requires further investigation.
3.3. Maintainers of Ca2+ Homeostasis during Migration: Store-Operated Ca2+ (SOC) Influx (Figure 3)
Figure 3.
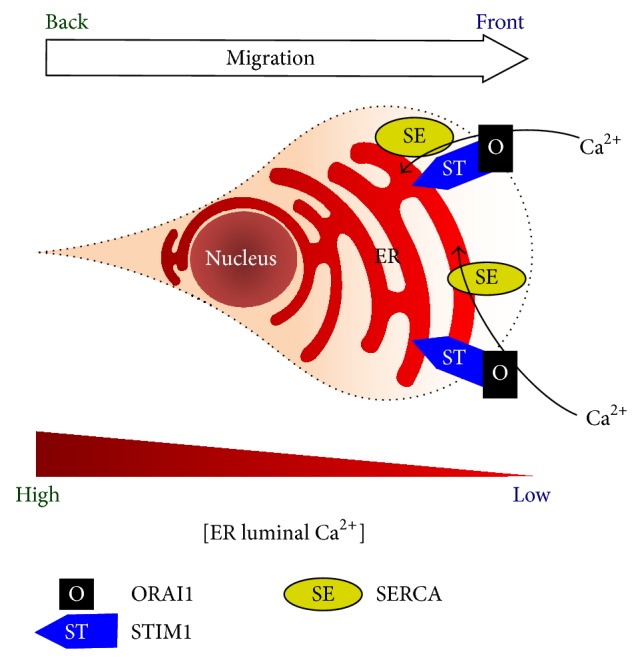
Internal Ca2+ storage is maintained through the differential activities of store-operated Ca2+ (SOC) influx during cell migration. Repetitive Ca2+ release from inositol triphosphate (IP3) receptors causes the depletion of Ca2+ in the endoplasmic reticulum (ER) near the leading edge. Such Ca2+ depletion activates STIM1 (shown as ST in the illustration), which is translocated to the ER-plasma membrane junction to open Ca2+ channels ORAI1 (shown as O in the illustration). The inward Ca2+ current through ORAI1 will further travel into the ER via sarcoplasmic/endoplasmic reticular Ca2+-ATPase (SERCA, shown as SE in the illustration). In migrating cells, STIM1 proteins are enriched in the front ER to maintain Ca2+ homeostasis, which is essential for proper polarity and motility.
SOC influx is an essential process to maintain internal Ca2+ storage [63] for IP3 receptor-based Ca2+ signaling, during which the luminal ER Ca2+ is evacuated. After IP3-induced Ca2+ release, although Ca2+ can be recycled back to the ER through SERCA, a significant amount of cytosolic Ca2+ will be pumped out of the cell through PMCA, resulting in the depletion of internal Ca2+ storage. To rescue this, low luminal Ca2+ activates STIM1 [55, 64], which is a membranous protein located at the ER and transported to the cell periphery by microtubules [65, 66]. Active STIM1 will be translocated to the ER-plasma membrane junction [67], opening the Ca2+ influx channel ORAI1 [68, 69]. Ca2+ homeostasis could therefore be maintained during active signaling processes including cell migration.
Since the identification of STIM1 and ORAI1 as the major players of SOC influx, numerous reports have emerged confirming their significant roles in cell migration and cancer metastasis (Tables 1 and 2). Although it is reasonable for those Ca2+-regulatory molecules to affect cell migration, the molecular mechanism is still not totally clear. Recent experimental evidence implied that STIM1 helped the turnover of cell-matrix adhesion complexes [7, 25], so SOC influx may assist cell migration by maintaining local Ca2+ pulses in the front of migrating cells. In a moving cell, local Ca2+ pulses near its leading edge result in the depletion of Ca2+ in its front ER. Such depletion subsequently activates STIM1 at the cell front. Compatible with the above assumption, more STIM1 was translocated to the ER-plasma membrane junction in the cell front compared to its back during cell migration [25]. Moreover, in addition to the ER and plasma membrane, STIM1 is also colocalized with EB1 [65, 66], the cargo protein located at the plus ends of microtubules. Further experiments revealed that STIM1 was actively transported to the front ER assisting cell migration [25]. Therefore, STIM1 together with other Ca2+ channels is meticulously regulated in a spatial manner maintaining cell polarity and motility.
Table 1.
Roles of store-operated Ca2+ (SOC) influx on cancer cell migration.
| Gene(s)/Protein(s) | Cell type | Highlight | Target(s) | Reference |
|---|---|---|---|---|
| ORAI1 | Esophageal squamous cell carcinoma (ESCC) | ORAI1 controls intracellular Ca2+ oscillations | N.A. | [105] |
|
| ||||
| ORAI1 and STIM1 | Clear cell renal cell carcinoma (ccRCC) | ORAI1 and STIM1 regulate cell proliferation and migration | N.A. | [106] |
|
| ||||
| ORAI1 and STIM2 | Melanoma cell lines | ORAI1 and STIM2 control melanoma growth and invasion in opposite manners | N.A. | [107] |
|
| ||||
| ORAI1 | Breast cancer cells | cAMP-PKA pathway decreases SK3 channel and SK3-ORAI1 complex activities, reducing Ca2+ entry and cancer cell migration | cAMP, PKA | [108] |
|
| ||||
| STIM1 | Breast cancer cell line MDA-MB-435s | Targeting SK3-ORAI1 in lipid rafts may inhibit bone metastasis | SK3 | [109] |
|
| ||||
| STIM1 | Cervical cancer cell lines (SiHa, HT-3, CaSki, and HeLa) | HDAC6 may disrupt STIM1-mediated SOC influx and block malignant cell behavior | HDAC6 | [110] |
|
| ||||
| ORAI1 and STIM1 | Glioblastoma multiforme (GBM) | STIM1 and ORAI1 affect the invasion of GBM cells | N.A. | [111] |
|
| ||||
| ORAI1 | Human T cell leukemia line, Jurkat cell | Monoclonal antibodies against ORAI1 reduce SOC influx, NFAT transcription, and cytokine release | N.A. | [112] |
|
| ||||
| ORAI1 | Human prostate cancer (PCa) cell | Bisphenol A pretreatment enhances SOC influx and ORAI1 protein in LNCaP cells; it also induces PCa cells migration | N.A. | [113] |
|
| ||||
| STIM1 | Cervical cancer cell | STIM1 regulates actomyosin reorganization and contractile forces to control cell migration | Actomyosin | [114] |
|
| ||||
| STIM1 | Hepatocellular carcinoma and hepatocyte cell lines | STIM1 level predicts prognosis in patients of liver cancer | N.A. | [115] |
|
| ||||
| STIM1 | Human epidermoid carcinoma A431 cells | STIM1 regulates SOC influx, cell proliferation, and tumorigenicity | N.A. | [116] |
|
| ||||
| STIM1 | Cervical cancer SiHa and CaSki cell lines | STIM1 regulates cervical cancer growth, migration, and angiogenesis | Focal adhesion, Pyk2 | [7] |
|
| ||||
| ORAI1 and STIM1 | MDA-MB-231 human breast cancer cells | Blocking STIM1 or ORAI1 using RNA interference or small molecule inhibitors decreased tumor metastasis in animal models | Focal adhesion | [82] |
Table 2.
