

Abstract
Hyaluronic acid (HA) is used in a wide range of medical applications, where its performance and therapeutic efficacy are highly dependent on its molecular weight. In the microbial production of HA, it has been suggested that a high level of intracellular ATP enhances the productivity and molecular weight of HA. Here, we report on heterologous HA production in an ε-poly-l-lysine producer, Streptomyces albulus, which has the potential to generate ATP at high level. The hasA gene from Streptococcus zooepidemicus, which encodes HA synthase, was refactored and expressed under the control of a late-log growth phase-operating promoter. The expression of the refactored hasA gene, along with genes coding for UDP-glucose dehydrogenase, UDP-N-acetylglucosamine pyrophosphorylase, and UDP-glucose pyrophosphorylase, which are involved in HA precursor sugar biosynthesis, resulted in efficient production of HA in the 2.0 MDa range, which is greater than typical bacterial HA, demonstrating that a sufficient amount of ATP was provided to support the biosynthesis of the precursor sugars, which in turn promoted HA production. In addition, unlike in the case of streptococcal HA, S. albulus-derived HA was not cell associated. Based on these findings, our heterologous production system appears to have several advantages for practical HA production. We propose that the present system could be applicable to the heterologous production of a wide variety of molecules other than HA in the case their biosynthesis pathways require ATP in vivo.
INTRODUCTION
Hyaluronic acid (HA) is a linear, uniformly repetitive glycosaminoglycan composed of up to 20,000 disaccharide units of d-glucuronic acid (GluUA) and N-acetylglucosamine (GlcNAc) (1) and ubiquitously exists as hydrogel in vertebrate tissues. HA is particularly abundant in cartilage, synovial fluid, dermis, and the vitreous humor of the eye. In these tissues, the polysaccharide has significant structural, physiological, and biological functions due to its viscoelastic property and ability to retain a large volume of water (2). These distinctive properties have been exploited in a wide range of medical applications, including in the areas of ophthalmology, orthopedics, and wound healing (3), where the performance and therapeutic efficacy of HA are highly dependent on its molecular weight. Product quality is also considered a key factor for these biomedical applications in order to avoid serious allergic and inflammatory reactions caused by contaminants, especially upon injection (4, 5). Thus, to meet the growing demand for HA in the field of biomedicine (6), it is critical to produce high-molecular-weight HA from a safe source that does not carry those concerns.
Traditionally, HA had been obtained commercially from rooster corms, but in recent years it has mainly been produced through the fermentation process of an attenuated pathogenic strain of group C streptococci that naturally synthesize the identical macromolecule as a part of the extracellular capsule in order to conceal it from the immune system of host organisms (7). Rooster corm-derived HA typically has a high molecular weight (up to 4 MDa), which meets the commercial demand for biomedical use, but carries concern of cross-species viral infections, while streptococcal fermentation has the potential to produce exotoxins and is challenging to genetically manipulate to produce high-molecular-weight HA. To overcome these issues, Widner et al. heterologously expressed the artificial HA biosynthetic operon in a well-investigated, exotoxin- and endotoxin-free microorganism, Bacillus subtilis, resulting in the production of HA in the 1-MDa range (8). Their strategy allowed the transition of HA production into more widely applicable modern biotechnology, but the system itself brought no benefit in terms of molecular weight, considering the fact that streptococcal HA is typically in the 0.5 to 2.0 MDa range. Despite the technical advantages of recombinant systems and the fact that many studies aiming at heterologous production of HA have been carried out with a wide range of bacteria, including Escherichia coli, recombinant systems commonly show fairly low productivity (9, 10).
In streptococci, the molecular mechanism by which HA synthase (HasA) controls the molecular weight of the product is not yet fully understood. Nielsen and coworkers reported that overexpression of genes involved in the precursor sugar pathway led to the production of high-molecular-weight HA (11). In addition, a fermentation study has demonstrated that enhanced ATP levels brought by changing fermentation parameters enhanced the production and improved molecular weight of HA, probably by increased fluxes of the sugar precursor pathways (12). Thus, metabolic engineering rather than HasA engineering would be beneficial for practical HA production. Technically, overexpression of the hasA gene along with genes for sugar pathways in heterologous hosts can be done conveniently with a wide variety of genetic tools. This approach itself, however, is not thought to have any major effect on HA production due to the limitation of the ATP-driving force. This could be a major cause of the low productivities in most conventional recombinant systems reported to date.
An actinomycete, Streptomyces albulus, produces an ε-poly-l-lysine (ε-PL), which consists of 25 to 35 l-lysine residues with isopeptide linkages between the α-carboxyl groups and ε-amino groups. This strain is known to produce the poly-amino acid at an extremely high level (>20 g/liter) (13). In light of the fact that one molecule of ATP is required for the formation of a single isopeptide bond in the polymer molecule during its biosynthesis (14), 24 to 34 molecules of ATP are needed to polymerize the l-lysine molecules. This implied that S. albulus has the ability to generate ATP at high level for the ε-PL production. In fact, our previous study demonstrated that the intracellular ATP level was increased during ε-PL production in S. albulus (15). In addition to this metabolic benefit, S. albulus has a long history in production of ε-PL that has been used as a food preservative in several countries, including Japan, South Korea, China, and the United States, and thus is recognized as safe. Accordingly, S. albulus could be a potent host for heterologous production of a high-molecular-weight HA. Here, we report on the heterologous production of HA in genetically engineered S. albulus via expression of a synthetic HA biosynthesis pathway mimicking that from group C streptococci. The present study shows that S. albulus has several advantages as a host for the practical production of HA.
MATERIALS AND METHODS
Bacterial strains and DNAs.
The strains and plasmids used in the present study are listed in Table 1. E. coli TOP10 (Invitrogen) was used for routine cloning. Streptomyces albulus CRM003 [pls::aac(3)IV] (14) was used as a host for heterologous production of HA. Transformation of S. albulus CRM003 was carried out by the method previously described (16). E. coli S17-1 was used as the plasmid donor for intergeneric conjugal DNA transfer (16, 17). The media and growth conditions for S. albulus recombinant strains have been described previously (16). The E. coli-Streptomyces shuttle expression vector pLAE008, a derivative of pLAE009 (18, 19), was used for the construction of a series of new expression vectors. Streptomyces lavendulae NBRC12340 was used as a source of the bpsA gene (GenBank accession no. AB240063) coding for the blue pigment indigoidine synthetase BpsA (20). Streptococcus zooepidemicus FERM BP878 was used as a source of the HA synthase gene (hasA). Genes coding for UDP-glucose dehydrogenase (UdgA), UDP-N-acetylglucosamine pyrophosphorylase (GlmU), and UDP-glucose pyrophosphorylase (GtaB) were amplified from the genomic DNA of Streptomyces avermitilis NBRC14893 by PCR and were used for the construction of synthetic HA biosynthesis gene clusters. Genomic DNA was isolated from Streptococcus and Streptomyces strains according to a standard procedure (21). Kanamycin (50 μg/ml), neomycin (50 μg/ml), and nalidixic acid (25 μg/ml) were used for the selection of E. coli and Streptomyces recombinant strains.
