

Abstract
Objective
Pancreatic beta-cells express three Munc18 isoforms. Much is known about the roles of Munc18a (pre-docked secretory granules-SGs) and Munc18b (newcomer SGs and SG–SG fusion) in insulin exocytosis. Although shown to influence glucose-stimulated insulin secretion (GSIS) in rodents the precise role of Munc18c in insulin SG exocytosis has not been elucidated. We here examined the role of Munc18c in human pancreatic beta-cells.
Methods
Munc18c-shRNA/RFP lenti-virus (versus control virus) was used to knock down the expression level of Munc18c in human islets or single beta-cells. Insulin secretion and granule exocytosis were measured by performing islets perifusion, single-cell patch clamp capacitance measurements and total internal reflection fluorescence microscopy (TIRFM).
Results
Munc18c is most abundant in the cytosol of human beta-cells. Endogenous function of Munc18c was assessed by knocking down (KD) its islet expression by 70% employing lenti-shRNA virus. Munc18c-KD caused reduction in cognate syntaxin-4 islet expression but not in other exocytotic proteins, resulting in the reduction in GSIS in first- (by 42%) and second phases (by 35%). Single cell analyses of RFP-tagged Munc18c-KD beta-cells by patch clamp capacitance measurements showed inhibition in both readily-releasable pool (by 52%) and mobilization from the reserve pool (by 57%). TIRFM to assess single SG behavior showed that Munc18c-KD inhibition of first phase GSIS was attributed to reduction in exocytosis of pre-docked and newcomer SGs, and second phase inhibition attributed entirely to reduction in newcomer SG fusion (SGs that undergo minimal residence or docking time at the plasma membrane before fusion).
Conclusion
Munc18c is involved in the distinct molecular machineries that affect exocytosis of both predocked and newcomer SG pools that underlie Munc18c's role in first and second phases of GSIS, respectively.
Keywords: Munc18c, Exocytosis, Newcomer insulin granules, Human islets
Abbreviations: Syn, syntaxin; SG, secretory insulin-containing granule; GSIS, glucose-stimulated insulin secretion; PM, plasma membrane; KD, knock down; CmPatch, clamp capacitance measurements; TIRFM, total internal reflection fluorescence microscopy; RRP, readily releasable pool; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; SM, Sec1/Munc18-like protein; v-, vesicle-; t-, target-; VAMPs, Vesicle Associated Membrane Proteins; SNAP25/23, synaptosomal-associated protein of 25/23 kD; GLP-1, glucagon-like peptide-1; T2DM, type 2 diabetes mellitus; Ad, adenovirus; EGFP, enhanced green fluorescent protein; NPY, neuropeptide Y
1. Introduction
Exocytosis underlying secretion is essential for human biology, playing a vital role in cellular processes as diverse as neurotransmission and blood glucose metabolic control. Two protein families are key to exocytosis: (1) Sec1p/Munc18 (SM) and (2) soluble N-ethylmaleimide-sensitive attachment protein receptor (SNARE) proteins [1]. Secretory granule (SG) exocytosis involves specific binding of vesicle(v)-SNARE vesicle-associated membrane protein (VAMP) with the binary cognate target membrane (t)-SNARE complex, composed of SNAP25/23 and syntaxin(Syn) proteins, to form the heterotrimeric SNARE core complex [2]. Plasma membrane (PM)–localized exocytotic syntaxins are further regulated by SM proteins (Sec1/Munc18) that bind with selectivity to cognate syntaxins. Both Munc18a and Munc18b bind Syn-1, Syn-2 and Syn-3, whereas only Munc18c binds to and regulates Syn-4 [3–5]. Whilst the essential function of these proteins is irrefutable, their exact regulatory roles in membrane fusion remain controversial and hotly pursued.
Whereas most cell types have one or two dominant Munc18s, all three Munc18 isoforms are expressed in islet beta-cells. The best-characterized SM protein is synaptic Munc18a [1,6], with diverse roles that include orchestrating SG docking and priming [7,8], chaperoning and stabilizing Syn-1A at the PM [9], inducing activated conformation of Syn-1A [10], and facilitating membrane fusion [11,12]. These functions of Munc18a, elucidated in neurons, are likely conserved in beta-cells to mediate exocytosis of predocked insulin SGs that underlie first-phase secretion by its interaction with Syn-1A SNARE complex (with SNAP25 and VAMP2) [11,12]. Recently, we reported that Munc18b activated and induced the formation of Syn-3 SM/SNARE complexes that mediated additional primary exocytosis as well as SG–SG fusion in rat pancreatic beta-cells [13]. We subsequently showed that Munc18b's cognate SNARE complex consisted of Syn-3 [14] and VAMP8 [15] in mediating exocytosis of newcomer SGs. Newcomer SGs, unlike predocked SGs, undergo minimal residence or docking time at the PM before fusion [15–17], which accounts for second-phase GSIS in its entirety and a considerable portion of first-phase GSIS.