Roles of store-operated SOC influx on the motility of nonmalignant cells.
| Gene(s)/Protein(s) | Cell type | Highlight | Target(s) | Reference |
|---|---|---|---|---|
| STIM1 | Endothelial progenitor cells (EPCs) | STIM1 affects EPCs proliferation and migration after vascular injury by regulating Ca2+ levels | N.A. | [117] |
|
| ||||
| ORAI1 | HEK293 | Selective activation of NFAT by ORAI1 | NFAT | [118] |
|
| ||||
| STIM1 | Endothelial leader cells | Cells employ an integrated and polarized Ca2+ signalling system for directed cell migration | PLC pathway | [25] |
|
| ||||
| ORAI1 | Keratinocytes | ORAI1-mediated Ca2+ entry enhances the turnover of focal adhesion through PKCβ, calpain, and focal adhesion kinase | PKC pathway | [119] |
|
| ||||
| ORAI1 and STIM1 | Retinal pigment epithelial cells (ARPE-19 cell line) | STIM1, ORAI1, ERK 1/2, and Akt determine EGF-mediated cell growth | MAPK pathway | [120] |
|
| ||||
| STIM1 | HEK293 | STIM regulates focal adhesion dynamics | Focal adhesion | [121] |
|
| ||||
| ORAI1 and STIM1 | Airway smooth muscle cell (ASMC) | STIM1 or ORAI1 controls PDGF-mediated ASMC proliferation and chemotactic migration | N.A. | [122] |
|
| ||||
| ORAI1 and STIM1 | ASMC | STIM1 and ORAI1 control PDGF-induced cell migration and Ca2+ influx | N.A. | [123] |
|
| ||||
| STIM1 | Intestinal epithelial cell (IEC) | Polyamines control TRPC1-mediated Ca2+ signaling and cell migration via differential STIM1 and STIM2 levels | TRPC1 | [124] |
|
| ||||
| ORAI1 and STIM1 | Vascular smooth muscle cells (VSMC) | STIM1- and ORAI1-mediated SOC influx regulates angiotensin II-induced VSMC proliferation | N.A. | [125] |
|
| ||||
| STIM1 | EPCs | STIM1 regulates the proliferation and migration of EPCs | N.A. | [126] |
|
| ||||
| ORAI1 and STIM1 | VSMC | STIM1 and ORAI1 regulate PDGF-mediated Ca2+ entry and migration in VSMC | N.A. | [127] |
|
| ||||
| ORAI1 and STIM1 | VSMC | Knockdown of STIM1 and ORAI1, but not STIM2, Orai2, or Orai3, reduces VSMC proliferation and migration | N.A. | [128] |
4. Ca2+ Effectors for Cell Migration (Figure 4)
Figure 4.
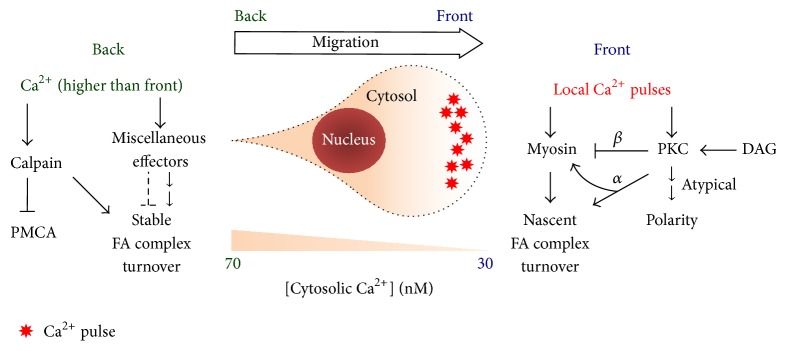
Ca2+ is temporally and spatially regulated to control the cell migration machinery via a wide variety of effectors. (Right) In the front, Ca2+ activates myosin and protein kinase C (PKC) for the maintenance of polarity and establishment of nascent cell-matrix adhesion. (Left) In the back, Ca2+ mediates calpain and miscellaneous focal-adhesion (FA) regulators, so proper disassembly of stable FA complexes can proceed. DAG: diacylglycerol; PMCA: plasma membrane Ca2+-ATPase.
As described above, intracellular Ca2+ is regulated locally and globally for effective cytoskeletal remodeling, cell migration, and cancer metastasis. Ca2+ pulses and spikes occur at the right place and right time, activating numerous downstream structural and signaling targets, which have been investigated separately over the past decades. The clarification of Ca2+ signaling in recent years has dramatically improved our understanding about how those components are regulated temporally and spatially in migrating cells. However, such advancement has revealed more questions than answers. More efforts are required to resolve those problems in the future.
4.1. Signaling-Related Targets
4.1.1. Protein Kinase C (PKC)
PKC is a typical downstream target of Ca2+ in receptor tyrosine kinase signaling pathways, during which the growth factor binds to the receptor and activates its tyrosine kinase through dimerization and autophosphorylation [70]. The resulting activation of PLC generates diacylglycerol (DAG) and IP3, which subsequently induces Ca2+ release from the ER. DAG and Ca2+ then bind separately to the C1 and C2 domains of classical PKC (PKCα, β, and γ) [71]. Depending on the substrate, classical PKC regulates a wide variety of physiological processes, including cell migration [72]. The action could be direct via phosphorylation or indirect through transcriptional activation.
The classical PKC family has direct and significant impact on cell migration. PKCα is enriched in the front of migrating cells [14]. It directly phosphorylates Rho GTPases and multiple components of focal adhesion complexes, regulating the remodeling of cell-matrix adhesion (see [73] for a more comprehensive review). PKCβ phosphorylates the heavy chains of myosin II, inhibiting myosin contraction and facilitating the process of directional determination in migrating cells [74–77]. How these PKCs respond to spatiotemporal Ca2+ signaling and coordinate for effective moving activities requires further investigation.
