

Abstract
Specific polyisoprene-cleaving activities of 1.5 U/mg and 4.6 U/mg were determined for purified Strep-tagged latex clearing protein (Lcp) of Streptomyces sp. strain K30 at 23°C and 37°C, respectively. Metal analysis revealed the presence of approximately one atom of iron per Lcp molecule. Copper, which had been identified in Lcp1VH2 of Gordonia polyisoprenivorans previously, was below the detection limit in LcpK30. Heme was identified as a cofactor in purified LcpK30 by (i) detection of characteristic α-, β-, and γ (Soret)-bands at 562 nm, 532 nm, and 430 nm in the visible spectrum after chemical reduction, (ii) detection of an acetone-extractable porphyrin molecule, (iii) determination of a heme b-type-specific absorption maximum (556 nm) after chemical conversion of the heme group to a bipyridyl-heme complex, and (iv) detection of a b-heme-specific m/z value of 616.2 via mass spectrometry. Spectroscopic analysis showed that purified Lcp as isolated contains an oxidized heme-Fe3+ that is free of bound dioxygen. This is in contrast to the rubber oxygenase RoxA, a c-type heme-containing polyisoprene-cleaving enzyme present in Gram-negative rubber degraders, in which the covalently bound heme firmly binds a dioxygen molecule. LcpK30 also differed from RoxA in the lengths of the rubber degradation cleavage products and in having a higher melting point of 61.5°C (RoxA, 54.3°C). In summary, RoxA and Lcp both are equipped with a heme cofactor and catalyze an oxidative C-C cleavage reaction but differ in the heme subgroup type and in several biochemical and biophysical properties. These findings suggest differences in the catalytic reaction mechanisms.
INTRODUCTION
The hydrocarbon natural rubber [poly(cis-1,4-isoprene)] is an important biopolymer that is produced by many plants and some fungi. Natural rubber, as well as chemically synthesized poly(cis-1,4-isoprene) (synthetic rubber), has been in use by mankind for more than 100 years. Huge amounts of rubber derived from the rubber tree Hevea brasiliensis are the basis for the manufacturing of tires, sealers, latex gloves, condoms, and many other items. Most of these materials are released to the environment in the form of waste or by abrasion in the case of tires. As a natural compound, rubber is a fully biodegradable material, and many rubber-degrading microorganisms have been isolated from various ecosystems in the past (1–8). The initial step of rubber biodegradation is the enzymatic cleavage of the polymeric carbon backbone to smaller products. Two types of rubber-cleaving enzymes have been characterized so far. One is rubber oxygenase A, RoxA, which is found in Gram-negative clearing zone formers. The other is latex clearing protein, Lcp, which has been identified only in Gram-positive organisms so far. RoxA first was isolated and biochemically characterized from Xanthomonas sp. strain 35Y (9). Biochemical and biophysical investigation revealed that RoxA is an extracellular dioxygenase with two covalently attached heme groups (10, 11) and is structurally but not functionally related to cytochrome c peroxidases (12, 13). RoxA cleaves poly(cis-1,4-isoprene) to 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al (ODTD; C15-tri-isoprenoid) as a main end product. Recently, several orthologs of RoxA were described in other Gram-negative bacteria (14). Lcp is widespread in Gram-positive rubber degraders and is unrelated to RoxA; Lcps have a significantly different molecular mass of about 42 kDa, compared to ≈72 kDa for RoxAs. The amino acid sequences of Lcps have no heme binding motifs that could serve as covalent attachment sites for heme groups, and Lcps cleave rubber to larger degradation products than RoxAs (6, 15, 16). Recently, the Lcp proteins of two species (Streptomyces sp. strain K30 [LcpK30] [17] and Gordonia polyisoprenivorans VH2 [Lcp1VH2]) (18, 19) were purified and biochemically characterized. Remarkably, copper was identified in recombinantly expressed hexa-His-tagged Lcp1VH2, and results of inhibitor studies were in agreement with the presence of Cu(II) ions in Lcp1VH2 (19). The authors suggested that Lcp1VH2 is a member of the so-called copper-containing white laccase family. In contrast to Lcp1VH2, copper or other metals could not be detected in LcpK30 (52%/69% amino acid identity/similarity), which was purified from a constructed ΔroxA Xanthomonas sp. mutant harboring a chromosomally anchored native lcpK30 gene. However, only a very low protein concentration was available for metal analysis; accordingly, the detection limit was high and a metal content of up to 0.8 mol of Fe per mol of Lcp or 0.2 mol of Cu per mol of Lcp could not be excluded in our previous study (17). The aim of the present study was to clarify the metal content of Lcp by increasing expression and yield of Lcp protein and repetition of metal analysis at higher fidelity. We decided to use a Strep-tagged Lcp variant to facilitate enzyme purification and to avoid the possibility of artificial binding of metal ions to a hexa-His tag. This construct was used for further characterization and to address the question of whether a cofactor is present or not in Strep-Lcp.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Table 1 shows strains and plasmids used in this study. Recombinant E. coli strains were grown in LB medium at 22°C or 37°C in the presence of appropriate antibiotics. Polyisoprene latex was kindly provided by Weber and Schaer, Hamburg (Germany), and was used after 3 washing steps in 0.1% (wt/vol) Nonidet P40. For purification of LcpK30, recombinant E. coli strains were grown in 4.8 liters of LB medium (8 individual cultures each in a 3-liter Erlenmeyer flask) supplemented with 0.1% (wt/vol) l-rhamnose for 20 h at 22°C by continuous shaking (120 rpm). Cells were harvested (4°C) by centrifugation, and Lcp was purified from soluble cell extracts as described below.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Reference or source |
|---|---|---|
| E. coli JM109 | Plasmid storage and expression of lcp | |
| E. coli XL1-blue | QuikChange transformation strain | Stratagene |
| Xanthomonas sp. strain 35Y ΔroxA-attB | No clear zone formation on latex agar | 12 |
| Xanthomonas sp. strain 35Y ΔroxA-attB pNH1-roxA-attP in chromosome (SN4230) | Expression of RoxA from rhamnose promoter in Xanthomonas sp. strain 35Y-CM, Kmr, Cmr; clearing zone formation in the presence of rhamnose plus latex | 12 |
| pMK::lcp (SN5314) | GeneArt vector, source of lcp, Kmr | 17 |
| pUC9::lcp (SN5339) | Cloning vector for lcp, Ampr | This study |
| p4782.1 (SN3513) | Mobilizable broad-host-range expression vector, Kmr | 21 |
| p4782.1::strep-lcp (SN5496) | Coding sequence of strep-lcp under rhamnose promoter control, Kmr | This study |
Kmr, kanamycin resistance; Ampr, ampicillin resistance; Cmr, chloramphenicol resistance.
