

Abstract
Cutinases have shown potential for hydrolysis of the recalcitrant synthetic polymer polyethylene terephthalate (PET). We have shown previously that the rate of this hydrolysis can be enhanced by the addition of hydrophobins, small fungal proteins that can alter the physicochemical properties of surfaces. Here we have investigated whether the PET-hydrolyzing activity of a bacterial cutinase from Thermobifida cellulosilytica (Thc_Cut1) would be further enhanced by fusion to one of three Trichoderma hydrophobins, i.e., the class II hydrophobins HFB4 and HFB7 and the pseudo-class I hydrophobin HFB9b. The fusion enzymes exhibited decreased kcat values on soluble substrates (p-nitrophenyl acetate and p-nitrophenyl butyrate) and strongly decreased the hydrophilicity of glass but caused only small changes in the hydrophobicity of PET. When the enzyme was fused to HFB4 or HFB7, the hydrolysis of PET was enhanced >16-fold over the level with the free enzyme, while a mixture of the enzyme and the hydrophobins led only to a 4-fold increase at most. Fusion with the non-class II hydrophobin HFB9b did not increase the rate of hydrolysis over that of the enzyme-hydrophobin mixture, but HFB9b performed best when PET was preincubated with the hydrophobins before enzyme treatment. The pattern of hydrolysis by the fusion enzymes differed from that of Thc_Cut1 as the concentration of the product mono(2-hydroxyethyl) terephthalate relative to that of the main product, terephthalic acid, increased. Small-angle X-ray scattering (SAXS) analysis revealed an increased scattering contrast of the fusion proteins over that of the free proteins, suggesting a change in conformation or enhanced protein aggregation. Our data show that the level of hydrolysis of PET by cutinase can be significantly increased by fusion to hydrophobins. The data further suggest that this likely involves binding of the hydrophobins to the cutinase and changes in the conformation of its active center.
INTRODUCTION
Polyethylene terephthalate (PET) is the best-known and most widespread synthetic polyester in the world. It is used for foils and bottles as well as for fibers for the textile industry (1). Recycling of PET and modification of its properties for different applications by traditional procedures involve harsh chemical and physicochemical treatments (2, 3). Enzymatic modification, particularly by cutinases, has been recognized as a powerful alternative in the past decade (4, 5) and, besides offering new avenues for PET recycling, has the additional advantage of creating a modified PET with increased dyeing efficacy and improved binding to polyvinyl chloride without altering the polymer's bulk properties (4, 5).
On the other hand, enzymatic hydrolysis of PET has the inherent disadvantage of occurring at a very low rate (5, 6). The reasons for this are not yet clearly understood. The access of the active center of the cutinase to the insoluble substrate is apparently one of the rate-limiting points, because enlarging the areas around the active sites of cutinases from Fusarium solani (Ascomycota, Fungi) and Thermobifida fusca (Actinobacteria, Bacteria) increased the rates of hydrolysis of PET and its oligomers (7, 8). In addition, the binding of the cutinase to the surface of PET also seems to limit the rate of hydrolysis (8). Many enzymes that act on insoluble substrates therefore contain protein domains for substrate binding. Such protein domains are well known for such enzymes as cellulases, hemicellulases, and chitinases that bind to insoluble carbohydrates (9, 10) but are also present on polyester hydrolases (11). In agreement with a positive role for surface binding modules, the fusion of the polyhydroxyalkanoate (PHA) binding module from Alcaligenes faecalis (Proteobacteria, Bacteria) to a cutinase from Thermobifida cellulosilytica was indeed able to increase the rate of PET hydrolysis (12).
We previously introduced yet another way to stimulate PET hydrolysis by cutinases, i.e., the addition of hydrophobins (13). These are small cysteine-rich proteins of exclusively fungal origin that can naturally adsorb to hydrophobic surfaces and to interfaces between hydrophobic (air, oil, and wax) and hydrophilic (water and cell wall) phases (14–16). Their addition stimulated the hydrolysis of PET by a Humicola insolens cutinase (13). The mechanism by which the hydrophobins stimulate the enzymatic activity of cutinases on PET is essentially unknown, however, and could involve the creation of a more hydrophilic surface, the binding and targeting of the cutinases to PET, or even the direct modulation of their activity.
The objective of this paper was to study the influence of hydrophobins on PET-hydrolyzing cutinase by comparing the effects of free hydrophobins to those of hydrophobins that are genetically fused to a cutinase.
MATERIALS AND METHODS
Chemicals and reagents.
Polyethylene terephthalate (PET) films were completely amorphous and were purchased from Goodfellow (United Kingdom). If not indicated differently, all chemicals were of analytical grade and were purchased from Sigma-Aldrich.
Microbial strains and plasmids.
Trichoderma reesei QM6a, T. virens Gv29-8, and T. harzianum CBS 226.95 were obtained from the Collection of Industrial Microorganisms of the Vienna University of Technology (TUCIM) and were used as sources of the hydrophobin genes used in this study. These strains were maintained and cultivated as described by Espino-Rammer et al. (13).
Escherichia coli Stellar (TaKaRa Bio Company, CA, USA) and E. coli XL-1 (Stratagene, Germany) competent cells were used for plasmid DNA maintenance. Vector pET-26b(+) (Novagen, Germany) was used for the expression of hydrophobins and fusion proteins in E. coli strain BL21-Gold(DE3) (GE Healthcare, Amersham, England). Stellar competent cells from E. coli HST04 were purchased from Clontech (TaKaRa Bio Company, CA, USA) and were used for the cloning of hydrophobin-encoding genes and the propagation of plasmid DNA. E. coli BL21 (protease deficient) and expression plasmid pGEX-4T-2 were purchased from GE Healthcare (Amersham, England). The strains were cultivated on Luria broth (LB) agar plates at 28°C.
General recombinant DNA techniques.