Besides classical PKCs, atypical PKCs [70] also regulate the polarity of migrating cells. Unlike classical PKCs, those PKCs do not require DAG or Ca2+ for activation [70]. Together with Rho GTPases [78, 79], these PKCs might be actively involved in the dynamic processes of cell protrusion and adhesion [78, 80]. How these actions synchronize with the Ca2+ dynamics during cell migration also awaits more research in the future.
4.1.2. Rho GTPases
Rho GTPases, including Rac1, RhoA, and Cdc42, have been known as the key components for the regulation of actin dynamics [81]. It is therefore not surprising to see their active involvement in cell migration. Spatially, in a simplified model, these GTPases are enriched at specific structures of a migrating cell, Rac1 in lamellipodia, RhoA around focal adhesion complexes, and Cdc42 near filopodia [8]. Temporally, activities of these GTPases are pulsatile and also synchronized to the cyclic lamellipodial activities in the front of migrating cells [29]. Therefore, Rho GTPases, similar to Ca2+ [24], exert actions at the right place and right time for proper actin remodeling and efficient cell migration.
Although the present data reveals no evidence of direct binding between Ca2+ and Rho GTPases, it is reasonable to expect their mutual interactions considering their perfect coordination during cell migration [24, 29, 30]. Such speculation is supported by the observation that blocking Ca2+ influx at the leading edges of polarized macrophages resulted in the disassembly of actin filaments and lamellipodia activities [14]. The facts that constitutively active Rac1 fully rescued the effects of SOC influx inhibition in migrating breast cancer cells [82] also indicate the regulatory role of Ca2+ on Rho GTPases. Moreover, the transamidation of Rac1 was shown to be dependent on intracellular Ca2+ and calmodulin in rat cortical cells, suggesting the biochemical link between Rho GTPases and Ca2+ signaling [83]. Hopefully more studies will be conducted in the near future to clarify the mechanism of how Ca2+ interacts with Rho GTPases.
4.2. Cytoskeleton-Related Targets
4.2.1. Myosin II
As mentioned above, local Ca2+ pulses at the junction of lamellipodia and lamella activate MLCK [24], which subsequently phosphorylates myosin light chain and triggers myosin contraction. It is worth noticing that the affinity between MLCK and myosin-calmodulin is extremely high, with the dissociation constant (K d) of about 1 nM [33]. Therefore, a slight increase of local Ca2+ concentration is sufficient to induce significant activation of MLCK and subsequent contraction of myosin II. Moreover, the high sensitivity of MLCK to Ca2+ implies that the front cytoplasm has to be free of Ca2+ at the basal status, so MLCK can be inactive at baseline but respond to small rises of Ca2+ promptly. Such design justifies the physiological importance of the front-low, back-high Ca2+ gradient in migrating cells.
In cell migration, the immediate effect of myosin contraction is the retraction of actin bundles, which not only facilitates the disassembly of F-actin at lamella but also allows the protruding front to attach to the extracellular matrix [28, 31]. In addition, myosin contraction also stabilizes nascent focal adhesion complexes in the front of migrating cells [32, 84]. This is probably because these contractions apply traction force on the complexes through actin bundles binding to them. Such force subsequently induces remodeling and stabilization of the components in focal adhesion. Therefore, through MLCK and myosin II, local Ca2+ pulses are tightly linked to the oscillatory dynamics of cell protrusion, retraction, and adhesion.
4.2.2. Actin
Besides myosin, Ca2+ also affects the dynamics of actin, the major component of cytoskeleton [85, 86]. Although Ca2+ does not directly bind to actin, it affects the activities of multiple actin regulators. First of all, Ca2+ activates protein kinase C and calmodulin-dependent kinases, both of which interact with actin affecting its dynamics [87–89]. Secondly, as also described above, Ca2+ signaling regulates the Rho GTPases [14], which are mandatory for the formation of actin bundles for lamellipodia, focal adhesion complexes, and filopodia [8], the major components for cell migration. In addition, the F-actin severing protein cofilin [90, 91] also depends on the cytosolic Ca2+ for its proper activity. Moreover, myosin, as one the major actin regulators, is totally dependent on Ca2+ for its proper activity [24]. Therefore, though not a direct regulator, Ca2+ modulates actin dynamics through multiple signaling pathways and structural molecules.
4.3. Adhesion-Related Targets
4.3.1. Calpain
In addition to kinase activities and physical force, Ca2+ also affects cell migration through protein cleavage and degradation. Calpain, as a Ca2+-dependent intracellular protease [92, 93], is involved in the turnover of stable focal adhesion complexes, probably at the rear end of migrating cells. Calpain has been revealed to cleave several components of the focal adhesion complex, including talin [94], paxillin [95], and focal adhesion kinases [96], compatible with previous reports showing that Ca2+ influx at the back of migrating cells facilitated retraction and detachment at their rear ends [97]. Beside focal adhesion, calpain also degrades PMCA [62]. Since there is an inverse correlation between the front-back gradients of Ca2+ and PMCA in migrating cells [25], decreased amount of PMCA in the cell back may result from the higher Ca2+ level and higher calpain activity in the back than in the front. However, such speculation requires more experimental data to be validated.
4.3.2. Pyk2 and Other Molecules
In addition to calpain, several adhesion-related proteins are also regulated by Ca2+, including Pyk2, plectin, and matrix metallopeptidases.
As a cytoplasmic protein tyrosine kinase, Pyk2 is activated by intracellular Ca2+ and protein kinase C [98]. It regulates the activities of focal adhesion kinase and GRB2, affecting focal adhesion complexes [99] and the MAP kinase signaling pathway [98]. In human cervical cancer cells, aberrant SOC influx changes focal adhesion dynamics through Pyk2 dysregulation [7].
Ca2+ also regulates the conveyance of integrin-based signaling into the cytoskeleton, with its interaction with plectin, the bridge between integrin complexes and actin filaments. Recent biochemical and biophysical evidence indicated that the binding of plectin 1a with Ca2+ effectively decreased its interactions with integrin β and with F-actin, decoupling cell-matrix adhesion with cytoskeletal structures [100, 101]. We may speculate that, with proper temporal and spatial Ca2+ regulation, cells could determine how many environmental signals would be conducted into the cells for cytoskeleton modification. More studies are required to clarify the above hypothesis.