Munc18c and cognate Syn-4 have been well studied to mediate GLUT4 vesicle translocation to PM and exocytosis in adipose tissue and muscle and also some aspects of GSIS [18,19]. Optimal levels of Munc18c play a pivotal role in maintaining peripheral insulin sensitivity and glucose uptake [20]. A transgenic mouse model of Munc18c protein overexpression in adipose, skeletal muscle, and pancreas, exhibits peripheral insulin resistance resulting from impaired insulin-stimulated glucose uptake and GLUT4 vesicle exocytosis, and decreased GSIS [21]. Islets isolated from Munc18c (−/+) heterozygous knockout mice were selectively deficient in second-phase GSIS, postulated to impact SG localization onto the PM and reduced Syn-4 availability for binding VAMP2 required to proceed to SG docking/fusion [22]. More recent studies show that stabilization of Syn-4 at the PM requires Munc18c's binding to its N-terminal peptide, but its basolateral targeting may be Munc18c-independent [23]. The precise role of Munc18c in each of these SG transportation stages is yet to be determined but likely involves Syn-4 activation and SG docking [24]. Another study suggests Munc18c, unlike Munc18a, binds not only t-SNAREs but also v-SNAREs (several VAMPs) to promote trans-SNARE zippering at the post-docking stage of the fusion reaction, thus accelerating fusion kinetics [25].
In this study, we found that depletion of endogenous Munc18c with consequent reduction of Syn-4 levels in human islet beta-cells reduced exocytosis of predocked and newcomer SGs in first-phase GSIS and newcomer SGs in second-phase GSIS. This is reminiscent and redundant to Munc18a/Syn-1A SNARE complex's role in predocked insulin SGs and Munc18b/Syn-3 SNARE complex's role in newcomer SGs.
2. Methods
2.1. Cell culture and lentivirus transduction
Human islets from institutional review board approved healthy donors (with either written informed consent from the donor or the next of kin obtained) were isolated and provided by the IsletCore (P. MacDonald) of the University of Alberta, Canada; and its use approved by the Institutional Review Board at the University of Toronto. The islets were dispersed into single cells using a Ca2+/Mg2+-free phosphate-buffered saline (at 5 mmol/l EDTA) with 0.25 mg/ml trypsin at 37 °C for 6 min with gentle shaking and then resuspended in enriched RPMI-1640 media containing 5 mmol/l d-glucose. The resulting cell suspensions were plated on glass coverslips and allowed to adhere approximately 48 h before experiments with 10% FBS and penicillin/streptomycin at 37 °C in an atmosphere of 5% CO2. Control (scrambled shRNA) and Munc18c shRNA lenti-viruses (CACCTTGGTGTTCCCATTGTT) are from GenTarget Inc (GenTarget Inc, San Diego, CA, USA). Human islets or single cells were transduced with lenti-control/RFP or lenti-Munc18c shRNA/RFP for 48 h. Cellular entry of viral particles after transduction was determined by RFP expression observed by epifluorescence imaging on Nikon TE2000U inverted microscopes when performing the patch clamp and TIRF imaging studies.
2.2. Western blotting
Western blots of human pancreatic islets and Wistar rat brain (as control) lysate samples were prepared and separated on 12–15% gradient SDS-PAGE, transferred to nitrocellulose membrane and individual proteins identified by specific primary antibodies (Synaptic Systems, Goettingen, Germany), unless otherwise indicated, against: Munc18c (gift from D. Thurmond, Indiana University, Indiana), Syn-3, Syn-4, SNAP25, SNAP23, VAMP2, VAMP8 (gift from W. Hong, Institute of Molecular and Cell Biology, Singapore) and tubulin (as loading control). Protein bands were visualized by chemiluminescence (Pierce, Nepean, ON, Canada). All of these antibodies have been validated and used in similar experiments done in the current work as in our previous reports [13,15].
2.3. Confocal microscopy
Samples (human single beta cells and C57BL/6 mouse beta cells) were washed with PBS, fixed in 4% paraformaldehyde for 10 min, and permeabilized in 0.2% Triton X-100 buffer for 10 min at room temperature. After incubation (30 min) with blocking buffer (10% goat serum), samples were incubated overnight at 4 °C with mouse anti-insulin (1:200, Sigma Aldrich) or rabbit anti-glucagon (1:200, Dako Diagnostics, Mississauga, ON, Canada), and coimmunostained with rabbit anti-Munc18c (1:200, Synaptic Systems, Goettingen, Germany) antibodies. Cells were washed with PBS, incubated with appropriate secondary antibodies, and then mounted with fluorescent mounting medium (Dako Cytomation, Carpinteria, CA, USA). Images were examined using a Leica DMIRE2 inverted fluorescence microscope (Leica Microsystems, Weitzlar, Germany) equipped with a Hamamatsu Back-Thinned EM-CCD camera (Hamamatsu Corp., Bridgewater, NJ, USA) and spinning disk confocal scan head. Data acquisition and analysis were performed using Volocity software (PerkinElmer, Waltham, MA, USA). In our experiments, we chose the 20X and 63X objectives and the 491, 561, and 638 nm lasers to excite the Fluorescein isothiocyanate (FITC), Texas Red or RFP, and Cy5 dyes, respectively. All the images were subjected to deconvolution to remove the background noise.
2.4. Islet perifusion
Human islets were infected with lenti-control/RFP and lenti-Munc18c shRNA/RFP for 48 h. Perifusion assays on human islets were performed as reported previously [15], with secreted insulin determined by a RIA kit (Linco, St Louis, MO, USA). Results are presented as insulin secretion normalized to total insulin content.