Molecular cloning of the genes was performed by standard methods (17). DNA digestion with restriction endonucleases (New England BioLabs, USA), dephosphorylation with alkaline phosphatase (Roche, Germany), and ligation with T4 DNA ligase (Fermentas, Germany) were performed according to the manufacturers' instructions. A plasmid minikit from Qiagen (Germany) was used to prepare plasmid DNA. Plasmids and DNA fragments were purified with Qiagen DNA purification kits (Qiagen, Germany).
Overexpression and purification of the free hydrophobins HFB4, HFB7, and HFB9b.
The primers listed in Table 1 were used to amplify the cDNA fragments of the class II hydrophobin-encoding genes hfb4 from T. reesei (GenBank accession no. XP_006964739.1) and hfb7 from T. harzianum (accession no. KP209450) as well as that of the pseudo-class I hydrophobin gene hfb9b from T. virens (accession no. EHK16817) The cDNA fragments were then fused between an N-terminal pelB leader in vector pET-26b(+) for direction of the proteins to the bacterial periplasm and a C-terminal 6×His tag for purification by affinity chromatography.
TABLE 1.
Oligonucleotide primers used for cloning of Trichoderma hydrophobins
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| hfb9b | CAACAACAACTGGCAGAGCAAC | GTAAACGACCTTGGACTGTCCG |
| hfb4 | CGCTGGGGAGACGACTACTCA | AAGAGCATCCTGGCACAAAAC |
| hfb7 | TGCTGGCGATGAAGGTTG | GGTTCCGACGGGCTTCTC |
Overexpression was performed in E. coli strain BL21-Gold(DE3). After E. coli was transformed by electroporation, it was cultivated in LB containing 40 μg/ml kanamycin at 37°C and was agitated at 170 rpm. Expression was induced by the addition of isothiopropyl-β-d-galactoside (IPTG) at a final concentration of 0.05 mM, and the cultures were then incubated for 5 h. Then the cells were harvested by centrifugation (5,000 rpm, 4°C, 10 min) and were lysed by ultrasonication (10 cycles; 30-s pulse with 1 min on ice between pulses). The supernatant was removed by centrifugation at 5,000 × g for 30 min at 4°C, and the pellet (which contained the inclusion bodies with the hydrophobins) was washed twice with the equilibration buffer (50 mM sodium phosphate, 300 mM sodium chloride [pH 7.0]) containing 2 M urea. After further centrifugation (5,000 × g, 10 min, 4°C), the pellet containing the inclusion bodies was solubilized in the equilibration buffer containing 8 M urea and was loaded onto 2 ml of Co-charged affinity resin (Talon metal affinity resin; TaKaRa Bio Company, CA, USA). Purification and on-column refolding were carried out essentially according to the manufacturer's instructions. The final protein elution was performed using 300 mM imidazole-HCl buffer (pH 7.0). PD-10 desalting columns (GE Healthcare, Amersham, England) were used to exchange the buffer to 100 mM K2HPO4–KH2PO4 buffer at pH 7.
Cloning, expression, and purification of fusion proteins.
The genes coding for Thc_Cut_hfb4, Thc_Cut_hfb7, and Thc_Cut_hfb9b were codon optimized for expression in E. coli and were synthesized by GeneArt (Invitrogen, Germany). The fusion proteins were constructed by connecting the hydrophobins over the linker region (P263 to P287) of cellobiohydrolase I from T. reesei (GenBank accession no. P62694.1 [18]) to Thc_Cut1 from Thermobifida cellulosilytica (GenBank accession no. ADV92526.1 [8]). The synthetic genes were cloned over the NdeI and HindIII restriction sites into pET26b(+), allowing expression of the enzymes without the pelB signal peptide. All fusion enzymes carried a C-terminal 6×His tag for purification by affinity chromatography.
Freshly transformed E. coli BL21-Gold(DE3) cells were used to inoculate 20 ml LB medium supplemented with 40 mg/ml kanamycin and were cultivated overnight at 37°C and 160 rpm. Then the overnight culture was used to inoculate 200 ml LB medium containing 40 mg/ml kanamycin. The culture was incubated until an optical density at 600 nm (OD600) of 0.6 to 0.8 was reached. After cooling to 20°C, the culture was induced by the addition of IPTG to a final concentration of 0.05 mM. After induction for 20 h at 25°C (this temperature was chosen to avoid the formation of inclusion bodies) and 160 rpm, the cells were harvested by centrifugation (25 min, 10°C, 4,000 rpm). Cell pellets from 100 ml cell culture were resuspended in 10 ml Ni-nitrilotriacetic acid (NTA) lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole [pH 8]). The resuspended cells were ultrasonified (Vibra-Cell; Sonics & Materials, Meyrin-Satigny, Switzerland) with three 30-s pulses with cooling on ice. The lysates were centrifuged (30 min, 10°C, 4,000 rpm) and were purified by affinity chromatography on Ni-NTA-Sepharose according to the manufacturer's protocol (IBA GmbH, Göttingen, Germany). Purity was confirmed by SDS-PAGE, and only preparations that yielded a single band were used for further analysis.
Assay of esterase activity.
Esterase activity was measured by using p-nitrophenyl acetate (PNPA) and p-nitrophenyl butyrate (PNPB) as substrates as described previously (19). Kinetic parameters were determined at 25°C and pH 7.0 with substrate concentrations in the range of 0.05 to 10 mM. Kinetic data were calculated with SigmaPlot, version 11.0, and Enzyme Kinetics, version 1.3 (both from Systat Software GmbH, USA), using the Michaelis-Menten equation.
Hydrolysis of PET films.
Prior to PET hydrolysis, all the films were washed in three consecutive steps: first in a solution of Triton X-100 (5 g/liter), then with 100 mM Na2CO3, and finally with deionized water. Each step lasted 30 min. Hydrolysis was performed by incubation of 1.0- by 0.5-cm strips of PET films with 5 μM enzyme or enzyme fusion protein in 2 ml 0.1 M potassium phosphate buffer, pH 7.0, in Eppendorf tubes at 50°C and 100 rpm. After different intervals, as indicated in the legend to Fig. 4, the samples were diluted with an equal volume of absolute methanol on ice. The products released—terephthalic acid (TA) and mono(2-hydroxyethyl) terephthalate (MHET)—were analyzed via reverse-phase high-performance liquid chromatography as described previously (8, 13).