TABLE 1.
Plasmids and strains used in this study
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| Streptomyces spp. | ||
| S. albulus CRM003 | Host strain for heterologous production of HA; pls::aac(3)IV (ε-poly–l-lysine production deficient) | 14 |
| S. albulus CR1 | ε-Poly–l-lysine producer strain, parental strain of CRM003 | 16 |
| S. lavendulae NBRC12340 | Source of the bpsA gene | NBRC |
| S. avermitilis NBRC14893 | Source of the udgA, glmU, and gtaB genes | NBRC |
| E. coli | ||
| Top10 | Host strain for routine cloning; F− Δ(araA-leu)7697 [araD139]B/r Δ(codB-lacI)3 ϕ80dlacZ58(M15) galK0 mcrA0 ΔgalU recA1 endA1 ΔnupG ΔrpsL (Strr) Δ(mcrC-mrr)715 | Invitrogen |
| S17-1 | Donor strain for conjugation; F− thi pro hsdR [RP4-2 Tc::Mu Km::Tn7 (Tp Sm)] | 17 |
| Streptococcus sp. | ||
| S. zooepidemicus FERM BP878 | Industrial HA producer strain | JNC Corp. (formerly Chisso Corp.) |
| Plasmids | ||
| pLAE009 | Growth phase-dependent inducible expression vector for S. albulus (E. coli-Streptomyces shuttle); pMB1 ori aph(3)II oriT(PR4) pNO33 rep Ppls | 18 |
| pLAE008 | pLAE009 derivative; no His tag coding sequence | This study |
| pLAE008b and -c | pLAE008 derivative; modified rbs (AGAGG→GGAGG) | This study |
| pJHA1 | pLAE008c-hasA | This study |
| pJHA2 | pLAE008c-hasAm | This study |
| pJHA3 | pLAE008c-hasAm-udgA | This study |
| pJHA4 | pLAE008c-hasAm-udgA-glmU-gtaB | This study |
| pJHA5 | pLAE008c-hasA-udgA | This study |
Strr, streptomycin resistance.
Refinement of the gene expression system in Streptomyces albulus.
As shown in Fig. 1a, pLAE008 is equipped with the pls promoter (Ppls), which was originally recognized as controlling the expression of the pls gene coding for ε-PL synthetase (Pls) in S. albulus, and with the ribosome-binding site (rbs; AGAGG) from the pls loci 8 bp upstream of the multiple cloning site (MCS) (18). The rbs sequence and its distance from MCS were explored to improve the expression level of a cloned gene as follows. An ∼450-bp region carrying the Ppls and rbs was amplified by PCR from the genomic DNA of S. albulus with the primers Ppls-fseF (5′-actatGGCCGGCCgttggtcgacgtcc-3′) and rbstypeB-ndeR (5′-ggaattcCATATGgccgcacctccctccgcccggtgggtggatgaccct-3′). Capitalized letters represent the restriction sites for FseI and NdeI, respectively, and the underlined region shows the rbs being replaced. The PCR-amplified fragment was digested and ligated with the larger fragment of FseI-NdeI doubly digested pLAE008, yielding pLAE008b, in which the rbs was replaced with GGAGG 10 bp upstream of the MCS (Fig. 1b). The same region was amplified with a second pair of primers, Ppls-fseF and rbstypeC-ndeR (5′-ggaattcCATATGgccgcacctccctccgcccggtgggtggatgaccct-3′), and the Ppls with the rbs of pLAE008 was replaced with the PCR fragment, yielding pLAE008c, which carries the same rbs (GGAGG) but at 8-bp upstream of the MCS (Fig. 1b).
FIG 1.

Refinement of the E. coli-Streptomyces shuttle expression vector pLAE008 aiming at improving translation efficiency. A physical map of pLAE008 (a) and the sequences around the ribosome-binding sites (rbs) in the pLAE008 derivatives (b) are shown schematically. To evaluate the functionality of the vectors, the heterologous indigoidine production caused by the expression of the bpsA gene cloned into the vectors was evaluated after second-stage fermentation for 6 h, and the results are presented in panel b.
To test the functionality of the newly constructed pLAE008 derivatives, a colorimetric heterologous production assay was carried out. The bpsA gene coding for the blue pigment indigoidine synthetase (BpsA) was amplified from the genome of Streptomyce lavendulae NBRC12340 with primers bpsA-ndeF (5′-ggaattcCATATGactcttcaggagaccagcgtgctcgag-3′) and bpsA-hindR (5′-cccAAGCTTtcactcgccgagcaggtagcggatgtgc-3′). The capitalized letters represent restriction sites for NdeI and HindIII, respectively. The PCR product was digested and was cloned into pLAE008 and its derivatives to yield pLAE008-bpsA, pLAE008b-bpsA, and pLAE008c-bpsA. The expression constructs were then individually introduced into S. albulus CRM003 by intergeneric conjugation with E. coli S17-1 as described previously (16). Exconjugants showing neomycin resistance were isolated and cultivated by the two-stage fermentation method as described previously (15). Briefly, S. albulus recombinant strains were cultivated in 2 ml of M3G medium [50 g of glucose, 5 g of yeast extract, 1 g of (NH4)2SO4, 1.6 g of Na2HPO4, 1.4 g of KH2PO4, 0.5 g of MgSO4·7H2O, 0.04 g of ZnSO4·7H2O, and 0.03 g of FeSO4·7H2O per liter (pH 6.8)] supplemented with Neo in mini-test tubes for 24 h at 30°C with reciprocal shaking at 150 rpm (first-stage fermentation). Grown cells were transferred and were again cultivated in the same volume of production buffer (pH 4.2) consisting of 100 mM citrate, 5% glucose (wt/vol), and 1% (NH4)2SO4 (wt/vol) (second-stage fermentation). After the second-stage fermentation at 30°C for 6 h, insoluble indigoidine produced was recovered along with cells by centrifugation (15,000 × g for 30 min). The blue precipitate was washed with water twice and then similarly washed with methanol. Subsequently, an excess amount of N,N′-dimethylformamide (DMF) was added to the blue precipitate to lead complete dissolution of indigoidine. After centrifugation and filtration (0.22-μm pore size), the resultant blue solution was optically analyzed in a spectrophotometer. Indigoidine produced was quantified by measuring the absorbance at 600 nm corresponding to the absorption maximum of indigoidine (log ε = 4.37 in DMF) (20).
Bioinformatic-based refactoring of the HA synthetase (hasA) gene.