2.5. Electrophysiology
Capacitance measurements were performed as previously reported [26]. Briefly, patch electrodes were pulled from 1.5-mm thin-walled borosilicate glass, coated close to the tip with orthodontic wax (Butler, Guelph, ON, Canada), and polished to a tip resistance of 2–3 MΩ when filled with intracellular solution. For measurement of membrane capacitance, the intracellular solution contains (in mmol/l): 125 Cesium glutamate, 10 CsCl, 10 NaCl, 1 MgCl2, 5 HEPES, 0.05 EGTA, 3 MgATP, and 0.1 cAMP (pH 7.2). The extracellular solution contains (in mmol/l): 118 NaCl, 5.6 KCl, 1.2 MgCl2, 10 CaCl2, 20 tetraethylammonium chloride, 5 HEPES, and 5 d-glucose (pH 7.4). Cm was estimated by the Lindau-Neher technique, implementing the “Sine-DC” feature of the Lock-in module (40 mV peak-to-peak and a frequency of 1 kHz) in the whole-cell configuration. Recordings were conducted using an EPC10 patch clamp amplifier and the Pulse and X-chart software programs (HEKA Electronik, Lambrecht, Germany). Exocytic events were elicited by a train of ten 500-ms depolarization pulses from −70 mV to 0 mV. All recordings were performed at 30 °C.
2.6. TIRFM and data analysis
Our TIRF microscope setup (Nikon, Toronto, ON, Canada) was constructed based on the prismless and through-the-lens configuration. Briefly, a condenser coupling multiple lasers (440 nm, 488 nm, 543 nm) was attached to the back port of our Nikon TE2000U inverted microscope, equipped with 60X oil immersion objective lens (NA = 1.49). We used a 488-nm beam to excite EGFP, and 488RDC longpass dichroic and 525/50-nm band-pass emission filters. Images were collected with a cooled 16-bit EM-CCD camera (Cascade 512, Roper Scientific, Martinsried, Germany). The penetration depth of the evanescent field (∼100 nm) was aligned by measuring the incidence angle of the 488-nm laser beam with a prism (n = 1.5163). Images were acquired at 5-Hz with a 100-ms exposure time. Fusion events, indicated by abrupt brightening of neuropeptide Y (NPY)-EGFP fluorescence, were manually selected as we previously reported in detail [15,27]. Insulin granule mobilization and exocytosis were analyzed by MatLab (MathWorks, Natickm, MA, USA), ImageJ (NIH, Bethesda, MD, USA), Igor Pro (WaveMetrics, Portland, OR, USA) softwares. A monolayer of human islet beta cells were infected with adenovirus NPY-EGFP and further cultured for 24–36 h before performing TIRFM. Before image acquisition, cells were pre-incubated for 30 min in KRB buffer containing 2.8 mmol/l glucose, and then were stimulated by KRB buffer containing 16.7 mmol/l glucose or 50 mmol/l KCl. All recordings were performed at 37 °C.
2.7. Statistical analysis
Data are presented as means ± SEMs. Statistical comparisons were performed using two-tailed Student's unpaired t-test, Manne Whitney test, one-way ANOVA or two-way ANOVA (SigmaStat. Systat Software Inc. Chicago, IL, USA) and considered significant when P < 0.05.
3. Results
3.1. Munc18c is present in human islet cells and its expression was effectively depleted by lenti-shRNA
Confocal microscopy imaging showed that Munc18c is present in dispersed human islet cells including all beta-cells and also all alpha-cells (Figure 1A). Magnified images show Munc18c is abundant in the cytosolic compartment in human and C57BL/6 mouse beta-cells with small portions colocalized with insulin SGs (Figure 1B). To assess the function of endogenous Munc18c, we employed lentivirus shRNA/RFP to deplete endogenous Munc18c protein (Figure 1C–E). Western blots showed that lenti-shRNA/RFP treatment depleted endogenous Munc18c levels by 68% in human islets (Figure 1D,E). Of note, the levels of cognate Syn-4 were reduced by 60%, whereas levels of VAMP2, VAMP8 and non-cognate Syn-1A were not altered. The coordinate reduction in Syn-4 with Munc18c depletion is not consistent with previous a report in Munc18c heterozygous(−/+) knockout mice [22], probably due to the more severe knockdown of Munc18c expression level in this study. Nonetheless, this is consistent with the current thinking that Munc18 proteins chaperone and stabilize their cognate syntaxins at their target membrane compartment [9]. When performing triple labeling of lenti-Munc18c-shRNA/RFP, Munc18c/FITC and Insulin/Cy5 in dispersed human islet-cells, lenti-Munc18c-shRNA/RFP infection depleted Munc18c in majority of human beta-cells (82.4% of all cells in average), and only a small residual Munc18c (weak Munc18c signal) was left in some RFP-positive beta-cells (Figure 1C). Therefore, single RFP-positive beta-cells exhibited near-total depletion of Munc18c, which is ideal for single cell analysis of Munc18c secretory function.
Figure 1.
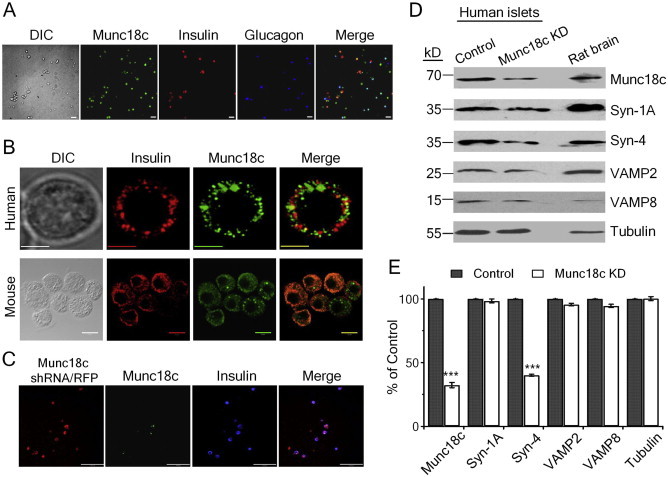
Munc18c is present in human islet beta-cells and alpha-cells and its expression is depleted by lenti-shRNA. (A) Representative confocal images show Munc18c (green) is present in both insulin-containing beta-cells (red) and glucagon-containing alpha-cells (blue). Scale bars: 100 μm. (B) Representative confocal images show Munc18c is abundant in the cell cytosol of human (top images, scale bars: 5 μm) and C57BL/6 mouse (bottom images; scale bars: 10 μm) beta-cells. (C) Triple labeling of lenti-Munc18c-shRNA/RFP, Munc18c/FITC and insulin/Cy5 in dispersed human islet cells. Scale bars: 100 μm. (D–E) Western blotting analysis of lenti-Munc18c-shRNA/RFP-induced knockdown of Munc18c expression in human islets. Representative blots of three separate experiments, where means ± SEMs are shown in (E).