FIG 4.
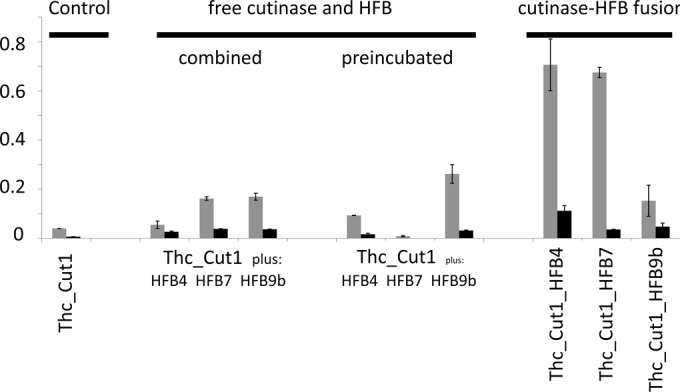
PET hydrolysis by Thc_Cut1 in the presence of hydrophobins or by Thc_Cut1 fused to hydrophobins. The release of products (TA [shaded bars] or MHET [filled bars]) after 24 h of incubation is shown. Equal concentrations of Thc_Cut1 were used in all experiments. Product release was linear versus time until at least this time point. Error bars indicate standard deviations (n ≥ 3).
In experiments where PET was first incubated with the hydrophobins, it was incubated in a 5 μM hydrophobin solution in 0.1 M potassium phosphate buffer (pH 7.0) at 50°C for 3 h. Thereafter, the PET film was transferred to a new Eppendorf tube, in which it was incubated with Thc_Cut1 under the same conditions as those for the native enzyme.
Analysis of surface activities of hydrophobins and hydrophobin fusion proteins.
PET films and glass pieces (1.5 by 0.7 cm) were defatted with 70% (wt/vol) ethanol. Thereafter, samples were washed first with 5 g of Triton X-100 per liter, then with 100 mM Na2CO3, and finally with deionized water (each step was carried out for 30 min at 50°C); the samples were then incubated with a Tris-HCl buffer (0.1 M; pH 7) containing a final concentration of 50 μg/ml of the hydrophobin or cutinase-hydrophobin protein/ml. After 12 h of incubation at 50°C, the PET films and glass pieces were washed twice with water and were dried for 10 min at 50°C. Water contact angles (WCAs) were determined in a drop shape analysis system (DSA100; Kruss GmbH, Hamburg, Germany) using deionized water as the test liquid with a drop volume of 2 μl. Water droplets were set on the surface of the PET film or glass piece, and the contact angles were determined after 3 s.
SAXS analysis.
All measurements were carried out at the Austrian small-angle X-ray scattering (SAXS) beamline at the Elettra Sincrotrone Trieste. The X-ray energy was 8.05099 keV. The beam was optimized with an aperture and guard slit for measurements with 1.5-mm glass capillaries. A 2-dimensional (2D) Pilatus 3 1M detector system was used. The sample-to-detector distance was 999.3 mm.
The hydrophobins, cutinase, and fusion proteins were put in capillaries with a diameter of 1.5 mm and a wall thickness of 10 μm (Hilgenberg GmbH, Malsfeld, Germany).
For each sample, at least 12 successive scattering images were recorded, each for 10 s, to check on possible radiation damage. The scattering images were integrated to obtain the intensity with respect to the scattering vector [I(Q)] and were background corrected before further evaluation.
The scattering intensities were normalized to each other in the Q range between 2.5 and 3 nm−1. In this region, the scattering intensity is flat and is dominated mainly by fluid scattering and an additional, smaller contribution from the glass capillary. The scattering intensity from the solution without protein was subtracted from that of the solution with protein. The consistency of this was cross-checked with the measured transmission of each sample.
The scattering intensity [I(Q)] was split into the form factor [P(Q)] and the apparent structure factor [S(Q)]. The protein form factor is the Fourier transform of the pair correlation of the crystallographic data of cutinase. The pair correlation was computed by taking into account all sites as possible sources of scattering (20–22).
LC-MS.
Liquid chromatography-tandem mass spectrometry (LC-MS) was performed in a Thermo Orbitrap Velos Pro mass spectrometer in positive-ion mode by alternating full-scan MS (m/z 380 to 2000). Protein bands were excised from gels and were chymotryptically cleaved according to the manufacturer′s instructions (Roche, Vienna, Austria). Peptide extracts were dissolved in 0.3% formic acid, 5% acetonitrile, and were separated by nano-HPLC on a Dionex UltiMate 3000 system equipped with a μ-precolumn (C18; particle size, 5 μm; pore size, 100 Å; length, 5 mm; inside diameter, 0.3 mm) and an Acclaim PepMap RSLC nanocolumn (C18; particle size, 2 μm; pore size, 100 Å; length, 150 mm; inside diameter, 0.075 mm) (all from Thermo Fisher Scientific, Vienna, Austria).
Biochemical techniques.
Protein concentrations were determined by using the Bio-Rad protein assay (Bio-Rad, CA, USA) with bovine serum albumin as a standard. The purified proteins were analyzed by SDS-PAGE, and staining was performed with Coomassie Brilliant Blue R-250 (23).
RESULTS
Properties of Trichoderma HFB4, HFB7, and HFB9b.
The hydrophobins HFB4 and HFB7 have been investigated previously (13) by using a glutathione S-transferase (GST) fusion, which enhances their solubility. In order to enable a comparison of their properties with those of hydrophobins fused to cutinase, we overproduced them here without a fusion but containing a C-terminal His tag to facilitate rapid purification. In addition, we included HFB9b (GenBank accession no. EHK16817) in this study, because it represents a pseudo-class I hydrophobin (24), and we were interested in whether it would show the same properties as those of class II members HFB4 and HFB7 or different properties.