Furthermore, matrix metallopeptidases (MMP), as facilitating factors for cancer metastasis, are also regulated by intracellular Ca2+. In prostate cancer, increased expression of TRPV2 elevated cytosolic Ca2+ levels, which enhanced MMP9 expression and cancer cell aggressiveness [102]. Further investigation in melanoma cells revealed that increased intracellular Ca2+ induced the binding of Ca2+-modulating cyclophilin ligand to basigin, stimulating the production of MMP [103]. Therefore, Ca2+ not only modulates the outside-in (integrin to actin) signaling but also regulates the inside-out (Ca2+ to MMP) signaling for cell migration and cancer metastasis.
5. Future: Interactions between Ca2+ and Other Signaling Pathways
Regarding the complicated temporal and spatial regulation of Ca2+ signaling in migrating cells, we would expect extensive interactions between Ca2+ and other signaling modules during cell migration. Indeed, though still preliminary, recent work has revealed potential cross talk between Ca2+ and other pathways controlling cell motility. These findings will shed new light on our pilgrimage toward a panoramic view of cell migration machinery.
5.1. Interactions between SOC Influx and Cell-Matrix Adhesion
In the present model, SOC influx maintains Ca2+ storage in the ER, which releases local Ca2+ pulses to enhance the formation of nascent focal adhesion complexes [25]. Therefore, the inhibition of SOC influx should weaken cell-matrix adhesion. Interestingly, STIM1, the Ca2+ sensor for the activation of the SOC influx, had been reported as an oncogene [82] or a tumor suppressor gene [104] by different groups. Furthermore, although most recent research suggested a positive role of STIM1 on cancer cell motility (Table 1), other reports revealed the opposite results in primary cells (Table 2). Therefore, effects of SOC influx on cell migration might vary under different circumstances.
One possible explanation of the confusing results uses the interaction between Ca2+ and basal cell-matrix adhesion. Primary cells are usually well attached to the matrix, so further enhancing their adhesion capability might trap them in the matrix and deter them from moving forward. In contrast, metastatic cancer cells often have weak cell-matrix adhesion, so strengthening their attachment to the matrix facilitates the completion of cell migration cycles. Indeed, recent evidence suggested that, in an in vitro cell migration assay [25], SOC influx might increase or decrease the motility of the same cell type depending on concentrations of fibronectin for the cells to attach. Though further explorations are required to validate the present data, the combination of SOC influx inhibition and cell-matrix adhesion blockage might be a novel approach to prevent cancer metastasis.
5.2. Coordination between the Oscillations of Ca2+ and Rho GTPases
Previous reports have revealed the oscillatory activities of Rho GTPases in the front of migrating cells, including Rac1, RhoA, and Cdc42 [29, 30]. These molecules regulate actin dynamics and coordinate with the pulsatile lamellipodial activities. Since the oscillation of local Ca2+ pulses synchronize with the retraction phases of lamellipodial cycles [24], there probably exists cross talk between Ca2+ signaling and Rho GTPases. Clarifying how these molecules are regulated to coordinate with each other will dramatically improve our understanding of lamellipodia and help developing better strategies to control physiological and pathological cell migration.
5.3. Link between Ca2+, RTK, and Lipid Signaling
The meticulous spatial control of Ca2+ signaling in migrating cells, together with the enrichment of RTK, phosphatidylinositol (3,4,5)-triphosphate (PIP3), and DAG in the cell front [25], reveals the complicated nature of the migration polarity machinery. How these signaling pathways act together to determine the direction for cells to move remains elusive and requires more research. In addition, understanding how nonpulsatile RTK and lipid signaling exert effects on oscillatory Ca2+ pulses will improve our knowledge about the spatial and temporal regulation of signal transduction inside the cells. Such information will further enhance our capability to develop novel strategies targeting pathological processes and manipulating diseases.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Masuyama R. Role of local vitamin D signaling and cellular calcium transport system in bone homeostasis. Journal of Bone and Mineral Metabolism. 2014;32(1):1–9. doi: 10.1007/s00774-013-0508-z. [DOI] [PubMed] [Google Scholar]
- 2.Calin-Jageman I., Lee A. Cav1 L-type Ca2+ channel signaling complexes in neurons. Journal of Neurochemistry. 2008;105(3):573–583. doi: 10.1111/j.1471-4159.2008.05286.x. [DOI] [PubMed] [Google Scholar]
- 3.Zhao X., Yamazaki D., Kakizawa S., Pan Z., Takeshima H., Ma J. Molecular architecture of Ca2+ signaling control in muscle and heart cells. Channels. 2011;5(5):391–396. doi: 10.4161/chan.5.5.16467. [DOI] [PubMed] [Google Scholar]
- 4.Clapham D. E. Calcium Signaling. Cell. 2007;131(6):1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 5.LaFerla F. M. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nature Reviews Neuroscience. 2002;3(11):862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 6.Shaw P. J., Feske S. Physiological and pathophysiological functions of SOCE in the immune system. Frontiers in Bioscience (Elite Edition) 2012;4(6):2253–2268. doi: 10.2741/540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y.-F., Chiu W.-T., Chen Y.-T., et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(37):15225–15230. doi: 10.1073/pnas.1103315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridley A. J., Schwartz M. A., Burridge K., et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 9.Lamalice L., Le Boeuf F., Huot J. Endothelial cell migration during angiogenesis. Circulation Research. 2007;100(6):782–794. doi: 10.1161/01.res.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- 10.Imhof B. A., Dunon D. Basic mechanism of leukocyte migration. Hormone and Metabolic Research. 1997;29(12):614–621. doi: 10.1055/s-2007-979112. [DOI] [PubMed] [Google Scholar]
- 11.Hanna S., Etzioni A. Leukocyte adhesion deficiencies. Annals of the New York Academy of Sciences. 2012;1250(1):50–55. doi: 10.1111/j.1749-6632.2011.06389.x. [DOI] [PubMed] [Google Scholar]
- 12.Barrientos S., Stojadinovic O., Golinko M. S., Brem H., Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair and Regeneration. 2008;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 13.Vicente-Manzanares M., Horwitz A. R. Cell migration: an overview. Methods in Molecular Biology. 2011;769:1–24. doi: 10.1007/978-1-61779-207-6_1. [DOI] [PubMed] [Google Scholar]