Construction of a recombinant E. coli strain for expression of the Streptomyces sp. strain K30 lcp gene (lcpK30).
The lcpK30 gene sequence was cloned into pUC9 via single NdeI and HindIII sites using pMK::lcp (Table 1) as the template. For substitution of the lcp TAT-dependent signal sequence (20) with the Strep-tag, the resulting vector pUC9::lcp was used as the template for PCR with the following primers: strep_f, GCTCATATGTGGAGCCACCCGCAGTTCGAAAAAGAGAACCTCTACTTCCAGGGCCTGCAGCGACCACTGTGGACGTGGTCACCGAGC; lcp-strep-r, GTCAATAAACCGGTAAGCTTTCAGGACG. The resulting Strep-tag-lcp DNA sequence (∼1.25 kbp) was cut with NdeI and HindIII and subcloned into p4782.1 (Table 1), yielding p4782.1::strep-lcp. This construct was transformed into the expression strain E. coli JM109.
Purification of recombinant RoxA and Lcp.
During all chromatic steps, the concentration of RoxA and Lcp was monitored by recording the absorption at 280 nm and 408 nm (RoxA) or 280 nm and 412 nm (Lcp), respectively. RoxA was purified from a ΔroxA Xanthomonas sp. with chromosomally integrated roxA plasmid (pNH1::roxA) under rhamnose control as described previously (12, 21). Lcp with the replacement of the TAT sequence by a Strep-tag was purified by using E. coli JM109 harboring p4782.1::lcp (volume, 4.8 liters) grown at 22°C for 20 h. The cells were harvested and resuspended in buffer A (1 ml buffer A/g cells, 100 mM potassium phosphate buffer [KPP], pH 7.7, 150 mM sodium chloride). After two subsequent French press lysis steps, the soluble fraction was obtained by ultracentrifugation (30 min, 40,000 × g, 4°C) and was applied to a 10-ml Strep-Tactin HC gravity flow column (IBA, Göttingen, Germany). Subsequently, the column was washed five times with 1 bed volume each time of buffer A, and Lcp was eluted with 3 applications (1 bed volume each) of buffer A plus 5 mM desthiobiotin. For further chromatographic steps, the ÄKTA fast protein liquid chromatography system (GE Healthcare, Sweden) was used. First, the Lcp preparation was rebuffered in 1 mM potassium phosphate buffer (pH 7.0) with a HiPrep Sephadex G25 26/10 column (bed volume, 53 ml). This step was necessary to avoid precipitation of Lcp during the following concentration step. The pooled Lcp-containing fractions were concentrated to a final volume of ≈4 ml via ultrafiltration (10-kDa cutoff) and then applied to a Superdex 200 16/600 size-exclusion chromatography column (2 separate runs, each loaded with 2 ml of the concentrated Lcp solution). The Lcp-containing fractions where pooled, concentrated via ultrafiltration (10-kDa cutoff), and stored on ice for up to 1 week. Alternatively, Lcp was frozen in liquid nitrogen and kept at −70°C for long-term storage.
Hemochrome pyridine assay.
The heme type of Lcp was determined spectroscopically with the bipyridyl assay according to Berry and Trumpower (22). RoxA, cytochrome c (horseheart, type III; Sigma, St. Louis, MO) (both c-type), and hemoglobin (b-type) (bovine; Sigma, St. Louis, MO) served as controls. A total of 25 μl of the respective protein stock solution (4 to 7 mg/ml) was added to 975 μl solution A (100 mM sodium hydroxide, 20% [vol/vol] pyridine, 0.3 mM potassium ferricyanide). Subsequently, 2 to 5 mg sodium dithionite was added, and the spectrum of the reduced cytochrome was recorded. The absorption maxima of the resulting α-bands are characteristic for α-type (584 to 588 nm), b-type (556 nm), and c-type (550 nm) cytochromes.
CD spectra.
Circular dichroism (CD) spectra were recorded with a J-815 CD spectrophotometer (Jasco, Gross-Umstadt, Germany). Eighty microliters of an Lcp solution (4.5 mg/ml in 1 mM KPP, pH 7.0) after the final Sephadex 200 gel filtration step was measured in a 0.2-mm cuvette in a wavelength range between 185 and 260 nm with standard sensitivity and a bandwidth of 1 nm. The spectra recorded with buffer were used as a baseline and subtracted from the spectrum of the protein solution in the same buffer. Twenty-five scans were collected and averaged. The melting curve was measured at the CD minimum of 230 nm with an increase of 1°C/minute from 20 to 80°C.
Assay of Lcp activity.