The surface-modulating activities of HFB4, HFB7, and HFB9b are shown in Table 2; in agreement with previous data using GST-fused hydrophobins, free HFB4 and HFB7 raised the hydrophobicity of glass considerably, and HFB4 and HFB9b resulted in a less pronounced but nevertheless clear increase in the hydrophilicity of PET. Interestingly, the free pseudo-class I hydrophobin HFB9b had the most pronounced effect on PET but was almost inactive on glass.
TABLE 2.
Changes in the water contact angle (WCA) upon incubation of glass or PET with free or fused hydrophobin proteins
| Hydrophobin | WCA (°) |
|||
|---|---|---|---|---|
| Glass |
PET |
|||
| Free hydrophobin | Hydrophobin fused to Thc_Cut1 | Free hydrophobin | Hydrophobin fused to Thc_Cut1 | |
| Blank | 12.7 (±0.5) | 0 | 70.4 (±0.3) | 66.1 |
| HFB4 | 67.7 (±0.4) | 61 (±9.6) | 63.4 (±0.3) | 68.2 (±2.3) |
| HFB7 | 72.5 (±1.6) | 47.5 (±6.4) | 69.3 (±0.3) | 59.2 (±4.0) |
| HFB9b | 13.3 (±5.4) | 28.7 (±14.1) | 60.1 (±3.6) | 67.2 (±2.1) |
Expression of cutinase-hydrophobin fusion proteins in E. coli.
The hydrophobins HFB4, HFB7, and HFB9b were C-terminally fused over the linker region of cellobiohydrolase I from T. reesei to the cutinase Thc_Cut1 from Thermobifida cellulosilytica. At different time points after induction, expression levels were examined by SDS-PAGE analysis of cleared cell lysates and cell pellets. Figure 1 shows the SDS-PAGE analysis of Thc_Cut1_hfb4, which represents the typical expression behavior of all fusion proteins. Expression of Thc_Cut1_hfb4 resulted in a strong protein band around 40 kDa in the soluble cell fraction after 5 h of induction (Fig. 1, lane 2), which corresponded well to the calculated mass of 38.7 kDa. Interestingly, this protein band disappeared with extended induction times, whereas a new protein band around 33 kDa emerged simultaneously (Fig. 1, lane 3). After 21 h of induction, only the smaller protein band was present (Fig. 1, lane 4). MS analysis of the dominant protein band in this lane revealed that it represented the fusion protein with only a truncated part of HFB4 (data not shown). After 10 h and 21 h of induction (Fig. 1, lanes 5 and 6), the fusion proteins were also directed into inclusion bodies. Consequently, the fusion proteins were expressed for only 5 h and were subsequently purified by affinity chromatography for further use.
FIG 1.
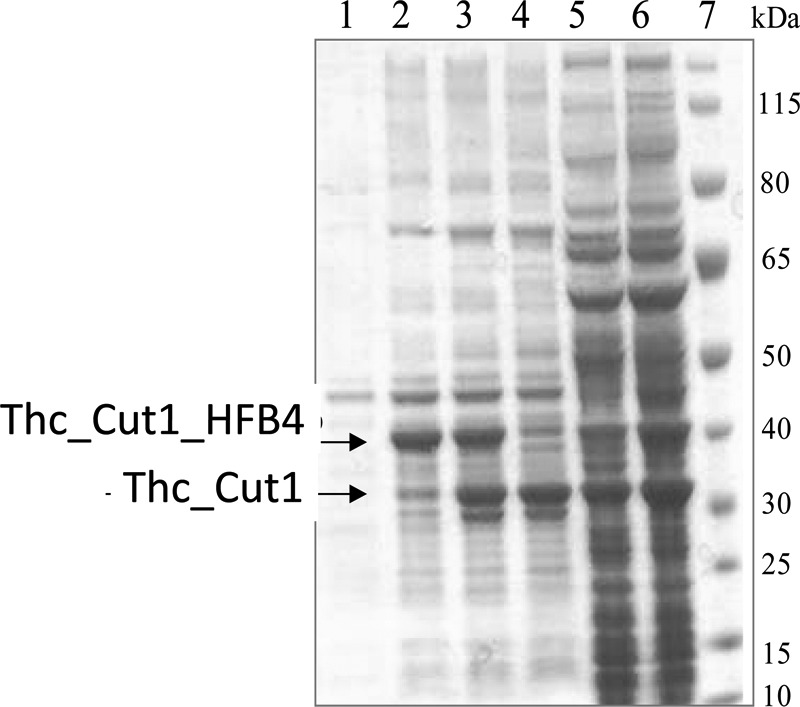
Expression of Thc_Cut1_hfb4 in E. coli BL21-Gold(DE3) at 25°C. Lane 1, uninduced cells; lanes 2 to 4, soluble cell fractions after 5 h, 10 h, and 21 h of induction; lanes 5 and 6, insoluble cell fractions after 10 h and 21 h of induction; lane 7, standard (PageRuler prestained protein ladder). Molecular masses are given on the right.
Biochemical properties of the cutinase-hydrophobin fusion proteins.
In order to learn whether fusion to any of the three hydrophobins had altered the biochemical properties of the cutinase, we first calculated the impact of these fusions on the isoelectric point (pI) by using ProtParam (25): the cutinase itself has a predicted pI of 6.3, and a similar value was obtained for the Thc_Cut1_hfb4 fusion protein. In contrast, the theoretical pIs of the Thc_Cut1_hfb7 and Thc_Cut1_hfb9b fusion proteins were elevated to 8.0 and 7.4, respectively. This would imply that the solubility of Thc_Cut1_hfb7 at our working pH of 7.0 is lowest and that Thc_Cut1_hfb4 should behave as an anion, whereas Thc_Cut1_hfb9b should behave as a cation.
To test whether the fusion proteins consisting of cutinases and hydrophobins had retained the biological activities of the respective fusion partners, we tested the enzymatic activity of the fused cutinase using the soluble model substrates p-nitrophenyl acetate (PNPA) and p-nitrophenyl butyrate (PNPB). The kinetic parameters thus obtained are given in Table 3. In general, fusion to hydrophobins resulted in a small increase in Km values with HFB4 and HFB7 but not with HFB9b, whereas the enzyme turnover number kcat was dramatically decreased for all three fusion proteins. Since fusion to the hydrophobins increases the molecular weight of the cutinase only by about 25%, this reduction in kcat cannot be explained only by the increase in the molecular weight. Consequently, the overall enzyme efficiency (kcat/Km) was significantly lower, too.