3.2. Munc18c depletion in human islets reduced biphasic GSIS
To examine the function of endogenous Munc18c on biphasic GSIS, human islets were transduced with lenti-control/RFP or lenti-Munc18c-shRNA/RFP, and GSIS assessed by islet perifusion assay. Basal insulin secretion in response to low glucose was similar. Biphasic secretory pattern in response to high glucose (16.7 mmol/l) was disrupted in Munc18c-KD islets with first-phase characteristic transient burst of insulin release inhibited, and subsequent sustained second-phase release also reduced (Figure 2A). Area under the curve (AUC) analysis showed that Munc18c depletion resulted in 42% reduction (Munc18c-KD: 2.48 ± 0.16; control: 4.24 ± 0.21; p < 0.001) in first-phase (8–21 min), 35% reduction (Munc18c-KD: 1.81 ± 0.14; control 2.79 ± 0.35; p < 0.01) in second-phase (22–46 min) (Figure 2B). There is no difference in total insulin content between control and Munc18c-KD islets (Munc18c-KD: 122.1 ± 1.6 μg; control: 129.3 ± 2.9 μg; p = 0.07). This result demonstrating that Munc18c depletion in human islets causing moderate reduction of both first- and second-phase GSIS suggests that first- and second-phase GSIS are mediated by both Munc18c-dependent or -independent pathways.
Figure 2.
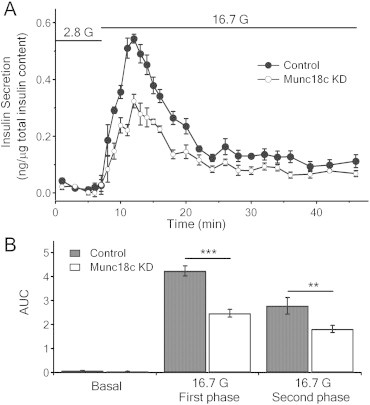
Depletion of Munc18c in human islets causes reduction in first- and second-phase GSIS. (A) Human islets infected with lenti-Munc18c-shRNA/RFP or lenti-/RFP (control) were subjected to islet perifusion assays. (B) Area under the curve (AUC) analysis of first-phase (8–21 min), second-phase (22–46 min) of GSIS. Basal secretion was not significantly affected. **p < 0.01; ***p < 0.001; N = 6.
3.3. Munc18c depletion reduced depolarization–induced exocytosis of SGs from RRP and reserve pool
It was postulated that exocytosis of RRP and mobilization from reserve pool correspond to first- and second-phase GSIS, respectively [28]. To examine endogenous Munc18c function on insulin SG pools and exocytotic kinetics, we performed patch clamp Cm measurement using lenti-control/RFP- and Munc18c-shRNA/RFP-expressing human beta-cells. Beta-cells were identified by their large size with the average of ∼10 μm as previously reported [29]. Insulin exocytosis was elicited by a train of ten 500-ms depolarization pulses, with the first two pulses approximating the size of RRP of primed and fusion-ready SGs; and subsequent pulses estimating the rate of SG refilling or mobilization from reserve pool(s) to RRP, where SGs are subsequently primed for fusion competence [30]. Cm increase in Munc18c-KD cells was greatly inhibited at every depolarizing pulse from the 2nd pulse to the 10th pulse (Figure 3A,B) compared with control cells, in which the size of RRP (ΔCm1st-2nd pulse) was markedly reduced by 52% (Munc18c-KD: 5.33 ± 1.24 fF/pF; control: 11.03 ± 2.52 fF/pF; p < 0.05; Figure 3C), and rate of SG refilling/mobilization was severely reduced by 57% (Munc18c-KD: 9.14 ± 2.24 fF/pF; control: 21.2 ± 4.89 fF/pF; p < 0.05; Figure 3C). This result suggests that Munc18c depletion greatly impairs depolarization-induced exocytosis by affecting RRP and mobilization of SGs from reserve pool(s).
Figure 3.