The amino acid sequence of HasA from S. Zooepidemicus was analyzed in a bioinformatic program SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui/) to predict the location of transmembrane domains. To express the hasA gene in the high G+C microorganism S. albulus, rare codons of the original hasA gene from S. zooepidemicus were replaced with preferred ones in Streptomyces strains. In addition, lysine residues adjacent to the predicted transmembrane domains in HasA were replaced with more basic arginine residues to enhance localization onto the membrane. After these refactorings, the overall G+C content of the resulting gene (hasAm) was increased from 39.9 to 63.4%, as shown in Fig. 2. The hasAm gene designed as described above was chemically synthesized.
FIG 2.

Construction of the refactored hasA (hasAm) gene. (a) Proposed domain structure of HasA from S. zooepidemicus. The location of the transmembrane (TM) domains was bioinformatically predicted using the online program SOSUI. (b) The hasAm gene was designed based on the amino acid sequence of HasA from S. zooepidemicus. In the gene design, all rare codons in Streptomycetes were replaced with preferred ones. Subsequently, lysine residues adjacent to the predicted TM domains (shown in shaded boxes) were replaced with more basic arginine residues as shown in the open boxes. Finally, the N- and C-terminal regions (underlined amino acid residues) were slightly changed to carry out the following cloning and assembling process. (c) Overall, the G+C content of the hasAm gene was significantly increased to 63.44% by these refactorings.
Construction of the hasA and hasAm gene expression vectors.
The hasA gene from S. zooepidemicus (FERM BP878) was amplified by PCR with the primers zhasA-ndeF(5′-ctcgtcgCATATGagaacattaaaaaacctcataactgttgtg-3′) and zhasA-xbaR (5′-gtcgacTCTAGAtcataataattttttacgtgttccccagtc-3′). Capitalized letters represent restriction sites used for subsequent cloning steps. The PCR product was digested and introduced into pLAE008c to yield pJHA1 (see Fig. 4). The synthetic hasAm gene was amplified by PCR with the primers mhasA-ndeF (5′-ctcgtcgCATATGcgctcgcgccgctggagc-3′) and mhasA-hindR (5′-cctccAAGCTTtcatgcgaggacctccttccgggtgc-3′) and was similarly cloned into pLAE008c to yield pJHA2. We further constructed two additional expression vectors, pJHA3 and pJHA4, carrying the hasAm gene, together with the gene(s) responsible for biosynthesis of the HA precursor sugars. For the construction of pJHA3 carrying the hasAm gene and the UDP-glucose 6-dehydrogenase gene (udgA), the hasAm gene was amplified by PCR with the primers mhasA-ndeF and mhasA-assembleR (5′-cgatcacggtgatcttgaggctcatgcgaggacctcc-3′). Next, the udgA was amplified from the genomic DNA of S. avermitilis NBRC14893 with the primers udgA-assembleF (5′-ctcgcatgagcctcaagatcaccgtgatcggcaccgg-3′) and udgA-hindR (5′-tcccAAGCTTctaggcggtggggcggcccat-3′). Both PCR products were combined and assembled into a single fragment by PCR with the primers mhasA-ndeF and udgA-hindR, generating the hasAm-udgA cassette. The cassette was then introduced into the NdeI-HindIII site of pLAE008c to yield pJHA3, in which the two cloned genes were translationally coupled under the control of Ppls. To enhance the precursor sugar supply for HA biosynthesis, pJHA4, which additionally carried the glmU and gtaB genes coding for UDP-N-acetylglucosamine pyrophosphorylase and UTP-glucose pyrophospholyase, respectively, was constructed. The glmU gene was amplified by PCR with glmU-ecorvF (5′-ctcgtcgGATATCgcatgagcgccatccgcccggcagc-3′) and glmU-assembleR (5′-atgcgactcagtcattcttcgccctccggcttccgggaagccacctc-3′), and the gtaB gene was amplified with gtaB-assembleF (5′-gccggagggcgaagaatgactgagtcgcatcccaggatcaccaaggctgt-3′) and gtaB-hindR (5′-cccAAGCTTctacatctcctcggctacgtaactgcgaagc-3′). Capitalized letters represent the restriction sites for EcoRV and HindIII, respectively. The glmU and gtaB genes were then combined and assembled by PCR with primers glmU-ecorvF and gtaB-hindR, yielding the glmU-gtaB cassette. The hasAm-udgA cassette was again similarly prepared by PCR to be flanked by XbaI and EcoRV restriction sites. The above two cassettes and XbaI-HindIII doubly digested pLAE008c were ligated to yield pJHA4, in which the four cloned genes were translationally coupled under the control of Ppls to form a synthetic HA biosynthesis gene cluster. To express the native hasA gene together with the udgA gene, an additional expression vector pJH5 was constructed as follows. The hasA gene from S. zooepidemicus was amplified by PCR with primers zhasA-ndeF(5′-ctcgtcgCATATGagaacattaaaaaacctcataactgttgtg-3′) and zhasA-assembleR (5′-cttgaggctcataataattcctcccgtgttccccagtcagcatt-3′). The udgA was similarly amplified from the genomic DNA of S. avermitilis NBRC14893 with primers udgA-assembleF (5′-aacacgggaggaattattatgagcctcaagatcaccgtgatcggcacc-3′) and udgA-xbaR (5′-tcccTCTAGActaggcggtggggcggcccat-3′). Both PCR products were combined and assembled into a single fragment by PCR with primers zhasA-ndeF and udgA-xbaR, generating the hasAz-udgA cassette. By introducing this cassette into pLAE008c, pJHA5 was constructed. All of the expression constructs generated as described above are schematically shown in Fig. 4 and were individually introduced into S. albulus CRM003 as described above.
FIG 4.

Gene organizations cloned into pLAE008c for heterologous HA production. These genes were individually cloned into pLAE008c under the control of the Ppls and expressed in S. albulus CRM003. To generate the synthetic HA biosynthesis gene cluster, the 3′ end of each open reading frame was redesigned to have a GGAGG rbs sequence 8 bp upstream of the start codon of its downstream gene. For the genes adjacent to the promoter, the rbs was provided from pLAE008c.
Expression of the HA biosynthesis genes in S. albulus CRM003.
The S. albulus CRM003 recombinants harboring pJHA1, pJHA2, pJHA3, pJHA4, and pJHA5 were individually cultivated by the two-stage fermentation method described earlier. To keep sufficient dissolved oxygen level throughout the fermentation, cultivations were carried out on a small scale (1.5 ml of culture broth) in 2-ml microcentrifuge tubes and breathable lids with vortexing at 1,800 rpm. After second-stage fermentation in the citrate-based production buffer at 30°C for 40 h, the amount of HA produced in the supernatant was measured as described below.