- 14.Evans J. H., Falke J. J. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(41):16176–16181. doi: 10.1073/pnas.0707719104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grynkiewicz G., Poenie M., Tsien R. Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. The Journal of Biological Chemistry. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- 16.Tsien R. Y., Rink T. J., Poenie M. Measurement of cytosolic free Ca2+ in individual small cells using fluorescence microscopy with dual excitation wavelengths. Cell Calcium. 1985;6(1-2):145–157. doi: 10.1016/0143-4160(85)90041-7. [DOI] [PubMed] [Google Scholar]
- 17.Brundage R. A., Fogarty K. E., Tuft R. A., Fay F. S. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science. 1991;254(5032):703–706. doi: 10.1126/science.1948048. [DOI] [PubMed] [Google Scholar]
- 18.Wei C., Wang X., Chen M., Ouyang K., Song L.-S., Cheng H. Calcium flickers steer cell migration. Nature. 2009;457(7231):901–905. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandeville J. T. H., Ghosh R. N., Maxfield F. R. Intracellular calcium levels correlate with speed and persistent forward motion in migrating neutrophils. Biophysical Journal. 1995;68(4):1207–1217. doi: 10.1016/S0006-3495(95)80336-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elf J., Li G.-W., Xie X. S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316(5828):1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers K. L., Martin J.-R., Renaud O., et al. Electron-multiplying charge-coupled detector-based bioluminescence recording of single-cell Ca2+ . Journal of Biomedical Optics. 2008;13(3) doi: 10.1117/1.2937236.031211 [DOI] [PubMed] [Google Scholar]
- 22.Mank M., Griesbeck O. Genetically encoded calcium indicators. Chemical Reviews. 2008;108(5):1550–1564. doi: 10.1021/cr078213v. [DOI] [PubMed] [Google Scholar]
- 23.Chen T.-W., Wardill T. J., Sun Y., et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499(7458):295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai F.-C., Meyer T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Current Biology. 2012;22(9):837–842. doi: 10.1016/j.cub.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai F.-C., Seki A., Yang H. W., et al. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nature Cell Biology. 2014;16(2):133–144. doi: 10.1038/ncb2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Small J. V., Stradal T., Vignal E., Rottner K. The lamellipodium: where motility begins. Trends in Cell Biology. 2002;12(3):112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- 27.Webb D. J., Parsons J. T., Horwitz A. F. Adhesion assembly, disassembly and turnover in migrating cells—over and over and over again. Nature Cell Biology. 2002;4(4):E97–E100. doi: 10.1038/ncb0402-e97. [DOI] [PubMed] [Google Scholar]
- 28.Burnette D. T., Manley S., Sengupta P., et al. A role for actin arcs in the leading-edge advance of migrating cells. Nature Cell Biology. 2011;13(4):371–382. doi: 10.1038/ncb2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacHacek M., Hodgson L., Welch C., et al. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461(7260):99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tkachenko E., Sabouri-Ghomi M., Pertz O., et al. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nature Cell Biology. 2011;13(6):660–667. doi: 10.1038/ncb2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giannone G., Dubin-Thaler B. J., Döbereiner H.-G., Kieffer N., Bresnick A. R., Sheetz M. P. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004;116(3):431–443. doi: 10.1016/s0092-8674(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 32.Giannone G., Dubin-Thaler B. J., Rossier O., et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell. 2007;128(3):561–575. doi: 10.1016/j.cell.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasturi R., Vasulka C., Johnson J. D. Ca2+, caldesmon, and myosin light chain kinase exchange with calmodulin. The Journal of Biological Chemistry. 1993;268(11):7958–7964. [PubMed] [Google Scholar]
- 34.Berridge M. J. Inositol trisphosphate and calcium signalling. Nature. 1993;361(6410):315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 35.Taylor C. W., Taufiq-Ur-Rahman, Pantazaka E. Targeting and clustering of IP3 receptors: key determinants of spatially organized Ca2+ signals. Chaos. 2009;19(3) doi: 10.1063/1.3127593.037102 [DOI] [PubMed] [Google Scholar]
- 36.Qin K., Dong C., Wu G., Lambert N. A. Inactive-state preassembly of Gq-coupled receptors and Gq heterotrimers. Nature Chemical Biology. 2011;7(10):740–747. doi: 10.1038/nchembio.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 4th. Garland Science; 2002. [Google Scholar]
- 38.Jacob R., Merritt J. E., Hallam T. J., Rink T. J. Repetitive spikes in cytoplasmic calcium evoked by histamine in human endothelial cells. Nature. 1988;335(6185):40–45. doi: 10.1038/335040a0. [DOI] [PubMed] [Google Scholar]
- 39.Giri L., Patel A. K., Karunarathne W. K. A., Kalyanaraman V., Venkatesh K. V., Gautam N. A G-protein subunit translocation embedded network motif underlies GPCR regulation of calcium oscillations. Biophysical Journal. 2014;107(1):242–254. doi: 10.1016/j.bpj.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levin E. G. Cancer therapy through control of cell migration. Current Cancer Drug Targets. 2005;5(7):505–518. doi: 10.2174/156800905774574048. [DOI] [PubMed] [Google Scholar]
- 41.Grahovac J., Wells A. Matrikine and matricellular regulators of EGF receptor signaling on cancer cell migration and invasion. Laboratory Investigation. 2014;94(1):31–40. doi: 10.1038/labinvest.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Angelucci A., Bologna M. Targeting vascular cell migration as a strategy for blocking angiogenesis: the central role of focal adhesion protein tyrosine kinase family. Current Pharmaceutical Design. 2007;13(21):2129–2145. doi: 10.2174/138161207781039643. [DOI] [PubMed] [Google Scholar]
- 43.Petersen O. H. Stimulus-secretion coupling: cytoplasmic calcium signals and the control of ion channels in exocrine acinar cells. Journal of Physiology. 1992;448:1–51. doi: 10.1113/jphysiol.1992.sp019028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thorn P., Lawrie A. M., Smith P. M., Gallacher D. V., Petersen O. H. Local and global cytosolic Ca2+ oscillations in exocrine cells evoked by agonists and inositol trisphosphate. Cell. 1993;74(4):661–668. doi: 10.1016/0092-8674(93)90513-p. [DOI] [PubMed] [Google Scholar]