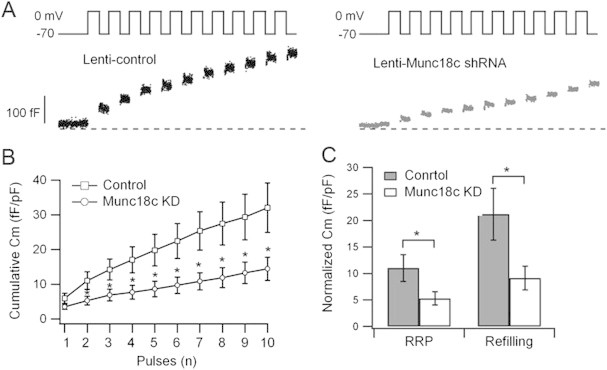
Depletion of Munc18c in human beta-cells causes reduction in RRP and SG pool mobilization. Patch clamp Cm performed on single human beta-cells (RFP-positive) infected with lenti-control shRNA/RFP or lenti-Munc18c-shRNA/RFP. (A) Representative recordings of exocytosis during a train of 500 ms depolarizations from −70 to 0 mV. (B) Cumulative changes in cell capacitance normalized to basal cell membrane capacitance (fF/pF) in control (n = 11 cells) and Munc18c-KD (n = 12 cells) beta-cells. (C) Size of RRP (ΔCm1st–2nd pulse) and rate of SG mobilization (ΔCm3rd–10th pulse). Values represent means ± SEMs. *p < 0.05.
3.4. Munc18c depletion diminishes biphasic GSIS by inhibiting pre-docked and newcomer SGs exocytosis
In order to visualize the spatio-temporal mobilization of populations of SGs and single SG fusion dynamics, we employed time-lapse TIRFM to monitor exocytosis of SGs tagged with NPY-EGFP. At basal unstimulated state, punctate fluorescence, indicating docked SGs, were not different between control (10.9 ± 0.77/100 μm2) and Munc18c-KD (10.5 ± 0.56/100 μm2) beta-cells (Figure 4A,B). On stimulation, single SG fusion events were observed as flashes of fluorescence that rapidly dissipated in a cloud-like diffusion pattern. We verified that fluorescence dissipation was not due to photobleaching [14,27]. Assessment of cumulative fusion events over time (Figure 4C) showed much less fusion events during the 13 min stimulation in Munc18c-KD beta-cells than control beta-cells. These exocytotic events, however, were not uniform but could be categorized into three distinct modes of exocytosis. ‘Pre-dock’ fusion mode refers to SGs that were already docked onto PM for a period of time prior to stimulation (black in Figure 4D,E). ‘Newcomer SGs’ were new SGs appearing de novo under evanescent field after stimulation and then underwent exocytosis; and appear in two distinct patterns as described previously [15,17,27,31]. ‘No-dock’ newcomer SGs (white in Figure 4D,E) were newly recruited by stimulation and immediately fused with PM (docking state of <200 ms, the minimal interval between two consecutive frames). ‘Short-dock’ newcomer SGs (grey in Figure 4D,E) were those newly recruited by stimulation, which first docked for some time varying from seconds to minutes at the PM, then fused with PM. Beta-cells were identified by their large size and response to the application of glucose.
Figure 4.
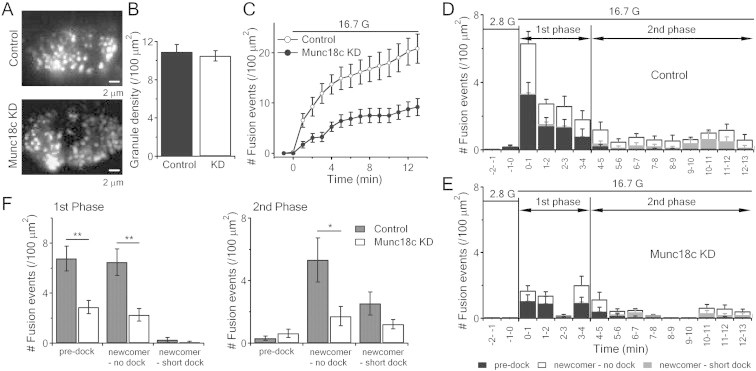
Munc18c depletion inhibits biphasic GSIS by reducing pre-docked and no-docked newcomer SGs exocytosis. (A) TIRF images of docked insulin SGs in control (top) or Munc18c-KD (bottom) human islet beta-cells. Scale bars, 2 μm. (B) Comparison of averaged SG densities before stimulation. (C) Normalized cumulative fusion events of SGs per unit area from control and Munc18c-KD beta-cells. (D,E) Histograms of fusion events in first-phase (first 4 min stimulation) and second-phase (5–13 min) showing insulin SG exocytosis dynamics caused by 16.7 mM glucose stimulation from lenti-control/RFP (D) and lenti-Munc18c-shRNA/RFP (E) -treated human beta-cells. Black, white and grey bars indicate pre-docked, no-dock and short-dock newcomer SGs, respectively; Control: 12 cells; Munc18c-KD: 11 cells, expressed as means ± SEMs. (F) Summary of the three modes of fusion events in first (left) and second phases (right), shown as means ± SEMs; *p < 0.05; **p < 0.01.
We assessed the pattern(s) of fusion events impacted by knockdown of Munc18c with glucose stimulation. At 2.8 mmol/l glucose, there were few spontaneous fusion events of mainly pre-docked SGs (black bars in Figure 4D,E). With high-glucose stimulation (16.7 mmol/l), both pre-docked and newcomers SGs had similar contributions to first-phase GSIS, but newcomer SGs accounted for almost all of second-phase GSIS. In first-phase GSIS, there was obvious inhibition of pre-docked (control: 6.76 ± 0.99, n = 12 cells vs Munc18c-KD: 2.89 ± 0.53 events/100 μm2, n = 11 cells; p < 0.01) and no-docked (control: 6.47 ± 1.05 vs Munc18c-KD: 2.27 ± 0.52 events/100 μm2; p < 0.01) newcomer SGs from Munc18c-KD cells (Figure 4D–F). In second-phase GSIS, there was reduction in only no-docked newcomer SGs (control: 5.33 ± 1.41 vs Munc18c-KD: 1.72 ± 0.62 events/100 μm2; p < 0.05) (Figure 4D–F). Munc18c depletion therefore affects pre-docked and newcomer SG fusion underlying first- and second-phase GSIS in human islet beta-cells, consistent with the islet perifusion (Figure 2) and Cm results (Figure 3).