S. albulus CRM003/pJHA3 and S. albulus CRM003/pJHA4 were selected and grown in a 3-liter-scale jar-fermentor (model MDL-8C; B. E. Marubishi). The strains were individually inoculated into 5 ml of SLB medium (103 g of sucrose, 10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl per liter) supplemented with neomycin and were cultivated until the stationary phase was reached (20 h at 30°C). Aliquots (1 ml) of the cultures were transferred to 100 ml of M3G medium (15) supplemented with neomycin and were grown at 30°C for 20 h. The culture broth was then transferred to 1.4 l of M3G medium containing glucose at 60 g/liter in the jar-fermentor and was cultivated at 30°C with agitation stirring at 500 rpm. The aeration was kept at 3.5 VVM (liters air/liters medium/min) throughout the fermentation. After the pH of the medium dropped to 4.2, 10% aqueous ammonia was fed to keep it at 4.2. During fermentation, a mixture of glucose and ammonium sulfate (10:1 ratio by weight) was continuously fed to keep the glucose concentration ca. 5% (wt/vol). After the fermentation, the viscous culture broth was diluted with 2 volumes of water, and the cells were removed by centrifugation (20,000 × g for 30 min), yielding a clear supernatant. The molecular weight and amount of HA produced in the supernatant were measured as described below. S. albulus strain CR1 (16), which is the parental strain of the recombination host CRM003, was also grown under the same condition, and ε-PL produced was quantified as reported previously (22).
Theoretical ATP consumptions for the production of HA and ε-PL were calculated from the amount of the products based on their biosynthetic mechanisms reported previously (14, 23) with the following parameters: molecular mass of the HA disaccharide unit = 376.35 (as dehydrated unit), and molecular mass of the l-lysine residue in ε-PL = 128.19 (as dehydrated residue). In the data processing, the fact that three molecules of ATP are consumed to synthesize every single HA disaccharide unit was taken into account. Similarly, the processing for the ε-PL production was carried out based on the fact that one molecule of ATP is required for the formation of a single peptide bond in the polymer molecule. The number of peptide bonds in a single ε-PL molecule was defined as 29 since the average number of l-lysine residues in ε-PL molecules is known to be 30 (13–15).
Purification and analysis of HA.
All manipulations were carried out at room temperature, and the HA-dissolving steps were taken 20 h unless otherwise described. Proteins contained in the clear supernatant were removed by chloroform treatment. Subsequently, HA in the water layer was precipitated with cetyl pyridinium chloride (0.65% [wt/vol]), and the resulting insoluble ionic complex was collected. The ionic complex was washed with water and then dissolved in 2 M NaCl. The pH of the crude HA was adjusted to around 9 with 2 N NaOH, and 2 volumes of ethanol was added to the crude HA. The resulting white fibrous HA was collected by centrifugation (2,500 × g for 5 min). The HA pellet was washed with 70% (vol/vol) ethanol and was again dissolved in 0.5 M NaCl. Ethanol was similarly added to the viscous solution. The resultant HA precipitant was collected and was finally dried in vacuo to give purified HA as a sodium salt.
The dried HA was dissolved in water and subjected to the following measurements. The HA concentration was measured by carbazole assay (24), which detects the glucuronic acid released from hydrolyzed HA. HA dissolved in 0.2 M NaCl at a concentration of 0.05% (wt/vol) was analyzed by size exclusion chromatography (SEC) to determine its molecular weight. The analytical conditions were as follows: a ligand exchange-size exclusion column (Shodex SUGAR KS-807; 8 mm by 300 mm) equipped with a guard column (Shodex SUGAR KS-807G; 8 mm by 50 mm) was used at 40°C in an Agilent 1100 HPLC system with an isocratic flow of 0.2 M NaCl at 0.5 ml/min. The eluent was monitored with UV at 210 nm and with a differential refractive index detector. The molecular mass of HA was calculated from a calibration curve generated by using commercially available HA standards with a known weight-average molecular weights determined by intrinsic viscosity (0.2, 0.8, and 1.2 MDa obtained from JNC Corp. [Tokyo, Japan], and 2.2 MDa from Sigma-Aldrich).
RESULTS
Refined gene expression system in S. albulus.
S. albulus shows a significant ε-PL production, but only under an acidic condition occurring naturally after the logarithmic growth phase (25). Therefore, industrial ε-PL production is achieved by controlling the pH of the medium within the optimum range (pH 4.0 to 4.5), resulting in the yield over 20 g/liter during a week of fed-batch fermentation (13). With feeding of glucose and ammonium sulfate (10:1 by weight) under the particular condition, S. albulus CR1 produced significant amount of ε-PL (see Fig. 5a), demonstrating that the microorganism has the potential to continuously generate a large amount of ATP for the ε-PL biosynthesis (see Fig. 5b). Our previous study, in which we showed that the wild strain NBRC14147 (the parental strain of the CR1) indeed accumulates a large amount of ATP in the cells when ε-PL is produced (15), also supports our observation. This could be a very important feature for the heterologous production of desired metabolites in cases in which ATP is required for their biosynthetic cascade in vivo. Since a fermentation study has demonstrated that increasing the supply of ATP to the HA pathway led to an enhancement of HA production and molecular weight, increasing the range from 1 to 2.4 MDa (6), we assumed that S. albulus could be a beneficial host for heterologous production of HA.
FIG 5.
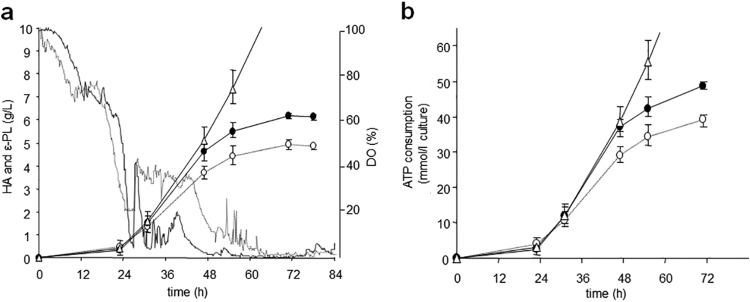
Fermentation profiles of ε-PL and HA in S. albulus strains. (a) ε-PL production in wild-type strain CR1 and HA production in S. albulus recombinant strains. The ε-PL productivity is shown with open triangles. The HA productivities and dissolved oxygen levels are shown with circles and continuous lines, respectively. The HA productivities in S. albulus CRM003/pJHA3 and S. albulus CRM003/pJHA4 are indicated with dotted lines and solid lines, respectively. These strains were cultivated in a mini-jar fermentor and error bars are given based on triplicate fermentations. (b) Theoretical ATP availability in S. albulus host and the actual ATP consumptions for the heterologous HA production. The ATP consumption for the ε-PL production in the strain CR1 indicated with open triangles is considered equivalent to the theoretical ATP availability in S. albulus host strain, CRM003, which was derived from the strain CR1. The values were calculated from the amount of the products at each time point based on their biosynthesis descried elsewhere. The values for 78-h cultures were omitted. Symbolic representations are same as in panel a.