- 45.Finch E. A., Turner T. J., Goldin S. M. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252(5004):443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- 46.Keizer J., Levine L. Ryanodine receptor adaptation and Ca2+(−)induced Ca2+ release- dependent Ca2+ oscillations. Biophysical Journal. 1996;71(6):3477–3487. doi: 10.1016/s0006-3495(96)79543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di A., Malik A. B. TRP channels and the control of vascular function. Current Opinion in Pharmacology. 2010;10(2):127–132. doi: 10.1016/j.coph.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 48.Nielsen N., Lindemann O., Schwab A. TRP channels and STIM/ORAI proteins: sensors and effectors of cancer and stroma cell migration. British Journal of Pharmacology. 2014;171(24):5524–5540. doi: 10.1111/bph.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao H., Chen X., Du X., Guan B., Liu Y., Zhang H. EGF enhances the migration of cancer cells by up-regulation of TRPM7. Cell Calcium. 2011;50(6):559–568. doi: 10.1016/j.ceca.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Middelbeek J., Kuipers A. J., Henneman L., et al. TRPM7 is required for breast tumor cell metastasis. Cancer Research. 2012;72(16):4250–4261. doi: 10.1158/0008-5472.can-11-3863. [DOI] [PubMed] [Google Scholar]
- 51.Chen J.-P., Luan Y., You C.-X., Chen X.-H., Luo R.-C., Li R. TRPM7 regulates the migration of human nasopharyngeal carcinoma cell by mediating Ca2+ influx. Cell Calcium. 2010;47(5):425–432. doi: 10.1016/j.ceca.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Roy B., Das T., Mishra D., Maiti T. K., Chakraborty S. Oscillatory shear stress induced calcium flickers in osteoblast cells. Integrative Biology. 2014;6(3):289–299. doi: 10.1039/c3ib40174j. [DOI] [PubMed] [Google Scholar]
- 53.Su L.-T., Agapito M. A., Li M., et al. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. The Journal of Biological Chemistry. 2006;281(16):11260–11270. doi: 10.1074/jbc.m512885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MacLennan D. H., Kranias E. G. Phospholamban: a crucial regulator of cardiac contractility. Nature Reviews Molecular Cell Biology. 2003;4(7):566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 55.Liou J., Kim M. L., Won D. H., et al. STIM is a Ca2+ sensor essential for Ca2+-store- depletion-triggered Ca2+ influx. Current Biology. 2005;15(13):1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gou W.-F., Niu Z.-F., Zhao S., Takano Y., Zheng H.-C. Aberrant SERCA3 expression during the colorectal adenoma-adenocarcinoma sequence. Oncology Reports. 2014;31(1):232–240. doi: 10.3892/or.2013.2837. [DOI] [PubMed] [Google Scholar]
- 57.Chung F.-Y., Lin S.-R., Lu C.-Y., et al. Sarco/endoplasmic reticulum calcium-ATPase 2 expression as a tumor marker in colorectal cancer. The American Journal of Surgical Pathology. 2006;30(8):969–974. doi: 10.1097/00000478-200608000-00006. [DOI] [PubMed] [Google Scholar]
- 58.Abell E., Ahrends R., Bandara S., Park B. O., Teruel M. N. Parallel adaptive feedback enhances reliability of the Ca2+ signaling system. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(35):14485–14490. doi: 10.1073/pnas.1018266108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strehler E. E., Treiman M. Calcium pumps of plasma membrane and cell interior. Current Molecular Medicine. 2004;4(3):323–335. doi: 10.2174/1566524043360735. [DOI] [PubMed] [Google Scholar]
- 60.James P., Maeda M., Fischer R., et al. Identification and primary structure of a calmodulin binding domain of the Ca2+ pump of human erythrocytes. The Journal of Biological Chemistry. 1988;263(6):2905–2910. [PubMed] [Google Scholar]
- 61.James P. H., Pruschy M., Vorherr T. E., Penniston J. T., Carafoli E. Primary structure of the cAMP-dependent phosphorylation site of the plasma membrane calcium pump. Biochemistry. 1989;28(10):4253–4258. doi: 10.1021/bi00436a020. [DOI] [PubMed] [Google Scholar]
- 62.James P., Vorherr T., Krebs J., et al. Modulation of erythrocyte Ca2+-ATPase by selective calpain cleavage of the calmodulin-binding domain. The Journal of Biological Chemistry. 1989;264(14):8289–8296. [PubMed] [Google Scholar]
- 63.Putney J. W., Jr. Capacitative calcium entry: sensing the calcium stores. Journal of Cell Biology. 2005;169(3):381–382. doi: 10.1083/jcb.200503161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brandman O., Liou J., Park W. S., Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131(7):1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grigoriev I., Gouveia S. M., van der Vaart B., et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Current Biology. 2008;18(3):177–182. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Honnappa S., Gouveia S. M., Weisbrich A., et al. An EB1-binding motif acts as a microtubule tip localization signal. Cell. 2009;138(2):366–376. doi: 10.1016/j.cell.2009.04.065. [DOI] [PubMed] [Google Scholar]
- 67.Liou J., Fivaz M., Inoue T., Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(22):9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feske S., Gwack Y., Prakriya M., et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 69.Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443(7108):230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 70.Rosse C., Linch M., Kermorgant S., Cameron A. J. M., Boeckeler K., Parker P. J. PKC and the control of localized signal dynamics. Nature Reviews Molecular Cell Biology. 2010;11(2):103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 71.Mellor H., Parker P. J. The extended protein kinase C superfamily. Biochemical Journal. 1998;332, part 2:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rang H. P., Dale M. M., Ritter J. M., Flower R. J., Henderson G. Rang & Dale's Pharmacology. 7th. Edinburgh, UK: Churchill Livingstone; 2011. [Google Scholar]
- 73.Fogh B. S., Multhaupt H. A. B., Couchman J. R. Protein kinase C, focal adhesions and the regulation of cell migration. Journal of Histochemistry and Cytochemistry. 2014;62(3):172–184. doi: 10.1369/0022155413517701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murakami N., Singh S. S., Chauhan V. P., Elzinga M. Phospholipid binding, phosphorylation by protein kinase C, and filament assembly of the COOH terminal heavy chain fragments of nonmuscle myosin II isoforms MIIA and MIIB. Biochemistry. 1995;34(49):16046–16055. doi: 10.1021/bi00049a019. [DOI] [PubMed] [Google Scholar]
- 75.Murakami N., Chauhan V. P. S., Elzinga M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry. 1998;37(7):1989–2003. doi: 10.1021/bi971959a. [DOI] [PubMed] [Google Scholar]