3.5. Munc18c depletion causes reduction in exocytosis of previously-docked SGs during high-K+ stimulation
Munc18c depletion affecting both phases of GSIS in our study seems inconsistent with a previous study showing heterozygous Munc18c-KO mice islets affecting predominantly the second-phase GSIS [22]. Since a major portion of first-phase GSIS is attributed to predocked SGs, which are preferentially released by high-K+ stimulation in the first few minutes [28,32], we employed this protocol on Munc18c-KD human beta-cells. Consistently, 50 mmol/l KCl triggered exocytosis of mainly previously-docked (pre-docked) SGs (black bars) in the first few minutes from control and Munc18c-KD cells (Figure 5A,B). Here, there was a 39% reduction in exocytosis in the first 3 min (Figure 5C) accounted for mostly from a reduction of exocytosis of pre-docked SGs in the Munc18c-KD cells, compared with control cells (summary analysis in Figure 5D; 6.65 ± 1.55 events/100 μm2 in Munc18c-KD vs 10.20 ± 2.84 events/100 μm2 in Control). High-K+ stimulation also evoked a release of a small number of newcomer-no dock SG fusion events (white bars, Figure 5A,B), that was much less than glucose stimulation (Figure 4); and which was not significantly affected by Munc18c-KD (Figure 5D). Newcomer short-dock SG fusion events was seldom observed during the 3-minute of acquisition. These results confirm that for high K+-induced exocytosis, initial docking of insulin SGs on PM is required. More importantly, these results further confirmed that Munc18c is involved in mediating exocytosis of previously-docked SGs, which may be associated with its cognate Syn-4-dependent pathway postulated to mediate first-phase GSIS in addition to second-phase GSIS [18,33–35].
Figure 5.
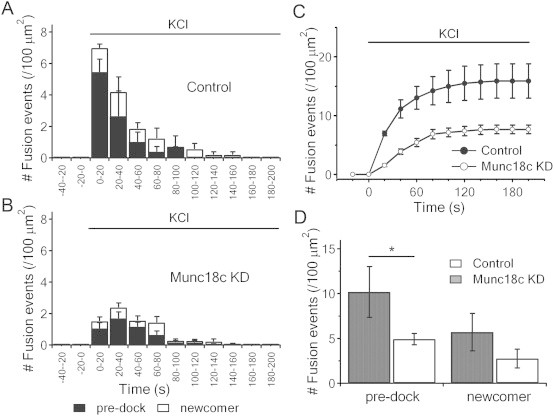
Munc18c depletion diminishes high-K+-evoked exocytosis of previously-docked insulin SGs. (A,B) Insulin SG exocytosis dynamics evoked by 50 mmol/l KCl from control (A) and Munc18c-KD (B) human beta-cells. Black and white bars indicate pre-docked and newcomer SGs, respectively. Data from 10 cells for each group, expressed as means ± SEMs. (C) Normalized cumulative fusion events of SGs per unit area from control (black circles) and Munc18c-KD (white circles) beta-cells. (D) Summary of fusion events from pre-docked and newcomer SGs in first- and second-phase after 50 mmol/l glucose stimulation. Values represent means ± SEMs. *p < 0.05.
3.6. GLP-1 potentiation rescues the reduction of pre-docked SG exocytosis but not that of newcomer SG fusion
With 10 nmol/L GLP-1 potentiation of high-glucose (16.7 mmol/L) stimulation, all exocytotic events were amplified including predocked and newcomer SGs in control and Munc18c-KD cells (Figure 6, compared Figure 4) as we had reported before [15,27,36]. In these GLP-1-treated human beta-cells, Munc18c depletion could still greatly reduce the no-docked (control: 33.0 ± 2.94 vs Munc18c-KD: 13.73 ± 1.73 events/100 μm2; p < 0.01; n = 10 cells for each) newcomer SGs fusion (Figure 6C,D). However, pre-docked SGs fusions were very similar between the two groups (Control: 11.52 ± 1.52 vs Munc18c-KD: 10.95 ± 1.23 events/100 μm2). Thus, while GLP-1 potentiation amplified the recruitment of pre-docked and newcomer-no dock SGs for exocytosis, GLP-1-activated cAMP/PKA pathways could only fully rescue the pre-docked SGs exocytotic deficiency caused by the Munc18c depletion. This result suggests that GLP-1 could activate an alternate Munc18c-independent pathway to overcome the Munc18c deficiency. This may in part explain why first-phase GSIS was less sensitive to the heterologous Munc18c deletion in mice [22], wherein the level of Munc18c depletion (∼50% in each beta-cell) was much less than the near-total depletion of Munc18c in the lenti-Munc18c-shRNA/RFP-infected human beta-cells in this study. Another explanation is that GLP-1 signaling might prefer the Munc18a/Syn-1A SNARE complex that likely has a more dominant effect on predocked SGs capable of overcoming the Munc18c deficiency. Second-phase GSIS, attributed almost entirely to newcomer SGs fusion, remained sensitive to the Munc18c deficiency that could not be rescued by GLP-1-activated pathways, supporting the study by D. Thurmond's group [22]. More work will be required to elucidate how GLP-1-triggered cAMP and PKA signaling or their activated accessory exocytotic molecules [37] distinctly affect Munc18c's actions on predocked SGs and their reduced effects on newcomer SGs.