As a heterologous host for HA production, we considered that the ε-PL production-deficient mutant of S. albulus, CRM003, in which the pls gene is inactivated (14), would be favorable, because generated ATP molecules should be utilized not for ε-PL biosynthesis but for the incoming HA biosynthetic pathway preferentially. In addition, we have developed an expression vector, pLAE009, for S. albulus strains (18, 19). This vector was equipped with the pls promoter (Ppls), which was originally identified as controlling the expression of the pls gene coding for ε-PL synthetase (Pls) after the late-log growth phase in S. albulus, during which the intracellular ATP level is significantly increased (15). Therefore, using the combination of S. albulus CRM003 and the late-log-phase operating vector, we set out to develop a heterologous HA production system.
In the present study, we used a derivative of the pLAE009 expression vector, pLAE008, which is identical to pLAE009 except that it lacks a His tag coding sequence (Fig. 1a). The expression system utilizing Ppls was originally designed to produce membrane-bound recombinant Pls in S. albulus CRM003 via moderate expression control at the late-log phase (18). We first tested whether this expression system would be available for other target proteins. To this end, we used indigoidine synthetase (BpsA) from Streptomyces lavendulae, because BpsA produces the readily detectable blue pigment indigoidine by the intermolecular condensation of two l-glutamine molecules using ATP (20). When the bpsA gene was cloned into pLAE008 and introduced into S. albulus CRM003, a moderate color change of the medium caused by the production of indigoidine was observed after induction of the expression of the bpsA gene by dropping the media pH. A spectrophotometric measurement revealed that the insoluble blue product isolated from the culture has a unique absorption maximum at 600 nm in DMF (data not shown), which shows good agreement with that of indigoidine (20), strongly suggesting that the expression system works with other target enzymes. In the present study, we further modified the expression system by changing the ribosome-binding site (rbs) sequence and its position to improve the translation initiation rate. Two newly constructed pLAE008 derivatives, pLAE008b and pLAE008c, were designed to have well known rbs consensus sequence (GGAGG) among Streptomyces strains located 10 and 8 bp upstream, respectively, of the multiple-cloning site (MCS) (Fig. 1b). Predictably, the expression of the bpsA gene from both of these derivatives resulted in increased indigoidine production. In particular, the expression level was much improved when using pLAE008c (Fig. 1b). Our strategy of modifying the rbs sequence and its distance from the MCS could successfully improve the gene expression level, and the results led us to decide to use pLAE008c for heterologous production of HA.
Heterologous HA production in S. albulus CRM003.
Only a few bacterial species are known to produce HA, namely, group A and group C streptococci (26) and Pasteurella multocida (27). It seemed logical to exploit the HA synthetase from Gram-positive bacteria rather than employing that from Gram-negative bacteria for heterologous HA production in the Gram-positive actinomycete, S. albulus. In addition, the HA biosynthetic pathway in streptococci has been well investigated (23), and thus it could be applicable to a synthetic biology approach. In streptococci, HA is synthesized by HA synthetase (HasA) from two nucleotide sugars, UDP-GlcUA and UDP-GlcNAc, generated via two parallel pathways starting from glucose (Fig. 3). To produce one molecule of the HA disaccharide unit, two molecules of glucose, two molecules of UTP, one molecule of acetyl coenzyme A, and three molecules of ATP are required. Thus, for HA biosynthesis, the intracellular ATP that drives the early steps of the sugar substrate pathways could be one of the most important factors. Some of the sugars from the parallel pathways and also their final products UDP-GlcUA and UDP-GlcNAc are known to be constituents of the bacterial cell wall. Therefore, we expected that the expression of the hasA gene would simply constitute the entire HA pathway in S. albulus. Surprisingly, however, the expression of the hasA gene from S. zooepidemicus resulted in no production of HA in the preliminary two-stage fermentation (data not shown). We suspected that the hasA gene with only 40% G+C content was not effectively translated in the high-G+C host organism, S. albulus. We next attempted to express the codon-optimized hasA gene (hasAm) that was designed and synthesized to have >60% G+C content based on Streptomyces codon usage. However, this attempt also failed to yield any HA production (data not shown).
FIG 3.

Proposed HA biosynthetic pathway in S. albulus. The pathway is based on the reported pathway in streptococci (23). To constitute the entire HA pathway in S. albulus, the refactored hyaluronic acid synthase (hasAm) was coexpressed with UDP-glucose dehydrogenase (UdgA), UDP-N-acetylglucosamine pyrophosphorylase (GlmU), and UDP-glucose pyrophosphorylase (GtaB) from S. avermitilis, which are homologous to HasB, HasD, and HasC in streptococci, respectively.
In streptococci, the hasA gene is known to form a gene cluster with several genes involved in precursor sugar biosynthesis (28). The HA biosynthetic gene cluster from S. zooepidemicus consists of the following five genes: hasA, hasB encoding UDP-Glc dehydrogenase, hasC encoding UDP-Glc pyrophosphorylase, hasD encoding UDP-GlcNAc pyrophosphorylase, and hasE encoding phosphoglucoisomerase. Thus, the HA-producing streptococci seem to have evolved the HA biosynthetic gene clusters in order to optimize HA production. We speculated that one or both of the UDP-sugar pathways was insufficient in S. albulus, and thus the reinforced sugar pathways would achieve HA production in our heterologous system. To examine this possibility, we constructed a synthetic HA biosynthesis gene cluster mimicking that of S. zooepidemicus by sequentially coupling the hasAm gene, together with the udgA, glmU, and gtaB genes from Streptomyces avermitilis, which are orthologs of the hasB, hasC, and hasD genes in streptococci, respectively. In the present study, we used two different strategies to reinforce sugar pathways, as follows. In the first approach, the hasAm gene was expressed with the udgA gene to reinforce the UDP-GlcUA pathway (pJHA3), and in the second approach, the hasAm gene was expressed with the udgA, glmU, and gtaB genes to reinforce both the UDP-GlcUA and UDP-GlcNAc pathways (pJHA4). In the expression constructs, all genes forming the gene clusters were designed to be translationally coupled together under the control of Ppls, as shown in Fig. 4. Remarkably, without a rigorous fermentation experiment, the HA production-proficient phenotype of S. albulus CRM003 transformants harboring pJHA3 and pJHA4 was readily apparent, as evidenced by the mucoid colony morphology on an M3G agar plate (data not shown). In small-scale two-stage fermentation, cultures of the two recombinant strains became very viscous due to the HA production. The maximum HA productivity was achieved with the transformant carrying pJHA4, and the amount of HA produced was 3.6 g per liter, which was comparable to the reported yield from S. zooepidemicus (12). Considering the fact that coexpression of the udgA gene already led to high-level production of HA (3.2 g/liter), UDP-GlcUA is the limiting factor. Even when the native hasA gene from S. zooepidemicus was coexpressed with the udgA gene from pJHA5, HA was produced, although its productivity was quite low (0.3 g/liter). These observations demonstrated that HasA was efficiently expressed in heterologous S. albulus through our genetic code refactoring strategy, and the synthetic HA biosynthesis gene clusters successfully constituted precursor pathways to lead to the heterologous production of HA. No other exogenous elements seemed to be required. Some Streptomyces strains are known to produce hyaluronidases, which are capable of breaking down HA polymer chains. S. albulus seemed not to produce such enzymes under the particular conditions used herein, as evidenced by the finding that exogenously administered HA was observed in the resting cell assay (data not shown).