Lcp activity was determined by the consumption of molecular oxygen with an OXY-4 mini apparatus and a corresponding optical oxygen sensor spot (PreSens, Regensburg, Germany). The sensor was attached to the inner surface of a 1.5-ml cuvette. The oxygen-dependent fluorescence of the sensor was continuously recorded via an optical fiber that had been fixed to the outside of the cuvette. The system was calibrated with oxygen-saturated (air) and anoxic (dithionite) buffer. Potassium phosphate buffer (100 mM, pH 7) supplemented with 0.2% (wt/vol) polyisoprene latex was added into the cuvette for determination of Lcp activity. The cuvette was sealed with a silicone lid to ensure the absence of a gas phase on top of the reaction mix that could replenish consumed dissolved dioxygen and would falsify the results (total volume, 1.5 ml). Homogeneity of the oxygen concentration in the cuvette was ensured by mixing with the aid of a magnetic stirrer and a microstirring bar. For determination of Lcp activity at temperatures above room temperature, the whole system was placed in a breeding incubator until a constant temperature, monitored in a reference cuvette, was reached. The baseline was recorded for approximately 7 min before the reaction was started by the addition of Lcp (final concentration, 4 μg/ml) using a Hamilton pipette. The resulting time-dependent decrease of oxygen concentration (100% to ≈80% oxygen saturation) was used to determine the specific activity of Lcp. One hundred percent oxygen in the reaction mix was equivalent to 260 μM at room temperature or 210 μM at 37°C. Triplicates and one control without Lcp were recorded simultaneously.
Inhibitor studies.
To examine the effect of potential inhibitors on Lcp activity, a 10 mM concentration of each test compound was added to Lcp. After an incubation period of 30 min at room temperature, the Lcp-inhibitor mixture was added to the reaction mix [100 mM potassium phosphate buffer, pH 7, supplemented with 0.2% poly(cis,1,4-isoprene), resulting in a final inhibitor concentration of 1 mM and an Lcp concentration of 4 μg/ml]. After incubation at room temperature for 2 h, the reaction mixture (700 μl in a 2-ml Eppendorf tube) was extracted with ethyl acetate (1 ml), dried, dissolved in 100 μl methanol, and further subjected to high-performance liquid chromatography (HPLC) analysis. An RP8 HPLC column (12 by 4 mm, 5-μm particle size) was operated at 0.7 ml/min with water (A) and methanol (B) as mobile phases. The concentration of B was increased from 50% (vol/vol) to 100% (vol/vol) within 15 min; products were detected at 210 nm. The C35 product peak (n = 6) was used for quantification and compared to a control without inhibitor (Fig. 1A).
FIG 1.
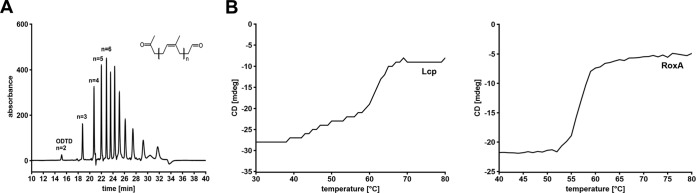
Degradation products of oxidative cleavage of poly(cis-1,4-isoprene). Rubber latex was incubated in the presence of purified Strep-LcpK30 for 2 h. Degradation products were extracted with ethyl-acetate and separated by HPLC. (B) Determination of the melting points of Strep-LcpK30 (left) and RoxA (right). Melting points were recorded at the CD minima of Lcp (230 nm) and RoxA (207 nm) and were at 61.5°C (Lcp) and 54.3°C (RoxA).
Matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS).
A Bruker Daltonics autoflex speed apparatus (Bruker Corporation, Billerica, MA) was employed, and 4-hydroxycinnamic acid (HCCA) was used as the matrix. To this end, the Bruker MTP anchor-chip target was prepared with protein calibration standard 2 (part number 222570) in 125 μl TA30 (30% [vol/vol] acetonitrile, 0.1% [wt/vol] trifluoric acid [TFA]) and mixed with the HCCA matrix solution (in TA30). Equal volumes of the samples were mixed with matrix solution, and 1 μl was spotted on the target. Data acquisition was conducted by employing reflection mode, and the flex analysis program was used for interpretation of the data. The following protein samples were used: cytochrome c, RoxA, hemoglobin, and LcpK30 (each at 10 mg/ml in 10 mM KPP buffer, pH 7). Determinations of m/z values of the heme groups were performed (i) with the untreated protein, (ii) with a trypsin-digested protein (10 μl protein stock mixed with 4 μl trypsin [0.5 μg/μl] and 86 μl 10 mM potassium phosphate buffer, pH 7, incubated overnight at 37°C), and (iii) with the supernatant of acetone-extracted protein. For extraction with acetone, the method of Morrison and Horie was used (23). In brief, 5 mg (NH4)2SO4 was dissolved in 20 μl of each protein solution, and 167 μl acetone was added. The formed precipitate was centrifuged at 12,000 rpm for 2 min in an Eppendorf centrifuge. The supernatant was discarded, another 167 μl acetone was added, and the centrifugation step was repeated. A total of 67 μl of acidic acetone (90 μl of 37% HCl–5 ml acetone) was added to the pellet, and the suspension was mixed thoroughly. The samples were centrifuged for 3 min at 12,000 rpm to separate the red supernatant containing the extracted heme from the white protein precipitate of Lcp or hemoglobin. A clear supernatant and a red precipitate were obtained for RoxA and cytochrome c, and this was in agreement with a covalent attachment of heme to the polypeptide chain in RoxA and cytochrome c.
Other techniques.
Protein concentration was determined by the bicinchoninic acid (BCA) method. The concentrations of purified Lcp samples also were determined by molar extinction coefficients of Lcp at 412 nm (ε412 = 80,000 M−1 cm−1). Heme staining for pseudoperoxidase activity and UV-visible (UV-vis) spectroscopy were conducted as described previously after separation of proteins via nonreducing SDS-PAGE (11). Metal content of purified Lcp was determined using inductively coupled plasma-MS (ICP-MS) by the Spuren-Analytisches Laboratorium Dr. Baumann (Germany).