- 76.Dulyaninova N. G., Malashkevich V. N., Almo S. C., Bresnick A. R. Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry. 2005;44(18):6867–6876. doi: 10.1021/bi0500776. [DOI] [PubMed] [Google Scholar]
- 77.Dulyaninova N. G., House R. P., Betapudi V., Bresnick A. R. Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Molecular Biology of the Cell. 2007;18(8):3144–3155. doi: 10.1091/mbc.e06-11-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Etienne-Manneville S., Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCζ . Cell. 2001;106(4):489–498. doi: 10.1016/S0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 79.Etienne-Manneville S., Hall A. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature. 2003;421(6924):753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 80.Rosse C., Formstecher E., Boeckeler K., et al. An aPKC-Exocyst complex controls paxillin phosphorylation and migration through localised JNK1 activation. PLoS Biology. 2009;7(11) doi: 10.1371/journal.pbio.1000235.e1000235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bustelo X. R., Sauzeau V., Berenjeno I. M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. BioEssays. 2007;29(4):356–370. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang S., Zhang J. J., Huang X.-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15(2):124–134. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 83.Dai Y., Dudek N. L., Li Q., Muma N. A. Phospholipase C, Ca2+, and calmodulin signaling are required for 5-HT2A receptor-mediated transamidation of Rac1 by transglutaminase. Psychopharmacology. 2011;213(2-3):403–412. doi: 10.1007/s00213-010-1984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kanchanawong P., Shtengel G., Pasapera A. M., et al. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468(7323):580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mostafavi E., Nargesi A. A., Ghazizadeh Z., et al. The degree of resistance of erythrocyte membrane cytoskeletal proteins to supra-physiologic concentrations of calcium: an in vitro study. The Journal of Membrane Biology. 2014;247(8):695–701. doi: 10.1007/s00232-014-9689-1. [DOI] [PubMed] [Google Scholar]
- 86.Wang F., Liu D.-Z., Xu H., et al. Thapsigargin induces apoptosis by impairing cytoskeleton dynamics in human lung adenocarcinoma cells. The Scientific World Journal. 2014;2014:7. doi: 10.1155/2014/619050.619050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohta Y., Nishida E., Sakai H. Type II Ca2+/calmodulin-dependent protein kinase binds to actin filaments in a calmodulin-sensitive manner. FEBS Letters. 1986;208(2):423–426. doi: 10.1016/0014-5793(86)81061-4. [DOI] [PubMed] [Google Scholar]
- 88.Larsson C. Protein kinase C and the regulation of the actin cytoskeleton. Cellular Signalling. 2006;18(3):276–284. doi: 10.1016/j.cellsig.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 89.Hoffman L., Farley M. M., Waxham M. N. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry. 2013;52(7):1198–1207. doi: 10.1021/bi3016586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guan C.-B., Xu H.-T., Jin M., Yuan X.-B., Poo M.-M. Long-range Ca2+ signaling from growth cone to soma mediates reversal of neuronal migration induced by slit-2. Cell. 2007;129(2):385–395. doi: 10.1016/j.cell.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 91.Huang Z.-H., Wang Y., Su Z.-D., et al. Slit-2 repels the migration of olfactory ensheathing cells by triggering Ca2+-dependent cofilin activation and RhoA inhibition. Journal of Cell Science. 2011;124(2):186–197. doi: 10.1242/jcs.071357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nagano M., Hoshino D., Koshikawa N., Akizawa T., Seiki M. Turnover of focal adhesions and cancer cell migration. International Journal of Cell Biology. 2012;2012:10. doi: 10.1155/2012/310616.310616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Glading A., Lauffenburger D. A., Wells A. Cutting to the chase: calpain proteases in cell motility. Trends in Cell Biology. 2002;12(1):46–54. doi: 10.1016/s0962-8924(01)02179-1. [DOI] [PubMed] [Google Scholar]
- 94.Franco S. J., Rodgers M. A., Perrin B. J., et al. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nature Cell Biology. 2004;6(10):977–983. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- 95.Cortesio C. L., Boateng L. R., Piazza T. M., Bennin D. A., Huttenlocher A. Calpain-mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. The Journal of Biological Chemistry. 2011;286(12):9998–10006. doi: 10.1074/jbc.m110.187294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan K. T., Bennin D. A., Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK) The Journal of Biological Chemistry. 2010;285(15):11418–11426. doi: 10.1074/jbc.m109.090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sjaastad M. D., Nelson W. J. Integrin-mediated calcium signaling and regulation of cell adhesion by intracellular calcium. BioEssays. 1997;19(1):47–55. doi: 10.1002/bies.950190109. [DOI] [PubMed] [Google Scholar]
- 98.Lev S., Moreno H., Martinez R., et al. Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376(6543):737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 99.Avraham S., London R., Fu Y., et al. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. The Journal of Biological Chemistry. 1995;270(46):27742–27751. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- 100.Kostan J., Gregor M., Walko G., Wiche G. Plectin isoform-dependent regulation of keratin-integrin α6β4 anchorage via Ca2+/calmodulin. The Journal of Biological Chemistry. 2009;284(27):18525–18536. doi: 10.1074/jbc.m109.008474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Song J.-G., Kostan J., Drepper F., et al. Structural insights into Ca2+-calmodulin regulation of plectin 1a-integrin β4 interaction in hemidesmosomes. Structure. 2015;23(3):558–570. doi: 10.1016/j.str.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Monet M., Lehen'kyi V., Gackiere F., et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Research. 2010;70(3):1225–1235. doi: 10.1158/0008-5472.can-09-2205. [DOI] [PubMed] [Google Scholar]
- 103.Long T., Su J., Tang W., et al. A novel interaction between calcium-modulating cyclophilin ligand and Basigin regulates calcium signaling and matrix metalloproteinase activities in human melanoma cells. Cancer Letters. 2013;339(1):93–101. doi: 10.1016/j.canlet.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 104.Suyama E., Wadhwa R., Kaur K., et al. Identification of metastasis-related genes in a mouse model using a library of randomized ribozymes. The Journal of Biological Chemistry. 2004;279(37):38083–38086. doi: 10.1074/jbc.c400313200. [DOI] [PubMed] [Google Scholar]