Based on the preliminary studies, two recombinant strains carrying pJHA3 and pJHA4 were selected and subjected to large-scale fermentation studies. During fermentation, the culture broth was collected at various time points, and the amount and molecular weight of the HA produced were measured. Immediately after the pH of the fermentation broth dropped to 4.2, HA production became apparent from the increasing media viscosity. As expected, while the transformant harboring pJHA3 accumulated a significant amount of HA (5.1 g/liter), the transformant expressing both sugar pathways (pJHA4) showed much higher productivity, with a maximum value of 6.2 g/liter (Fig. 5a). An HA heterologously produced in Bacillus subtilis as a host strain is commercially available, although there has been no report on its productivity (8). In general, recombinant strains are recognized to produce a considerably smaller amount of HA than that produced in native streptococci, although a genetically engineered E. coli showed an exceptionally high titer of 3.8 g/liter (9, 10). In spite of the significant numbers of fermentation studies, HA productivity in the 5 to 7 g/liter range seems to the practical maximum because of the extreme viscosity of HA (9). In contrast, the HA production in our recombinant system with pJHA4 was comparable to the reported yields from industrial S. zooepidemicus mutant strains, and was 1.6- to 32-fold higher than those of the existing recombinant systems previously reported (9, 10).
S. albulus produces more than 20 g/liter of ε-PL during a week of fermentation (13). Although ATP is consumed for numerous cellular processes, the fact that one molecule of ATP is required for the formation of a single isopeptide bond in the polymer molecule in ε-PL biosynthesis implies that more than 150 mmol/liter culture of ATP is consumed directly for the production (14). In the HA production in S. albulus CRM003/pJHA4, the ATP consumption calculated from the amount of produced HA based on its biosynthesis showed good agreement with that for the ε-PL production in its parental CR1 strain until drastic media viscosity inhibited oxygen supply, while S. albulus CRM003/pJHA3 showed lower ATP consumption (Fig. 5b). These observations suggested that, without additional expression of the hasE gene coding for phosphoglucoisomerase, almost all of theoretically available ATP in the S. albulus cells was consumed in the UDP-sugar biosynthesis pathways enhanced by the expression of the exogenous udgA, glmU, and the gtaB genes. Thus, our strategy using expression of the hasAm gene, along with the genes for the UDP-GlcUA and UDP-GlcNAc pathways in S. albulus, allowed us to produce HA at a high level.
Enhancement of the UDP-GlcNAc pathway modestly influenced the molecular weight of HA.
There are several available methods for determining the molecular weight of HA (e.g., laser light scattering, intrinsic viscosity, and size exclusion chromatography). Despite the abundance of literature on HA fermentation, size exclusion chromatography (SEC) has frequently been used for the characterization of HA because of its generality, but it has been shown to be inaccurate for molecular weights greater than 2.0 MDa due to the limited pore size of traditional SEC media (12). To provide a more reliable characterization, we used a ligand exchange-SEC column (see Materials and Methods) that shows sufficient resolution for HA with a molecular weight >3.0 MDa (http://www.shodex.com/en/dc/03/06/31.html). Although the use of non-HA polymer standards could also lead to inaccuracy because of the difference in polymer shapes, several HA characteristics estimated with pullulan and other calibration standards have been reported. Therefore, to avoid potential inaccuracies, we used commercially available HA polymer standards for an accurate molecular weight estimation.
As described above, expression of the udgA gene, along with the hasAm gene from pJHA3, successfully allowed us to heterologously produce HA by boosting UDP-GlcUA synthesis, and further coexpression of the gtaB and glmU genes whose protein products are involved in the synthesis of precursor sugars, UDP-GlcUA and UDP-GlcNAc, respectively, led to additional augmentation of the HA production. Since, in streptococci, it has been reported that the UDP-GlcNAc supply has a dominant impact on the molecular weight of HA (11), we expected that increased flux of the UDP-GlcNAc pathway in addition to the UDP-GlcUA pathway would also allow us to produce high-molecular-weight HA. Upon determination of the molecular weight, as expected, we observed a modest but clearly consistent difference between the HA produced by the two recombinant strains (Fig. 6b). Although the molecular weight of the HA produced by the transformant coexpressing both sugar pathways from pJHA4 was consistently found to be equivalent to the high-molecular-weight HA standard (2.2 MDa) that has been the preferred choice for biomedical applications (29), the molecular weight of the HA derived from the partial pathway on pJHA3 appeared to be slightly smaller. We observed consistent chromatograms irrespective of the cultivation time, indicating that the mechanical shear stress caused by agitation had a negligible effect on the molecular weight of HA throughout the fermentation.
FIG 6.
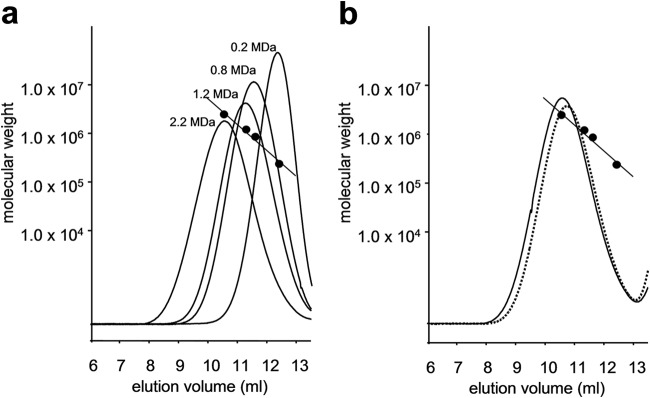
Estimation of molecular weight of HA. SEC elution profiles of HA standards (a) and S. albulus-derived HAs (b) monitored in a differential refractive index detector are shown. The elution profiles of the recombinant HA shown in panel b were from 71-h cultures.