RESULTS AND DISCUSSION
Purification of LcpK30.
Strep-LcpK30 was purified from soluble cell extracts of a recombinant E. coli JM109 culture via Strep-tag affinity chromatography and subsequent size-exclusion chromatography as described in Materials and Methods. About 26 mg of an almost homogeneous LcpK30 preparation was obtained from the cells present in a culture volume of 4.8 liters (5.4 mg Lcp/liter culture). This result corresponds to an over 40-fold increase of Lcp yield compared to that of our previous expression system in recombinant Xanthomonas sp. strain 35Y (17). Specific activities of 1.5 U/mg were determined for freshly purified LcpK30 at 23°C using an oxygen consumption assay. Remarkably, a considerably higher specific activity of 4.6 U/mg was determined at 37°C. Purified Strep-LcpK30 cleaved poly(cis-1,4-isoprene) to a mixture of oligonucleotide-isoprenoids with terminal keto and aldehyde groups (Fig. 1A), as shown previously for native Lcp (17). A melting temperature of 61.5°C was determined for Strep-LcpK30 (Fig. 1B); thus, it was remarkably higher than that of RoxA (54.3°C).
SDS-PAGE analysis of a purified LcpK30 preparation showed a prominent band at an apparent molecular mass of ≈48 kDa that roughly corresponded with the theoretical value of 43.3 kDa for a Strep-tagged version of LcpK30 (Fig. 2A). No or only very weak positive staining was detected for the LcpK30 band when the SDS-PAGE gel was subjected to heme staining for pseudoperoxidase activity, while RoxA, which was used as a positive control, showed a very strong signal (not shown). Remarkably, a band with positive pseudoperoxidase activity was detected near the bottom of the PAGE gel and indicated the presence of a very small heme compound. All attempts to remove this molecule from the LcpK30 preparation by different chromatography methods (anion exchange, hydrophobic interaction, hydroxyapatite, and gel filtration) failed. In all cases, the LcpK30 protein (A280) coeluted with a compound having a strong absorption at ≈412 nm, which is typical for a heme molecule (Fig. 2B). When purified Lcp was run on a native PAGE and subjected to heme staining, a strong pseudoperoxidase-positive band was detected (not shown), suggesting that Lcp has a noncovalently bound heme group. Thus, the absence of positive heme staining in the 48-kDa Lcp band in SDS-PAGE analysis could be explained by removal of the noncovalently bound heme molecule during the denaturation step prior to electrophoresis.
FIG 2.
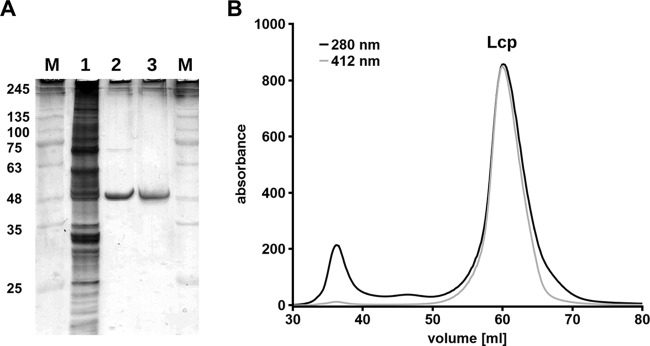
Purification of Strep-LcpK30. (A) Reducing SDS-PAGE of Lcp preparations. M, marker proteins with molecular masses (in kilodaltons) indicated; lane 1, soluble crude extract; lane 2, Lcp pool after affinity chromatography (Strep-Tactin HC); lane 3, Lcp pool after gel filtration on Superdex 200. (B) Coelution of Lcp (A280) and heme (A412) during gel filtration on Superdex 200.
LcpK30 is an Fe-containing protein.
As pointed out in Introduction, controversial reports were published on the metal content of Lcp protein in Streptomyces sp. strain K30 and in Gordonia polyisoprenivorans (17, 19). Therefore, it was necessary to repeat the metal analysis of purified Lcp at higher sensitivity. Two LcpK30 preparations were subjected to metal analysis by inductively coupled plasma-MS (ICP-MS) analysis. Substantial amounts of Fe and trace amounts of Ni were identified in both preparations. The ratio of metal atoms per molecule of Lcp was 1.16 mol Fe and 0.12 mol Ni in one sample or 1.04 mol Fe and 0.05 mol Ni in the second Lcp sample. Copper was identified at a very low concentration of 0.056 mol Cu/mol LcpK30 in only one sample but was below the detection limit in the other sample (<0.01 mol Cu per mol LcpK30). No other metals (vanadium to zinc were tested) were detected. We conclude that LcpK30 is an iron-containing protein and apparently harbors one atom of Fe per molecule of LcpK30 peptide. The traces of nickel in both preparations and of copper in one Lcp preparation are far below stoichiometric amounts and presumably represent contaminations. We have no plausible explanation for the detection of copper in Lcp1VH2 of G. polyisoprenivorans, except that copper ions could have artificially bound to the hexa-His tag (19). However, in this case one would have expected the presence of other heavy metals in the Lcp preparation as well. Because of substantial amino acid similarity of LcpVH1 and LcpK30 (52% identity/69% similarity, 99% overlap), the presence of different metals as cofactors in the two related Lcps is unlikely.
UV-vis spectroscopy of LcpK30.