TABLE 3.
Kinetic parameters of cutinase from T. cellulosilytica fused to hydrophobins from Trichoderma on soluble substrates
| Cutinase | PNPA |
PNPB |
||||
|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) | Km (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) | |
| Thc_Cut1 | 1.4 ± 0.1 | 363 | 291 | 0.8 ± 0.1 | 325 | 406 |
| Thc_Cut1_hfb4 | 2.4 ± 0.2 | 99 | 42 | 1.3 ± 0.1 | 86 | 68 |
| Thc_Cut1_hfb7 | 2.3 ± 0.2 | 129 | 55 | 0.9 ± 0.1 | 112 | 130 |
| Thc_Cut1_hfb9b | 1.5 ± 0.1 | 105 | 71 | 0.8 ± 0.1 | 49 | 62 |
Surface-modulating activities of the cutinase-hydrophobin fusion proteins.
To test whether the three fused hydrophobins had retained their biological activities, we compared their abilities to modify the surface hydrophilicity or hydrophobicity, using glass or PET as the solid phase, with those of the free hydrophobins. As shown in Table 2, HFB4 and HFB7 rendered the hydrophilic surface of glass more hydrophobic, whereas HFB9b was almost inactive in this respect. When fused to cutinase, HFB4 retained this property and HFB7 also resulted in increased hydrophobicity, albeit to a smaller degree than free HFB7. Interestingly, the fusion protein containing HFB9b also caused a small increase in the hydrophobicity of glass. The ability to render PET more hydrophilic was poor with free HFB4 and HFB9b, and HFB7 was practically inactive. Interestingly, this low activity of HFB4 and HFB9b was completely lost when the hydrophobin was fused to the cutinase, whereas the fusion protein with HFB7 exhibited a small increase in hydrophilicity.
Behavior of the hydrophobin-cutinase fusion proteins in solution.
We used small-angle X-ray scattering (SAXS) analysis to determine whether the fusion of the hydrophobins to cutinase either alters the 3D protein structure, leads to aggregation, or both. HFB4 and HFB9b were used for this analysis. The hypothetical form factor calculated from the crystallographic data of cutinase (Fig. 2 and 3, bold blue lines) served as a reference. Figure 2 shows the result for free HFB4, alone or in combination with cutinase, and for the cutinase-hydrophobin fusion protein. HFB4 alone exhibited the lowest scattering contrast, which was only slightly, yet significantly, above zero. Similarly low scattering was observed for HFB7 (data not shown) and HFB9b (see below). On the other hand, analysis of a mixture of HFB4 and cutinase showed a higher scattering contrast, suggesting binding of the two proteins. A still higher scattering contrast, however, was obtained for the Thc_Cut1_hfb4 fusion protein. The stronger scattering may be due partially to the larger size of the fusion protein (38.7 versus 28 kDa) but may instead indicate a change in protein conformation or enhanced aggregation.
FIG 2.
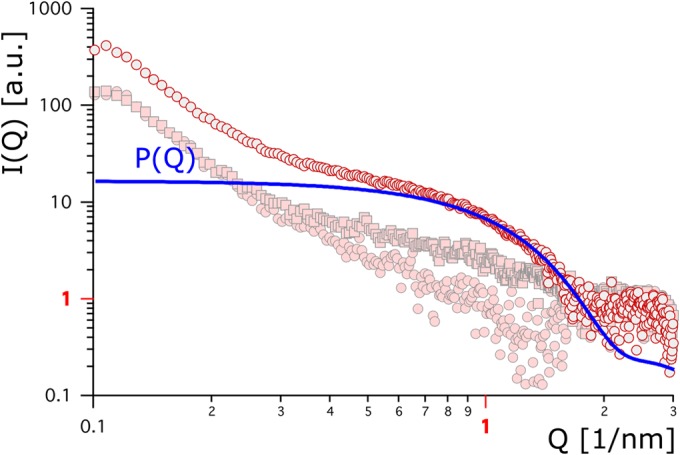
Background-corrected scattering data for Thc_Cut1_hfb4 (gray circles outlined in red), Thc_Cut1 plus HFB4 (pink squares outlined in gray), and HFB4 (pink circles outlined in gray) are plotted against the scattering vector. The bold blue line refers to a hypothetical protein form factor.
FIG 3.
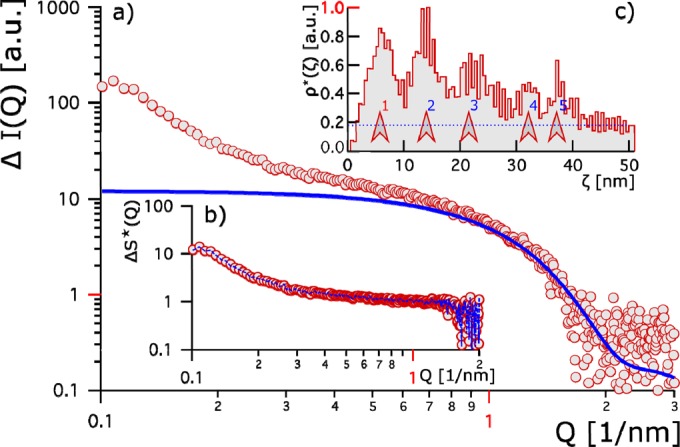
(a) Red circles show the relative change in the scattering contrast of Thc_Cut1 plus HFB4 from that of Thc_Cut1 plus HFB9. The bold blue line represents the hypothetical Thc_Cut1 form factor. (b) Changes in the apparent structure factor between the two systems. Red circles indicate the apparent structure factor, and the blue line gives the fit thereto. (c) The apparent pair correlation [ρ*(ζ)] is given as a function of the relative distance ζ. It is normalized to 1. Five peaks (marked by red arrows) are identified and give evidence for stronger clustering of Thc_Cut1 plus HFB9b than of Thc_Cut1 plus HFB4. The first peak (1 [red]) evolves at a distance equivalent to a protein size of 5 nm; blue-numbered peaks 2 to 5 indicate longer-range interactions and hint of stronger aggregation.