DISCUSSION
S. albulus is a producer strain of ε-PL that has been used as a food preservative in several countries, including Japan, South Korea, China, and the United States. This particular strain has potential for supplying a high level of ATP in vivo after the late-log phase, when ε-PL is produced (15). Under such a condition, our gene expression system utilizing the pls promoter (Ppls) specifically induces expression of cloned genes (15). This expression control would help to avoid the competitive ATP consumption between cell growth and production of desired metabolites. In fact, in the colorimetric test experiment with the refined expression vector pLAE008c, cells grew normally until the stationary phase was reached, and subsequently a significant amount of blue pigment indigoidine was produced (Fig. 1b). Because the gene expression in this genetic system was designed to synchronize with the ATP accumulation (15), no medium optimization was necessary to achieve high levels of production. This feature should be one of the biggest advantages for the heterologous production of desired molecules. Having successfully applied our genetic platform to production of the exogenous molecule indigoidine, we next turned our attention to the production of HA as a more practical case.
Group A and group C streptococci and Pasteurella multocida are known to produce HA naturally (26, 27). It has been reported that HasA from group A and C streptococci is a membrane-associated enzyme that forms pores for the transport of growing HA polymer chains through the membrane (30). In contrast, HasA from Pasteurella multocida seems to require a multicomponent system to transport HA polymer chains to the outside of the cell (31). We used HasA from Gram-positive group C streptococci for heterologous production of HA in the Gram-positive S. albulus. HA with high molecular weight was efficiently produced in S. albulus via a genetic code refactoring strategy combined with assembly of the synthetic pathway, indicating that HasA was, as expected, folded and localized properly into the cytoplasmic membrane and the exogenous UDP-sugar pathways were sufficiently active. In addition, by enhancing both UDP-sugar pathways, ATP utilization for the HA production was successfully maximized (Fig. 5b), which in turn led to the increased HA productivity (Fig. 5a). Thus, high level of ATP availability in S. albulus cells must have facilitated the HA production. With these approaches, we successfully constructed an S. albulus recombinant strain that produced HA in yields comparable to that of the industrial S. zooepidemicus mutant strains. As shown in Fig. 5a, although a significant HA production rate was observed in the early stage of the fermentations, the velocity after 48 h was stalled, likely due to a reduction in the oxygen supply caused by increasing medium viscosity. The agitation appeared to have no effect at those time points, suggesting that a reduction in the oxygen supply caused the reduced turnover of the tricarboxylic acid cycle generating ATP that drives both sugar substrate pathways. Therefore, the HA production in our recombinant system might be further improved with an optimized agitation apparatus that maintains a sufficient level of dissolved oxygen throughout the fermentation.
While the molecular weight of bacterial HA varies from less than 1.0 to 2.0 MDa (9), a range of 2.0 to 2.5 MDa is preferable for intra-articular injection and other biomedical uses (29). The polydisparity and weight-average molecular weight of the S. albulus-derived HA remains controversial, since the comparative SEC analysis used in the present study did not provide this information. However, as apparent from the SEC chromatograms shown in Fig. 6b, the molecular weight of the HA derived from S. albulus CRM003 harboring pJHA4 was equivalent to that of the high-molecular-weight HA standard (2.2 MDa). Although, in general, it is quite challenging to obtain a higher molecular weight HA with satisfactory levels of production because the viscoelastic property of HA highly depends on its molecular weight, an HA compatible with biomedical use could be efficiently produced by the heterologous expression system described here. The genome of S. albulus strains has been sequenced recently (32, 33). From another standpoint, although dozens of potential secondary metabolite gene clusters can be found in the genome, no secondary metabolite other than ε-PL has thus far been reported. This implies that these gene clusters in the genome except one for the ε-PL biosynthesis are silent and thus constitutes a benefit in terms of product quality and safety, which are important parameters in HA biomanufacturing. In addition, unlike streptococcal HA, the S. albulus-derived HA is not cell associated, which is advantageous for the purification process. Given these facts, the heterologous HA production system we show here provides several advantages for practical HA manufacturing.
Taken together, these findings show that our present recombinant system was highly efficient for the production of high-molecular-weight HA via the expression of synthetic HA biosynthetic gene clusters in S. albulus. The present study suggests the possibility that S. albulus could be a superior host for heterologous production of a wide variety of molecules other than HA in the case ATP is required in their biosynthetic cascade. In addition, the genome of this particular strain could provide an opportunity for further genetic engineering in tailor-made molecule biomanufacturing (32, 33). Thus, we propose that this superb biological property of S. albulus in combination with the strict gene expression control by Ppls would open a new avenue for the production of valuable molecules.
ACKNOWLEDGMENTS
This study was supported in part by Kakenhi grants 23108520 and 25660064 (to Y.H.).
T.Y. and K.Y. designed the research. T.Y., N.S., and K.Y. performed the experiments. Y.H. and K.Y. analyzed the data. Y.H. and K.Y. wrote the paper.