A concentrated LcpK30 (>2 mg/ml) preparation showed a red color, and this is in agreement with the presence of a heme group in LcpK30. UV-vis spectroscopy of purified LcpK30, as isolated, revealed a strong absorption band at 412 nm that corresponds roughly with the absorption maximum at ≈420 nm published for Lcp1VH2 (19). Reduction of LcpK30 by the addition of sodium dithionite resulted in a red shift of the absorption band to 430 nm and in the appearance of new absorption bands at 532 and 562 nm (Fig. 3). The presence of one absorption band around 412 nm in the as-isolated state and the presence of three absorption bands (α-band, β-band, and γ [Soret]-band) in the visible spectrum after chemical reduction is typical for heme-containing proteins of the cytochrome family. To determine the cytochrome subgroup type, the hemochrome pyridine assay according to Berry and Trumpower (22) was used. To this end, purified LcpK30 was incubated in 0.1 M NaOH in the presence of 20% (vol/vol) pyridine, and the absorption spectrum of the formed bipyridyl-hemochromes was recorded after reduction by dithionite (Fig. 4A). RoxA and cytochrome c (both c-type cytochromes) as well as hemoglobin (b-cytochrome) were used as controls. The absorption maxima of the resulting α-bands of the bipyridyl-heme complexes are characteristic for a-type (584 to 588 nm), b-type (556 nm), and c-type (550 nm) cytochromes. The absorption spectra of the hemochromes of the three controls were identical to those reported in the literature (22). The absorption spectrum of the LcpK30 bipyridyl-heme complex was identical to that of hemoglobin with a maximum at 556 nm (Fig. 4A), and this indicated that LcpK30 is a member of the b-type cytochrome family.
FIG 3.
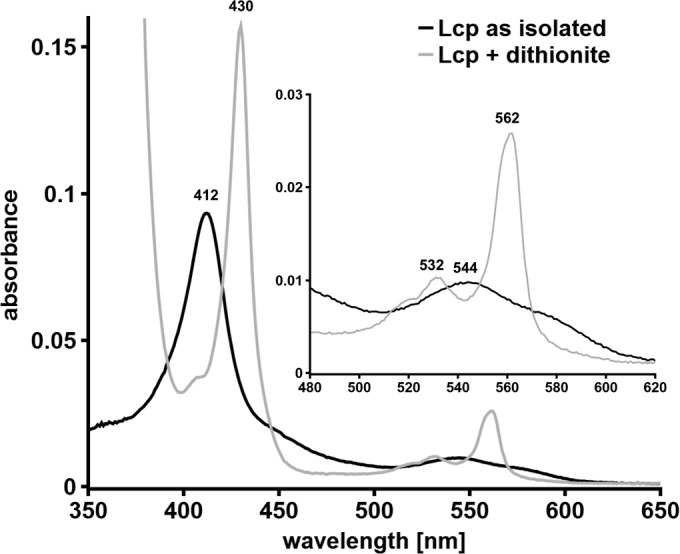
UV-vis spectroscopy of Strep-LcpK30 as isolated and after chemical reduction with dithionite. Wavelength absorption maxima are indicated above each peak.
FIG 4.
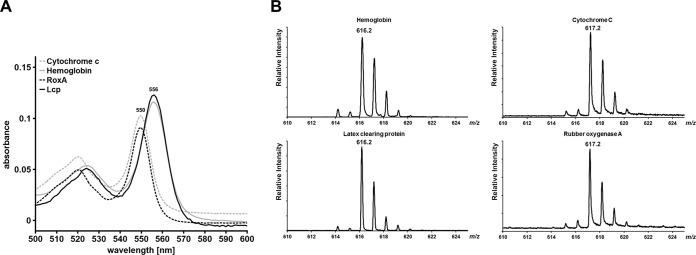
Determination of heme subgroup by bipyridyl assay (A) and MALDI-TOF MS analysis (B). Cytochrome c, hemoglobin, and RoxA were used as controls with known subgroup types. Lcp gave essentially the same results as those obtained for b-cytochrome hemoglobin both in bipyridyl assay and in MALDI-TOF MS analysis.
Heme is not covalently bound to Lcp.
In contrast to c-type cytochromes, in which the heme is covalently attached to sulfur atoms of cysteines of so-called heme binding motifs in the polypeptide chain of the respective protein (e.g., C191SAC194H in the case of N-terminal heme of RoxA), b-type cytochromes have noncovalently bound heme groups. Therefore, heme groups of b-cytochromes can be released from the protein by precipitation or denaturation of the protein. To test Lcp for the presence of noncovalently bound heme, purified LcpK30 was extracted with acidic acetone. The same procedure was performed with commercial hemoglobin (bovine; Sigma, St. Louis, MO), with purified RoxA (c-type cytochrome), and with commercial cytochrome c (horseheart, type III; Sigma, St. Louis, MO). The supernatants of Lcp and of hemoglobin were red and indicated the liberation of heme groups from the proteins. Accordingly, a typical heme spectrum was detectable for the supernatants of acetone-extracted Lcp and hemoglobin and confirmed that heme was liberated by the acidic extraction procedure. In contrast, the supernatants of RoxA and cytochrome c were pale, and no heme spectrum could be detected; instead, the precipitates were red and indicated that the heme groups had coprecipitated with the polypeptide moiety. These results are in agreement with a covalent attachment of heme to RoxA and cytochrome c and with a noncovalent attachment of heme in Lcp and hemoglobin.