In order to distinguish between the two possibilities—i.e., that the protein fusion leads to a conformational change or to aggregation—we compared the scattering contrast of cutinase to which HFB4 had been added with that of cutinase to which HFB9b had been added (Fig. 3). For this purpose, we assumed identical form factors [P(Q) = P1(Q) = P2(Q), where subscript numbers 1 and 2 correspond to HFB4 and HFB9b, respectively]. We computed the form factors from crystallographic data (Fig. 3a, blue line) and accessed the change in the apparent structure factor [ΔS*(Q)] between the two systems (Fig. 3b). This enabled us to determine the change in the apparent pair correlation of the two systems (Fig. 3c). The characteristic slope in the relative change of scattering contrast at small Q values revealed stronger aggregation of cutinase with HFB9b than with HFB4 (Fig. 3c). The aggregates with HFB9b had a characteristic size of 35 to 40 nm.
Comparison of the effects of free and covalently bound hydrophobins on PET hydrolysis by cutinase.
As a prerequisite for studying the respective cutinase-hydrophobin fusion proteins used in this study, we tested whether HFB4, HFB7, and HFB9b, expressed without GST fusion, were also capable of stimulating the hydrolysis of PET by cutinase 1 from T. cellulosilytica. As shown in Fig. 4, each of the three hydrophobins was able to stimulate PET hydrolysis when added simultaneously with cutinase; HFB7 and HFB9b were superior to HFB4. It was noteworthy that the release of MHET was more strongly stimulated than that of TA. Interestingly, this picture changed when PET was preincubated with the hydrophobins: this increased the stimulating effects of HFB4 and HFB9b, whereas that of HFB7 was reduced to almost zero. Under these conditions, the ratio of TA to MHET was similar to that obtained with the cutinase in the absence of hydrophobins. Washing the PET films after incubation with the hydrophobins did not yield different results (data not shown), indicating strong binding of HFBs to PET.
When similar experiments were performed with the cutinase-hydrophobin fusion proteins, fusion with HFB4 or HFB7 led to a >16-fold-higher rate of hydrolysis product release from PET (as seen by the values for 24 h), and the TA/MHET was lowest for the fusion to HFB4 (Fig. 4). In contrast, fusion to HFB9b resulted in a level of stimulation comparable to that achieved by the simultaneous addition of cutinase and HFB9b, and this level of stimulation was even lower than that achieved when PET was preincubated with HFB9b before the addition of cutinase, again indicating the different behavior of this pseudo-class I hydrophobin.
DISCUSSION
In this paper, we have investigated whether the fusion of hydrophobins to a PET-hydrolyzing cutinase would enhance the activity of the enzyme against PET. The rationale for this was the hypothesis that the previously observed stimulation by hydrophobins of PET degradation by cutinase (13) could be due to targeting of the cutinases to the surface of PET. Consequently, fusion of the cutinase with the hydrophobin should result in an increase in the number of cutinase molecules per PET surface unit and should thus increase the rate of hydrolysis, as has recently been documented for a cutinase fused to the polyhydroxyalkanoate (PHA)-binding module from A. faecalis (12). Indeed, the present data generally verified this hypothesis, since the fusion of two of the three hydrophobins investigated (HFB4 and HFB7) to the cutinase resulted in a dramatic (16-fold) increase in the rate of PET hydrolysis over that shown by the enzyme alone or by the cutinase coincubated with a hydrophobin. However, the stimulatory effects of the individual hydrophobins differed depending on whether they were incubated in combination with cutinase, preincubated with PET, or fused to the cutinase. For example, HFB9b, which was most stimulatory when added separately to the cutinase, showed no further stimulation as part of a fusion protein and even stayed below the level obtained by preincubation of PET with the hydrophobin. In addition, although fusion with HFB7 resulted in excellent stimulation of the cutinase, preincubation of HFB7 with PET almost completely eliminated cutinase activity, suggesting that the interaction of HFB7 with the cutinase must be different from that of HFB4. Thus, it seems that the positive impact of hydrophobins on cutinase activity against PET is not a monocausal effect but is due to several interacting events.
In theory, the stimulation of cutinase activity by soluble hydrophobins could be due either to an interaction with the surface, to binding to the cutinase, or to a mixture of both. We note that the presence of the 6×His tag could affect the way in which hydrophobins interact with proteins when bound to surfaces, but this effect should not be responsible for the differences seen here, because all preparations contained the 6×His tag. Further, fusion of the hydrophobins to cutinase introduces several additional levels of influence: as we have shown here, the fusion proteins by themselves exhibit different isoelectric points, and thus, their hydration is different and likely results in different solubilities, but it is not clear whether this impacts other properties. Their charges at the pH of hydrolysis will also be different. In addition, we have also shown that the catalytic efficacy of the cutinase against soluble substrates is significantly decreased in the fusion proteins, implying that the function (and likely the stereogeometry) of the active center is altered, too, and this also fits the findings that the fusion proteins formed a higher ratio of MHET to TA in the course of hydrolysis. These effects on soluble substrates and on the differences in the PET release product ratio were also observed in our previous study, in which two binding domains of different natures were fused to Thc_Cut1 (12).