REFERENCES
- 1.Weissmann B, Meyer K. 1954. The structure of hyalobiuronic acid and of hyaluronic acid from umbilical cord. J Am Chem Soc 76:1753–1757. [Google Scholar]
- 2.Swann DA, Kuo JW. 1991. Hyaluronic acid, p 286–305. In Byrom D. (ed), Biomaterials: novel materials from biological sources. Stockton Press, New York, NY. [Google Scholar]
- 3.Goa KL, Benfield P. 1994. Hyaluronic acid. A review of its pharmacology and use as a surgical aid in ophthalmology, and its therapeutic potential in joint disease and wound healing. Drugs 47:536–566. [DOI] [PubMed] [Google Scholar]
- 4.Puttick MP, Wade JP, Chalmers A, Connell DG, Rangno KK. 1995. Acute local reactions after intra-articular hylan for osteoarthritis of the knee. J Rheumatol 22:1311–1314. [PubMed] [Google Scholar]
- 5.Ottaviani RA, Wooley P, Song Z, Markel DC. 2007. Inflammatory and immunological responses to hyaluronan preparations: study of a murine biocompatibility model. J Bone Joint Surg Am 89:148–157. doi: 10.2106/JBJS.F.00293. [DOI] [PubMed] [Google Scholar]
- 6.Chong BF, Blank LM, Mclaughlin R, Nielsen LK. 2005. Microbial hyaluronic acid production. Appl Microb Biotechnol 66:341–351. doi: 10.1007/s00253-004-1774-4. [DOI] [PubMed] [Google Scholar]
- 7.Wessels MR, Moses AE, Goldberg JB, DiCesare TJ. 1991. Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proc Natl Acad Sci U S A 88:8317–8321. doi: 10.1073/pnas.88.19.8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Widner B, Behr R, Von Dollen S, Tang M, Heu T, Sloma A, Sternberg D, DeAngelis PL, Weigel PH, Brown S. 2005. Hyaluronic acid production in Bacillus subtilis. Appl Environ Microbiol 71:3747–3752. doi: 10.1128/AEM.71.7.3747-3752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu L, Liu Y, Li J, Du G, Chen J. 2011. Microbial production of hyaluronic acid: current state, challenges, and perspectives. Microb Cell Fact 10:99. doi: 10.1186/1475-2859-10-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao Z, Shin HD, Chen R. 2009. A recombinant E. coli bioprocess for hyaluronan synthesis. Appl Microbiol Biotechnol 84:63–69. doi: 10.1007/s00253-009-1963-2. [DOI] [PubMed] [Google Scholar]
- 11.Chen WY, Marcellin E, Hung J, Nielsen LK. 2009. Hyaluronan molecular weight is controlled by UDP-N-acetylglucosamine concentration in Streptococcus zooepidemicus. J Biol Chem 284:18007–18014. doi: 10.1074/jbc.M109.011999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armstrong DC, Johns MR. 1997. Culture conditions affect the molecular weight properties of hyaluronic acid produced by Streptococcus zooepidemicus. Appl Environ Microbiol 63:2759–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hiraki J, Hatakeyama M, Morita H, Izumi Y. 1998. Improved epsilon-poly-l-lysine production of an S-(2-aminoethyl)-l-cysteine resistant mutant of Streptomyces albulus. Seibutu Kougaku Kaishi 76:487–493. (In Japanese.) [Google Scholar]
- 14.Yamanaka K, Maruyama C, Takagi H, Hamano Y. 2008. ε-Poly-l-lysine dispersity is controlled by a highly unusual nonribosomal peptide synthetase. Nat Chem Biol 4:766–772. doi: 10.1038/nchembio.125. [DOI] [PubMed] [Google Scholar]
- 15.Yamanaka K, Kito N, Imokawa Y, Maruyama C, Utagawa T, Hamano Y. 2010. Mechanism of ε-poly-l-lysine production and accumulation revealed by identification and analysis of an ε-poly-l-lysine-degrading enzyme. Appl Environ Microbiol 76:5669–5675. doi: 10.1128/AEM.00853-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamano Y, Nicchu I, Hoshino Y, Kawai T, Nakamori S, Takagi H. 2005. Development of gene delivery systems for the ε-poly-l-lysine producer, Streptomyces albulus. J Biosci Bioeng 99:636–641. doi: 10.1263/jbb.99.636. [DOI] [PubMed] [Google Scholar]
- 17.Simon R, Priefer U, Pühler A. 1983. A Broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 18.Yamanaka K, Kito N, Kita A, Imokawa Y, Maruyama C, Utagawa T, Hamano Y. 2011. Development of a recombinant ε-poly-l-lysine synthetase expression system to perform mutational analysis. J Biosci Bioeng 111:646–649. doi: 10.1016/j.jbiosc.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 19.Kito N, Maruyama C, Yamanaka K, Imokawa Y, Utagawa T, Hamano Y. 2012. Mutational analysis of the three tandem domains of ε-poly-l-lysine synthetase catalyzing the l-lysine polymerization reaction. J Biosci Bioeng 115:523–526. doi: 10.1016/j.jbiosc.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi H, Kumagai T, Kitani K, Mori M, Matoba Y, Sugiyama M. 2007. Cloning and characterization of a Streptomyces single module type non-ribosomal peptide synthetase catalyzing a blue pigment synthesis. J Biol Chem 282:9073–9081. doi: 10.1074/jbc.M611319200. [DOI] [PubMed] [Google Scholar]
- 21.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical streptomyces genetics. John Innes Foundation, Norwich, United Kingdom. [Google Scholar]
- 22.Itzhaki RF. 1972. Colorimetric method for estimating polylysine and polyarginine. Anal Biochem 50:569–574. doi: 10.1016/0003-2697(72)90067-X. [DOI] [PubMed] [Google Scholar]
- 23.O'Regan M, Martini I, Crescenzi F, De Luca C, Lansing M. 1994. Molecular mechanisms and genetics of hyaluronan biosynthesis. Int J Biol Macromol 16:283–286. doi: 10.1016/0141-8130(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 24.Bitter T, Muir HM. 1962. A modified uronic acid carbazole reaction. Anal Biochem 4:330–334. doi: 10.1016/0003-2697(62)90095-7. [DOI] [PubMed] [Google Scholar]
- 25.Shima S, Sakai H. 1977. Polylysine produced by Streptomyces. Agric Biol Chem 41:1807–1809. doi: 10.1271/bbb1961.41.1807. [DOI] [Google Scholar]
- 26.MacLennan AP. 1956. The production of capsules, hyaluronic acid and hyaluronidase by group A and group C streptococci. J Gen Microbiol 14:134–142. doi: 10.1099/00221287-14-1-134. [DOI] [PubMed] [Google Scholar]
- 27.Carter GR, Annau E. 1953. Isolation of capsular polysaccharides from colonial variants of Pasteurella multocida. Am J Vet Res 14:475–478. [PubMed] [Google Scholar]
- 28.Blank LM, Hugenholtz P, Nielsen LK. 2008. Evolution of the hyaluronic acid synthesis (has) operon in Streptococcus zooepidemicus and other pathogenic streptococci. J Mol Evol 67:13–22. doi: 10.1007/s00239-008-9117-1. [DOI] [PubMed] [Google Scholar]
- 29.Adam N, Ghosh P. 2001. Hyaluronan molecular weight and polydispersity in some commercial intra-articular injectable preparations and in synovial fluid. Inflamm Res 50:294–299. doi: 10.1007/PL00000247. [DOI] [PubMed] [Google Scholar]
- 30.Heldermon C, DeAngelis PL, Weigel PH. 2001. Topological organization of the hyaluronan synthase from Streptococcus pyogenes. J Biol Chem 276:2037–2046. doi: 10.1074/jbc.M002276200. [DOI] [PubMed] [Google Scholar]
- 31.Townsend KM, Boyce JD, Chung JY, Frost AJ, Adler B. 2001. Genetic organization of Pasteurella multocida cap loci and development of a multiplex capsular PCR typing system. J Clin Microbiol 39:924–929. doi: 10.1128/JCM.39.3.924-929.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dodd A, Swanevelder D, Featherston J, Rumbold K. 2013. Draft genome sequence of Streptomyces albulus strain CCRC 11814, an ε-poly-l-lysine-producing actinomycete. Genome Announc 1:e00696-13. doi: 10.1128/genomeA.00696-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Z, Xia J, Feng X, Li S, Xu H, Bo F, Sun Z. 2014. Genome sequence of Streptomyces albulus PD-1, a productive strain for epsilon-poly-l-lysine and poly-l-diaminopropionic acid. Genome Announc 2:e00297-14. doi: 10.1128/genomeA.00297-14. [DOI] [PMC free article] [PubMed] [Google Scholar]