The molecular masses of the heme groups of hemoglobin, Lcp, RoxA, and cytochrome c were determined by MALDI-TOF MS analysis as described in Materials and Methods. 4-Hydroxycinnamic acid (HCCA) was used as the matrix and led to good cocrystallization, laser desorption, and flying properties. According to a detailed mass-spectrometric characterization of b- and c-type hemes, the main peaks of b- and c-type heme groups have different heme-type characteristic m/z values of 616 and 617, respectively (24). Figure 4B shows the results of a MALDI-TOF MS analysis of hemoglobin, Lcp, cytochrome c, and RoxA. While RoxA and cytochrome c gave main peaks at m/z values of 617, the m/z value of Lcp and hemoglobin was 616. These results confirmed that cytochrome c and RoxA both have a c-type heme and that hemoglobin and Lcp are members of the b-type cytochrome family. The same results were obtained when trypsin-digested protein samples were used in MALDI-TOF analysis, and the presence of heme b in the acidic acetone extract of LcpK30 and hemoglobin was shown as well (data not shown). In summary, multiple and independent pieces of evidence show that LcpK30 is a member of the b-type cytochrome family.
Inhibitor studies.
The effect of metal chelators and other compounds that could act as inhibitors was studied for purified Lcp. In contrast to our previous study, in which we had added the inhibitor at a final concentration of 1 mM to the latex assay solution and had started the reaction by the addition of Lcp, in this study we preincubated Lcp with a 10-fold concentration (10 mM) of the inhibitor for 30 min before the Lcp-inhibitor mixtures were added to the assay solution. We used the direct product assay (HPLC-based detection of polyisoprene cleavage products) instead of the indirect oxygen consumption assay. Remarkably, except for diethyl-thiocarbamate (56% inhibition), none of the chelators (disodium 4,5-dihydroxy-1,3-benzenedisulfonate [Tiron], potassium ethyl-xanthogenate, or phenanthroline) had a remarkable effect on Lcp-catalyzed cleavage of polyisoprene (Table 2). This result was in agreement with our previous analysis (17) and showed that even preincubation of the enzyme with a high inhibitor concentration was not able to substantially inhibit the activity of the enzyme. This result is surprising and suggests that the heme group, with its central Fe ion, must be deeply buried in the protein so that none of the chelators, with the exception of diethyl-dithiocarbamate, could affect Lcp activity. Remarkably, after addition of diethyl-dithiocarbamate to Lcp, no decrease of the Soret band or other changes could be observed in the UV-vis spectrum (not shown). This indicates that diethyl-dithiocarbamate does not bind to Lcp-iron and, moreover, is not able to remove the iron ion from the heme center. Thus, it is possible that diethyl-dithiocarbamate is able to inhibit the enzyme by another (unknown) mechanism. Surprisingly, the reductant dithionite had only a minor effect on the polyisoprene cleaving activity of Lcp. UV-vis-spectroscopic analysis clearly showed that 10 mM dithionite completely reduced Lcp and also removed dioxygen from an aqueous solution. However, determination of the oxygen concentration in our reaction chamber filled with Lcp assay solution showed that dissolved oxygen reappears about 20 to 30 min after the addition of dithionite. Since our Lcp activity assay for inhibitors takes 120 min and must be performed in an oxic atmosphere (dioxygen is a cosubstrate), the small effect of dithionite on Lcp activity can be explained by an inhibition of the reaction only in the first 20 to 30 min.
TABLE 2.
Sensitivity of Lcp to potential inhibitors
| Compound | Relative activitya (%) |
|---|---|
| None (control) | 100 |
| Dithionite | 85 |
| Disodium 4,5-dihydroxy-1,3-benzenedisulfonate (Tiron) | 110 |
| Ethyl xanthogenate | 93 |
| Diethyl-dithiocarbamate | 44 |
| Imidazole | 52 |
| Phenanthroline | 80 |
Activity was determined using the HPLC-based detection of cleavage products after 2 h of incubation of polyisoprene latex with Lcp and subsequent solvent extraction of the products. Lcp was preincubated with the inhibitor (10 mM) for 30 min at room temperature before the Lcp-inhibitor mixture was added to the assay solution. The final concentration of inhibitors was 1 mM. The 100% value corresponds to the area of the C35 product peak (Fig. 1A) (17). All data show representative means from a duplicate experiment.
The oxidation states of Fe in Lcp and RoxA are different.
Previous structural and biophysical analysis had revealed that the heme group of the RoxA active site has a dioxygen molecule bound to the heme iron atom of the active heme site (11, 13). Because of the strong electronegativity of oxygen, one electron of the Fe2+ ion is relocated to the dioxygen molecule, resulting in an Fe3+-O2− state. This is the reason why the UV-vis spectrum of RoxA as isolated gives a typical oxidized heme spectrum. However, application of anoxic conditions (N2-atmosphere) or removal of dioxygen from RoxA by chemicals (e.g., pyrogallol) resulted in increases of the α-band at 549 nm and in other changes of the UV-vis spectrum that were characteristic for the removal of previously heme-bound dioxygen (11). This showed that the Fe atom of the active-site heme is in a reduced form (Fe2+) in the absence of bound oxygen. To determine the oxidation state of the heme in Lcp, spectral analysis of purified Lcp as isolated was performed and gave a typical oxidized spectrum similar to that of RoxA (Fig. 3). However, application of anoxic conditions (N2-atmosphere) did not result in the detection of an α-band in Lcp. In agreement with this, chemical reduction by dithionite and anaerobic reoxidation of Lcp did not result in the appearance of absorption maxima around 539 and 573 nm, as in RoxA, that would have indicated the removal of a previously bound dioxygen molecule (data not shown).
Addition of 1 mM imidazole to Lcp had a weak effect on the UV-vis spectrum of Lcp (Fig. 5A). The UV-vis spectrum of RoxA, however, showed a prominent increase of the α-band of the active-site heme group upon the addition of imidazole that was not observed for Lcp (Fig. 5B). In the case of RoxA, the bound dioxygen is replaced by imidazole, leading to an Fe2+-imidazole state that can be observed in the UV-vis spectrum. The weak changes in the UV-vis spectrum for Lcp are consistent with the interaction of the heme group with imidazole, but the reduction bands that can be seen for RoxA are missing, suggesting an oxidized (Fe3+) state without bound dioxygen. Another indication for this state was determined when carbon monoxide was added to RoxA and Lcp: no change of the visible spectrum was detected for LcpK30, whereas a prominent new absorption maximum at 415 nm and two minor signals at 521 nm and 550 nm appeared in the spectrum of RoxA in the presence of CO (Fig. 6). It is known that CO efficiently binds to a reduced Fe2+-heme species but is unable to bind to Fe3+ (25–27). These results support our conclusion that the oxidation states of the heme groups in RoxA and Lcp are different.