In order to interpret these findings, it is necessary to consider the structure of cutinase. The structure of cutinase from T. cellulosilytica has not yet been determined, but in silico analysis confirms that it also has the typical α/β hydrolase fold and that its amino acid sequence shows the catalytic triad (S-D-H) and an associated oxyanion hole (26). In contrast to lipases, which undergo significant conformational changes, including the movement of a lid over the active site to expose a hydrophobic substrate binding pocket, the active site of a cutinase with the preformed oxyanion hole is permanently exposed to the surface and is protected only by two loops close to the active site, which are moved away upon binding to the substrate (6, 27). It is commonly believed that this topical arrangement of the active center enables the binding of, and reaction with, very large substrates. It might therefore be speculated that a conformational shift that results in a yet wider opening of the active center could increase the activity against insoluble substrates such as PET, whereas the affinity and turnover number for small soluble substrates would be decreased. In fact, engineering of the active center of the cutinase from F. solani f. sp. pisi for the accommodation of larger substrate molecules enhanced activity on PET (7, 28). Since the SAXS analysis also supports the occurrence of a conformational shift, we speculate that one of the reasons for the stimulation of cutinase activity by covalently bound hydrophobins could be an alteration in the conformation of the cutinase active center. However, more-detailed structural analyses would be needed to support this hypothesis.
It is well known that hydrophobins can self-assemble into a monolayer on hydrophobic surfaces with their hydrophobic patch binding to the surface and exposing the hydrophilic side outward, thus reversing the hydrophobicity of the surface. However, the mechanisms for the assembly of hydrophobins on hydrophilic surfaces are not yet fully understood (29). It was therefore rational to assume that the stimulatory effect of hydrophobins on PET hydrolysis by cutinase is due to the creation of a hydrophobic surface that targets the enzyme to its substrate. However, as has been clearly shown, the cutinases bind directly to the hydrophobic surfaces of their substrates (6, 27). Also, as we have shown in this paper, the three hydrophobins tested hardly changed the hydrophobicity of PET. On the other hand, the experiments in which PET was preincubated with hydrophobins clearly showed that they must bind to PET. An exception was HFB7, where preincubation failed to show any stimulation of hydrolysis by the cutinase, and it is possible that HFB7 indeed does not bind to PET. A refined explanation for the stimulatory effect of hydrophobins on PET hydrolysis by cutinase may be offered by the recent data of Peng et al. (30), who showed that the T. reesei hydrophobin HFB1 (class II) adsorbs to a hydrophobic surface through its hydrophobic patch and adopts a nearly vertical hydrophobic dipole relative to the surface. If such an orientation would also occur on PET, the hydrophobins could physically interact with the cutinases, while leaving enough surface of the polymer for cutinase binding. Yet in order to understand the mechanism of cutinase stimulation by hydrophobins, the mode of binding of the latter to PET needs further investigation.
Nevertheless, this study shows that enzymatic PET hydrolysis can be considerably enhanced by the fusion of hydrophobins to cutinases. The levels of stimulation observed in this study are higher than those in systems where the hydrophobins and cutinase were mixed (13; this study) and are also higher than those reached by covalent fusion to a PHA-binding module from A. faecalis (12). The use of cutinase-hydrophobin fusion proteins is thus an important step toward increasing the rate of PET modification and recycling.
ACKNOWLEDGMENTS
This study was supported by the Federal Ministry of Science, Research and Economy (BMWFW), the Federal Ministry of Traffic, Innovation, and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol, and the Government of Lower Austria and Business Agency Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG.
We are grateful to Hinrich Grothe and Florian Handle, Vienna University of Technology (Austria), for making their equipment available for this study.
REFERENCES
- 1.Köpnick H, Schmidt M, Brügging W, Rüter J, Kaminsky W. 2005. Polyesters, p 233–238. Ullmann's encyclopedia of industrial chemistry, vol A21 John Wiley, New York, NY. [Google Scholar]
- 2.Ignatyev IA, Thielemans W, Vander Beke B. 2014. Recycling of polymers: a review. ChemSusChem 7:1579–1593. doi: 10.1002/cssc.201300898. [DOI] [PubMed] [Google Scholar]
- 3.Carta D, Cao G, D'Angeli C. 2003. Chemical recycling of poly(ethylene terephthalate) (PET) by hydrolysis and glycolysis. Environ Sci Pollut Res Int 10:390–394. doi: 10.1065/espr2001.12.104.8. [DOI] [PubMed] [Google Scholar]
- 4.Guebitz GM, Cavaco-Paulo A. 2008. Enzymes go big: surface hydrolysis and functionalization of synthetic polymers. Trends Biotechnol 26:32–38. doi: 10.1016/j.tibtech.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Wei R, Oeser T, Zimmermann W. 2014. Synthetic polyester-hydrolyzing enzymes from thermophilic actinomycetes. Adv Appl Microbiol 89:267–305. doi: 10.1016/B978-0-12-800259-9.00007-X. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Su L, Chen J, Wu J. 2013. Cutinase: characteristics, preparation, and application. Biotechnol Adv 31:1754–1767. doi: 10.1016/j.biotechadv.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Silva C, Da S, Silva N, Matamá T, Araújo R, Martins M, Chen S, Chen J, Wu J, Casal M, Cavaco-Paulo A. 2011. Engineered Thermobifida fusca cutinase with increased activity on polyester substrates. Biotechnol J 6:1230–1239. doi: 10.1002/biot.201000391. [DOI] [PubMed] [Google Scholar]