- 105.Zhu H., Zhang H., Jin F., et al. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget. 2014;5:3455–3471. doi: 10.18632/oncotarget.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim J.-H., Lkhagvadorj S., Lee M.-R., et al. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochemical and Biophysical Research Communications. 2014;448(1):76–82. doi: 10.1016/j.bbrc.2014.04.064. [DOI] [PubMed] [Google Scholar]
- 107.Stanisz H., Saul S., Müller C. S. L., et al. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell and Melanoma Research. 2014;27(3):442–453. doi: 10.1111/pcmr.12222. [DOI] [PubMed] [Google Scholar]
- 108.Clarysse L., Guéguinou M., Potier-Cartereau M., et al. cAMP-PKA inhibition of SK3 channel reduced both Ca2+ entry and cancer cell migration by regulation of SK3-Orai1 complex. Pflügers Archiv. 2014;466(10):1921–1932. doi: 10.1007/s00424-013-1435-5. [DOI] [PubMed] [Google Scholar]
- 109.Chantôme A., Potier-Cartereau M., Clarysse L., et al. Pivotal role of the lipid raft SK3-orai1 complex in human cancer cell migration and bone metastases. Cancer Research. 2013;73(15):4852–4861. doi: 10.1158/0008-5472.can-12-4572. [DOI] [PubMed] [Google Scholar]
- 110.Chen Y.-T., Chen Y.-F., Chiu W.-T., et al. Microtubule-associated histone deacetylase 6 supports the calcium store sensor STIM1 in mediating malignant cell behaviors. Cancer Research. 2013;73(14):4500–4509. doi: 10.1158/0008-5472.CAN-12-4127. [DOI] [PubMed] [Google Scholar]
- 111.Motiani R. K., Hyzinski-García M. C., Zhang X., et al. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Archiv European Journal of Physiology. 2013;465(9):1249–1260. doi: 10.1007/s00424-013-1254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Greenberg M. L., Yu Y., Leverrier S., Zhang S. L., Parker I., Cahalan M. D. Orai1 function is essential for T cell homing to lymph nodes. Journal of Immunology. 2013;190(7):3197–3206. doi: 10.4049/jimmunol.1202212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Derouiche S., Warnier M., Mariot P., et al. Bisphenol A stimulates human prostate cancer cell migration via remodelling of calcium signalling. SpringerPlus. 2013;2, article 54 doi: 10.1186/2193-1801-2-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen Y.-T., Chen Y.-F., Chiu W.-T., Wang Y.-K., Chang H.-C., Shen M.-R. The ER Ca2+ sensor STIM1 regulates actomyosin contractility of migratory cells. Journal of Cell Science. 2013;126(5):1260–1267. doi: 10.1242/jcs.121129. [DOI] [PubMed] [Google Scholar]
- 115.Yang N., Tang Y., Wang F., et al. Blockade of store-operated Ca2+ entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Letters. 2013;330(2):163–169. doi: 10.1016/j.canlet.2012.11.040. [DOI] [PubMed] [Google Scholar]
- 116.Yoshida J., Iwabuchi K., Matsui T., Ishibashi T., Masuoka T., Nishio M. Knockdown of stromal interaction molecule 1 (STIM1) suppresses store-operated calcium entry, cell proliferation and tumorigenicity in human epidermoid carcinoma A431 cells. Biochemical Pharmacology. 2012;84(12):1592–1603. doi: 10.1016/j.bcp.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 117.Cong X.-P., Wang W.-H., Zhu X., Jin C., Liu L., Li X.-M. Silence of STIM1 attenuates the proliferation and migration of EPCs after vascular injury and its mechanism. Asian Pacific Journal of Tropical Medicine. 2014;7(5):373–377. doi: 10.1016/s1995-7645(14)60058-4. [DOI] [PubMed] [Google Scholar]
- 118.Kar P., Samanta K., Kramer H., Morris O., Bakowski D., Parekh A. B. Dynamic assembly of a membrane signaling complex enables selective activation of NFAT by Orai1. Current Biology. 2014;24(12):1361–1368. doi: 10.1016/j.cub.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vandenberghe M., Raphaël M., Lehen'kyi V., et al. ORAI1 calcium channel orchestrates skin homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(50):E4839–E4848. doi: 10.1073/pnas.1310394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang I.-H., Tsai Y.-T., Chiu S.-J., et al. Involvement of STIM1 and Orai1 in EGF-mediated cell growth in retinal pigment epithelial cells. Journal of Biomedical Science. 2013;20, article 41 doi: 10.1186/1423-0127-20-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schäfer C., Rymarczyk G., Ding L., Kirber M. T., Bolotina V. M. Role of molecular determinants of store-operated Ca2+ entry (Orai1, phospholipase A2 group 6, and STIM1) in focal adhesion formation and cell migration. The Journal of Biological Chemistry. 2012;287(48):40745–40757. doi: 10.1074/jbc.M112.407155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Spinelli A. M., González-Cobos J. C., Zhang X., et al. Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration. Pflügers Archiv. 2012;464(5):481–492. doi: 10.1007/s00424-012-1160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Suganuma N., Ito S., Aso H., et al. STIM1 regulates platelet-derived growth factor-induced migration and Ca2+ influx in human airway smooth muscle cells. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0045056.e45056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rao J. N., Rathor N., Zhuang R., et al. Polyamines regulate intestinal epithelial restitution through TRPC1-mediated Ca2+ signaling by differentially modulating STIM1 and STIM2. The American Journal of Physiology—Cell Physiology. 2012;303(3):C308–C317. doi: 10.1152/ajpcell.00120.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guo R.-W., Yang L.-X., Li M.-Q., Pan X.-H., Liu B., Deng Y.-L. Stim1-and Orai1-mediated store-operated calcium entry is critical for angiotensin II-induced vascular smooth muscle cell proliferation. Cardiovascular Research. 2012;93(2):360–370. doi: 10.1093/cvr/cvr307. [DOI] [PubMed] [Google Scholar]
- 126.Kuang C.-Y., Yu Y., Guo R.-W., et al. Silencing stromal interaction molecule 1 by RNA interference inhibits the proliferation and migration of endothelial progenitor cells. Biochemical and Biophysical Research Communications. 2010;398(2):315–320. doi: 10.1016/j.bbrc.2010.06.088. [DOI] [PubMed] [Google Scholar]
- 127.Bisaillon J. M., Motiani R. K., Gonzalez-Cobos J. C., et al. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. The American Journal of Physiology—Cell Physiology. 2010;298(5):C993–C1005. doi: 10.1152/ajpcell.00325.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Potier M., Gonzalez J. C., Motiani R. K., et al. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. The FASEB Journal. 2009;23(8):2425–2437. doi: 10.1096/fj.09-131128. [DOI] [PMC free article] [PubMed] [Google Scholar]