FIG 5.
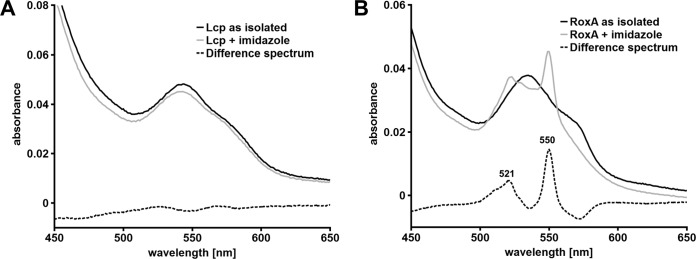
Effect of imidazole on UV-vis spectra of Lcp and RoxA. The UV-vis spectra of Lcp (A) and RoxA (B), as isolated, were recorded in the absence and presence of 1 mM imidazole. The respective difference spectra are indicated by the dotted lines. Only a part of the spectra (450 to 650 nm) is shown.
FIG 6.
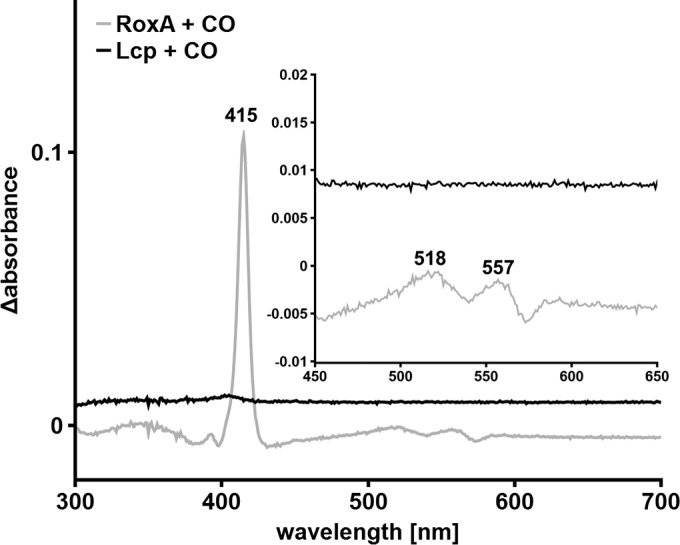
Effect of carbon monoxide (CO) on the UV-vis spectrum of RoxA and Strep-LcpK30. The UV-vis difference spectra of RoxA and Lcp, as isolated, in the absence and presence of CO-saturated buffer were recorded. A strong reaction is detectable for RoxA compared to no change of the spectrum in the case of Strep-LcpK30.
In summary, we have clarified that Lcp is a copper-free but iron-containing b-type cytochrome. In contrast to the active heme site of RoxA, the heme group of Lcp is in the oxidized state (Fe3+), and this explains the absence of heme-bound dioxygen in Lcp.
ACKNOWLEDGMENTS
This work was supported by a grant of the Deutsche Forschungsgemeinschaft to D.J.
We thank Weber and Schaer Company for providing polyisoprene. The help of T. Jurkowski and of S. Kudithipudi (Institute of Biochemistry, University Stuttgart) in MALDI-MS and CD analysis is greatly acknowledged.
REFERENCES
- 1.Heisey RM, Papadatos S. 1995. Isolation of microorganisms able to metabolize purified natural rubber. Appl Environ Microbiol 61:3092–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jendrossek D, Tomasi G, Kroppenstedt RM. 1997. Bacterial degradation of natural rubber: a privilege of actinomycetes? FEMS Microbiol Lett 150:179–188. doi: 10.1016/S0378-1097(97)00072-4. [DOI] [PubMed] [Google Scholar]
- 3.Linos A, Berekaa MM, Reichelt R, Keller U, Schmitt J, Flemming HC, Kroppenstedt RM, Steinbüchel A. 2000. Biodegradation of cis-1,4-polyisoprene rubbers by distinct actinomycetes: microbial strategies and detailed surface analysis. Appl Environ Microbiol 66:1639–1645. doi: 10.1128/AEM.66.4.1639-1645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rose K, Steinbüchel A. 2005. Biodegradation of natural rubber and related compounds: recent insights into a hardly understood catabolic capability of microorganisms. Appl Environ Microbiol 71:2803–2812. doi: 10.1128/AEM.71.6.2803-2812.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imai S, Ichikawa K, Muramatsu Y, Kasai D, Masai E, Fukuda M. 2011. Isolation and characterization of Streptomyces, Actinoplanes, and Methylibium strains that are involved in degradation of natural rubber and synthetic poly(cis-1,4-isoprene). Enzyme Microb Technol 49:526–531. doi: 10.1016/j.enzmictec.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 6.Yikmis M, Steinbüchel A. 2012. Historical and recent achievements in the field of microbial degradation of natural and synthetic rubber. Appl Environ Microbiol 78:4543–4551. doi: 10.1128/AEM.00001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imai S, Yoshida R, Endo Y, Fukunaga Y, Yamazoe A, Kasai D, Masai E, Fukuda M. 2013. Rhizobacter gummiphilus sp. nov., a rubber-degrading bacterium isolated from the soil of a botanical garden in Japan. J Gen Appl Microbiol 59:199–205. doi: 10.2323/jgam.59.199. [DOI] [PubMed] [Google Scholar]
- 8.Chia KH, Nanthini J, Thottathil GP, Najimudin N, Haris MRHM, Sudesh K. 2014. Identification of new rubber-degrading bacterial strains from aged latex. Polym Deg Stab 109:354–361. doi: 10.1016/j.polymdegradstab.2014.07.027. [DOI] [Google Scholar]
- 9.Braaz R, Fischer P, Jendrossek D. 2004. Novel type of heme-dependent oxygenase catalyzes oxidative cleavage of rubber (poly-cis-1,4-isoprene). Appl Environ Microbiol 70:7388–7395. doi: 10.1128/AEM.70.12.7388-7395.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braaz R, Armbruster W, Jendrossek D. 2005. Heme-dependent rubber oxygenase RoxA of Xanthomonas sp. cleaves the carbon backbone of poly(cis-1,4-isoprene) by a dioxygenase mechanism. Appl Environ Microbiol 71:2473–2478. doi: 10.1128/AEM.71.5.2473-2478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmitt G, Seiffert G, Kroneck PMH, Braaz R, Jendrossek D. 2010. Spectroscopic properties of rubber oxygenase RoxA from Xanthomonas sp., a new type of dihaem dioxygenase. Microbiology 156:2537–2548. doi: 10.1099/mic.0.038992-0. [DOI] [PubMed] [Google Scholar]