- 8.Herrero Acero E, Ribitsch D, Dellacher A, Zitzenbacher S, Marold A, Steinkellner G, Gruber K, Schwab H, Guebitz GM. 2013. Surface engineering of a cutinase from Thermobifida cellulosilytica for improved polyester hydrolysis. Biotechnol Bioeng 110:2581–2590. doi: 10.1002/bit.24930. [DOI] [PubMed] [Google Scholar]
- 9.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. 2004. Carbohydrate-binding modules: fine tuning polysaccharide recognition. Biochem J 382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Várnai A, Mäkelä MR, Djajadi DT, Rahikainen J, Hatakka A, Viikari L. 2014. Carbohydrate-binding modules of fungal cellulases: occurrence in nature, function, and relevance in industrial biomass conversion. Adv Appl Microbiol 88:103–165. doi: 10.1016/B978-0-12-800260-5.00004-8. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima-Kambe T, Shigeno-Akutsu Y, Nomura N, Onuma F, Nakahara T. 1999. Microbial degradation of polyurethane, polyester polyurethanes and polyether polyurethanes. Appl Microbiol Biotechnol 51:134–140. doi: 10.1007/s002530051373. [DOI] [PubMed] [Google Scholar]
- 12.Ribitsch D, Oracal Yebra A, Zitzenbacher S, Wu J, Nowitsch S, Steinkellner G, Greimel K, Doliska A, Oberdorfer G, Gruber CC, Gruber K, Schwab H, Stana-Kleinschek K, Herrero Acero E, Guebitz GM. 2013. Fusion of binding domains to Thermobifida cellulosilytica cutinase to tune sorption characteristics and enhancing PET hydrolysis. Biomacromolecules 14:1769–1776. doi: 10.1021/bm400140u. [DOI] [PubMed] [Google Scholar]
- 13.Espino-Rammer L, Ribitsch D, Przylucka A, Marold A, Greimel KJ, Herrero Acero E, Guebitz GM, Kubicek CP, Druzhinina IS. 2013. Two novel class II hydrophobins from Trichoderma spp. stimulate enzymatic hydrolysis of poly(ethylene terephthalate) when expressed as fusion proteins. Appl Environ Microbiol 79:4230–4238. doi: 10.1128/AEM.01132-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wösten HA, de Vocht ML. 2000. Hydrophobins, the fungal coat unraveled. Biochim Biophys Acta 1469:79–86. doi: 10.1016/S0304-4157(00)00002-2. [DOI] [PubMed] [Google Scholar]
- 15.Whiteford JR, Spanu PD. 2002. Hydrophobins and the interaction between fungi and plants. Mol Plant Pathol 3:391–400. doi: 10.1046/j.1364-3703.2002.00129.x. [DOI] [PubMed] [Google Scholar]
- 16.Linder MB, Szilvay GR, Nakari-Setala T, Penttilä ME. 2005. Hydrophobins: the protein-amphiphiles of filamentous fungi. FEMS Microbiol Rev 29:877–896. doi: 10.1016/j.femsre.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook JE, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 18.Mattinen M, Kontteli M, Kerovuo J, Linder M, Annila A, Lindeberg G, Reinikainen T, Drakenberg T. 1997. Three-dimensional structures of three engineered cellulose-binding domains of cellobiohydrolase I from Trichoderma reesei. Protein Sci 6:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ribitsch D, Heumann S, Trotscha E, Herrero Acero E, Greimel K, Leber R, Birner-Gruenberger R, Deller S, Eiteljoerg I, Remler P, Weber T, Siegert P, Maurer K, Donelli I, Freddi G, Schwab H, Guebitz GM. 2011. Hydrolysis of polyethylene terephthalate by para-nitrobenzylesterase from Bacillus subtilis. Biotechnol Prog 27:951–960. doi: 10.1002/btpr.610. [DOI] [PubMed] [Google Scholar]
- 20.Shin SH, Comolli LC, Tscheliessnig R, Wang C, Nam KT, Hexemer A, Siegerist CE, De Yoreo JJ, Bertozzi CR. 2013. Self-assembly of “S-bilayers,” a step toward expanding the dimensionality of S-layer assemblies. ACS Nano 7:4946–4953. doi: 10.1021/nn400263j. [DOI] [PubMed] [Google Scholar]
- 21.Horejs C, Gollner H, Pum D, Sleytr UB, Peterlik H, Jungbauer A, Tscheliessnig R. 2011. Atomistic structure of monomolecular surface layer self-assemblies: toward functionalized nanostructures. ACS Nano 5:2288–2297. doi: 10.1021/nn1035729. [DOI] [PubMed] [Google Scholar]
- 22.Mueller M, Loh MQ, Tscheliessnig R, Tee DH, Tan E, Bardor M, Jungbauer A. 2013. Liquid formulations for stabilizing IgMs during physical stress and long-term storage. Pharm Res 30:735–750. doi: 10.1007/s11095-012-0914-2. [DOI] [PubMed] [Google Scholar]
- 23.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Stuhl K. 2006. Current protocols in molecular biology. Greene Publishing Associates/Wiley Interscience, New York, NY. [Google Scholar]
- 24.Seidl-Seiboth V, Gruber S, Sezerman U, Schwecke T, Albayrak A, Neuhof T, von Döhren H, Baker SE, Kubicek CP. 2011. Novel hydrophobins from Trichoderma define a new hydrophobin subclass: protein properties, evolution, regulation and processing. J Mol Evol 72:339–351. doi: 10.1007/s00239-011-9438-3. [DOI] [PubMed] [Google Scholar]
- 25.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Barioch A. 2005. Protein identification and analysis tools on the ExPASy server, p 571–607. In Walker JM. (ed), The proteomics protocols handbook. Humana Press, Totowa, NJ. [Google Scholar]
- 26.Roth C, Wei R, Oeser T, Then J, Föllner C, Zimmermann W, Sträter N. 2014. Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca. Appl Microbiol Biotechnol 98:7815–7823. doi: 10.1007/s00253-014-5672-0. [DOI] [PubMed] [Google Scholar]
- 27.Pio TF, Macedo GA. 2009. Cutinases: properties and industrial applications. Adv Appl Microbiol 66:77–95. doi: 10.1016/S0065-2164(08)00804-6. [DOI] [PubMed] [Google Scholar]
- 28.Araújo R, Silva C, O'Neill A, Micaelo N, Guebitz GM, Soares CM, Casal M, Cavaco-Paulo A. 2007. Tailoring cutinase activity towards polyethylene terephthalate and polyamide 6,6 fibers. J Biotechnol 128:849–857. doi: 10.1016/j.jbiotec.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 29.Grunér MS, Szilvay GR, Berglin M, Lienemann M, Laaksonen P, Linder MB. 2012. Self-assembly of class II hydrophobins on polar surfaces. Langmuir 28:4293–4300. doi: 10.1021/la300501u. [DOI] [PubMed] [Google Scholar]
- 30.Peng C, Liu J, Zhao D, Zhou J. 2014. Adsorption of hydrophobin on different self-assembled monolayers: the role of the hydrophobic dipole and the electric dipole. Langmuir 30:11401–11411. doi: 10.1021/la502595t. [DOI] [PubMed] [Google Scholar]