Figure 6.
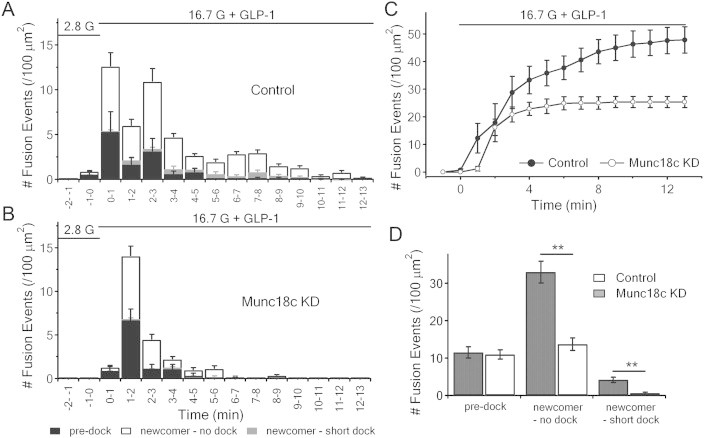
GLP-1-potentiated GSIS rescues only pre-docked SGs exocytosis. (A,B) Insulin SG exocytosis dynamics caused by 16.7 mmol/l glucose and 10 nmol/l GLP-1 from lenti-control/RFP and lenti-Munc18c-shRNA/RFP-treated human beta-cells. Data obtained from 10 cells for each, and expressed as means ± SEMs. (C) Normalized cumulative fusion events of insulin SGs per unit area from control (black circles) and Munc18c-KD (white circles) human beta-cells. (D) Summary of fusion events from pre-docked SGs and newcomer SGs after 16.7 mmol/l glucose and 10 nmol/l GLP-1 stimulation. Values represent the means ± SEMs. **p < 0.01.
4. Discussion
In this study, we have illuminated in human beta-cells that Munc18c acts on distinct SG pools to mediate biphasic GSIS. Depletion of endogenous Munc18c employing lenti-shRNA/RFP in human pancreatic islets (Figure 1) inhibited GSIS in both first- and second-phase (Figure 2), which is due to reduction in the RRP and mobilization from the reserve pool (Figure 3). TIRFM illustrated that Munc18c-KD inhibition of first-phase GSIS was attributed to reduction in exocytosis of both pre-docked and newcomer (no-docked) SGs, and second-phase inhibition was attributed to reduction in exocytosis of no-docked newcomer SGs (Figure 4). Munc18c's role in mediating pre-docked SGs exocytosis was verified by employing high-K+ stimulation known to preferentially act on predocked SGs (Figure 5). Finally, we found that GLP-1 potentiation can rectify the defective pre-docked SGs fusion caused by Munc18c depletion, but cannot rescue the reduction of newcomer SGs exocytosis induced by Munc18c-KD (Figure 6). We conclude that Munc18c regulates exocytosis of both pre-docked and newcomer SG pools underlying biphasic GSIS in human islet beta-cells.
Distinct SM/SNARE complexes and regulators confer distinct exocytoses [16]. The synaptic SM protein Munc18a (known also as Munc18-1) acts to prime Syn-1A to form the SNARE complex with SNAP25 and VAMP2 that mediates docking and fusion of SGs with the PM [1,6]. These pre-docked SGs sit on the PM for a long time until Ca2+ release is evoked to trigger exocytosis [1,6,28], accounting for first-phase GSIS [1,6,28]. More recent work showed that primary exocytosis is contributed by newcomer SGs [16,17,31], which in fact exceeded pre-docked SGs in the overall GSIS [16]. Remarkably, these newcomer SG are recruited to PM to undergo exocytosis with minimal to no residence time on the PM [16,17]. We had identified the exocytotic machinery mediating newcomer SGs [14,15,26,27], which is a second SM/SNARE complex comprised of Munc18b, Syn-3, VAMP8 and SNAP25. In that body of work, we showed that depletion of endogenous Syn-3 [14] or VAMP8 [15] reduced newcomers SGs; and when exogenously expressed, restored the deficient newcomers SGs, without affecting predocked SGs.
Reduced protein and/or mRNA levels of key exocytotic syntaxins (Syn-1A, Syn-4) and their cognate SM proteins (Munc18a, Munc18c) have been reported in islets and skeletal muscle of diabetic and obese human patients [38–40]. It has long been assumed that these SM and syntaxins are faithful partners, Munc18c with Syn-4 and Munc18a with Syn-1A. This is actually not the case as shown by two studies, whereby both Munc18a and Munc18c were discovered to be promiscuous in binding multiple syntaxin partners [25,34]. In the Munc18c report that employed in vitro membrane fusion assays, Munc18c could activate multiple t-SNARE pairs, including Syn-2/SNAP23, Syn-3/SNAP23 and Syn-4/SNAP23 [25]; we supposed that SNAP25 could be exchanged for SNAP23 in beta-cells. It is possible that Munc18c actions on newcomer SGs could be attributed to its possible binding and activation of Syn-3 shown to mediate newcomer SG recruitment and fusion [22], a function redundant to Munc18b [12]. Whereas Munc18c activation of Syn-4 prefers VAMP2 to form the SNARE complex that mediates exocytosis of predocked SGs [13], Munc18c/Syn-4 complex could also potentially assemble with VAMP8 as well described to occur in pancreatic acini to mediate exocytosis at the basolateral PM [41]; and through VAMP8 mediate newcomer SG exocytosis [15]. A very recent study demonstrated that Doc2b may act as a PM scaffold for Munc18c and Munc18a to dock on to form a macromolecular complex, to perhaps then promote their respective SNARE complex assemblies in pancreatic beta-cells [42]. Much further work will be required to dissect these and other possibilities on how distinct Munc18c-activated molecular machineries mediate the recruitment and/or exocytosis of both predocked and newcomer SGs. To this end, Munc18c was found to be rather different from Munc18a in its ability to directly bind VAMPs and to activate syntaxins without undergoing the initial step of binding syntaxins in the inactive ‘closed’ conformation that prevents fusion [20]. Furthermore, while Munc18a- and Munc18b-activated SNARE complexes seem to prefer predocked and newcomer SGs, respectively [reviewed in ref. 35], Munc18c-activated exocytotic machineries are capable of acting on both SG pools. Taken together, restoring or even overexpressing Munc18c into diabetic islets could be a new and preferred avenue to treat type-2 diabetes in effectively rescuing both first- and second-phase GSIS.