- 12.Birke J, Hambsch N, Schmitt G, Altenbuchner J, Jendrossek D. 2012. Phe317 is essential for rubber oxygenase RoxA activity. Appl Environ Microbiol 78:7876–7883. doi: 10.1128/AEM.02385-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seidel J, Schmitt G, Hoffmann M, Jendrossek D, Einsle O. 2013. Structure of the processive rubber oxygenase RoxA from Xanthomonas sp. Proc Natl Acad Sci U S A 110:13833–13838. doi: 10.1073/pnas.1305560110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birke J, Röther W, Schmitt G, Jendrossek D. 2013. Functional identification of rubber oxygenase (RoxA) in soil and marine myxobacteria. Appl Environ Microbiol 79:6391–6399. doi: 10.1128/AEM.02194-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rose K, Tenberge KB, Steinbüchel A. 2005. Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly(cis-1,4-isoprene) rubber degradation. Biomacromolecules 6:180–188. doi: 10.1021/bm0496110. [DOI] [PubMed] [Google Scholar]
- 16.Yikmis M, Steinbüchel A. 2012. Importance of the latex-clearing protein (Lcp) for poly(cis-1,4-isoprene) rubber cleavage in Streptomyces sp. K30. Microbiologyopen 1:13–24. doi: 10.1002/mbo3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birke J, Jendrossek D. 2014. Rubber oxygenase (RoxA) and latex clearing protein (Lcp) cleave rubber to different products and use different cleavage mechanisms. Appl Environ Microbiol 80:5012–5020. doi: 10.1128/AEM.01271-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiessl S, Schuldes J, Thürmer A, Halbsguth T, Broker D, Angelov A, Liebl W, Daniel R, Steinbüchel A. 2012. Involvement of two latex-clearing proteins during rubber degradation and insights into the subsequent degradation pathway revealed by the genome sequence of Gordonia polyisoprenivorans strain VH2. Appl Environ Microbiol 78:2874–2887. doi: 10.1128/AEM.07969-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiessl S, Böse D, Oetermann S, Eggers J, Pietruszka J, Steinbüchel A. 2014. Latex clearing protein-an oxygenase cleaving poly(cis-1,4-isoprene) rubber at the cis double bonds. Appl Environ Microbiol 80:5231–5240. doi: 10.1128/AEM.01502-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yikmis M, Arenskotter M, Rose K, Lange N, Wernsmann H, Wiefel L, Steinbüchel A. 2008. Secretion and transcriptional regulation of the latex-clearing protein, Lcp, by the rubber-degrading bacterium Streptomyces sp. strain K30. Appl Environ Microbiol 74:5373–5382. doi: 10.1128/AEM.01001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hambsch N, Schmitt G, Jendrossek D. 2010. Development of a homologous expression system for rubber oxygenase RoxA from Xanthomonas sp. J Appl Microbiol 109:1067–1075. doi: 10.1111/j.1365-2672.2010.04732.x. [DOI] [PubMed] [Google Scholar]
- 22.Berry EA, Trumpower BL. 1987. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal Biochem 161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 23.Morrison M, Horie S. 1965. Determination of heme a concentration in cytochrome preparations by hemochromogen method. Anal Biochem 12:77–82. doi: 10.1016/0003-2697(65)90144-2. [DOI] [PubMed] [Google Scholar]
- 24.Yang HJ, Park KH, Sin S, Lee J, Park S, Kim HS, Kim J. 2013. Characterization of heme ions using MALDI-TOF MS and MALDI FT-ICR MS. Int J Mass Spectrom 343-344:37–44. doi: 10.1016/j.ijms.2013.03.014. [DOI] [Google Scholar]
- 25.Yoshioka S, Kobayashi K, Yoshimura H, Uchida T, Kitagawa T, Aono S. 2005. Biophysical properties of a c-type heme in chemotaxis signal transducer protein DcrA. Biochemistry 44:15406–15413. doi: 10.1021/bi0513352. [DOI] [PubMed] [Google Scholar]
- 26.Cooper CE. 1999. Nitric oxide and iron proteins. Biochim Biophys Acta 1411:290–309. doi: 10.1016/S0005-2728(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 27.Hirata F, Ohnishi T, Hayaishi O. 1977. Indoleamine 2,3-dioxygenase. Characterization and properties of enzyme O2− complex. J Biol Chem 252:4637–4642. [PubMed] [Google Scholar]