Acknowledgments
This work was supported by grants to H. Y. Gaisano from the Canadian Institute of Health Research (MOP 86544 and MOP 89889). Some of the equipment used in this study was supported by the 3D (Diet, Digestive Tract and Disease) Centre funded by the Canadian Foundation for Innovation and Ontario Research Fund, project number 19442.
Conflict of interest
The authors declare that there is no conflict of interest associated with this manuscript.
References
- 1.Sudhof T.C., Rothman J.E. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323(5913):474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jahn R., Scheller R.H. SNAREs–engines for membrane fusion. Nature Reviews Molecular Cell Biology. 2006;7(9):631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 3.Garcia E.P., Gatti E., Butler M., Burton J., De Camilli P. A rat brain Sec1 homologue related to Rop and UNC18 interacts with syntaxin. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(6):2003–2007. doi: 10.1073/pnas.91.6.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamori Y., Kawanishi M., Niki T., Shinoda H., Araki S., Okazawa H. Inhibition of insulin-induced GLUT4 translocation by Munc18c through interaction with syntaxin4 in 3T3-L1 adipocytes. The Journal of Biological Chemistry. 1998;273(31):19740–19746. doi: 10.1074/jbc.273.31.19740. [DOI] [PubMed] [Google Scholar]
- 5.Tellam J.T., McIntosh S., James D.E. Molecular identification of two novel Munc-18 isoforms expressed in non-neuronal tissues. The Journal of Biological Chemistry. 1995;270(11):5857–5863. doi: 10.1074/jbc.270.11.5857. [DOI] [PubMed] [Google Scholar]
- 6.Burgoyne R.D., Barclay J.W., Ciufo L.F., Graham M.E., Handley M.T., Morgan A. The functions of Munc18-1 in regulated exocytosis. Annals of the New York Academy of Sciences. 2009;1152:76–86. doi: 10.1111/j.1749-6632.2008.03987.x. [DOI] [PubMed] [Google Scholar]
- 7.Deak F., Xu Y., Chang W.P., Dulubova I., Khvotchev M., Liu X. Munc18-1 binding to the neuronal SNARE complex controls synaptic vesicle priming. The Journal of Cell Biology. 2009;184(5):751–764. doi: 10.1083/jcb.200812026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voets T., Toonen R.F., Brian E.C., de Wit H., Moser T., Rettig J. Munc18-1 promotes large dense-core vesicle docking. Neuron. 2001;31(4):581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- 9.Arunachalam L., Han L., Tassew N.G., He Y., Wang L., Xie L. Munc18-1 is critical for plasma membrane localization of syntaxin1 but not of SNAP-25 in PC12 cells. Molecular Biology of the Cell. 2008;19(2):722–734. doi: 10.1091/mbc.E07-07-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerber S.H., Rah J.C., Min S.W., Liu X., de Wit H., Dulubova I. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science. 2008;321(5895):1507–1510. doi: 10.1126/science.1163174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oh E., Kalwat M.A., Kim M.J., Verhage M., Thurmond D.C. Munc18-1 regulates first-phase insulin release by promoting granule docking to multiple syntaxin isoforms. The Journal of Biological Chemistry. 2012;287(31):25821–25833. doi: 10.1074/jbc.M112.361501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen J., Tareste D.C., Paumet F., Rothman J.E., Melia T.J. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128(1):183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Lam P.P., Ohno M., Dolai S., He Y., Qin T., Liang T. Munc18b is a major mediator of insulin exocytosis in rat pancreatic beta-cells. Diabetes. 2013;62(7):2416–2428. doi: 10.2337/db12-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu D., Koo E., Kwan E., Kang Y., Park S., Xie H. Syntaxin-3 regulates newcomer insulin granule exocytosis and compound fusion in pancreatic beta cells. Diabetologia. 2013;56(2):359–369. doi: 10.1007/s00125-012-2757-0. [DOI] [PubMed] [Google Scholar]
- 15.Zhu D., Zhang Y., Lam P.P., Dolai S., Liu Y., Cai E.P. Dual role of VAMP8 in regulating insulin exocytosis and islet beta cell growth. Cell Metabolism. 2012;16(2):238–249. doi: 10.1016/j.cmet.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Gaisano H.Y. Here come the newcomer granules, better late than never. Trends in Endocrinology & Metabolism. 2014;25(8):381–388. doi: 10.1016/j.tem.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Shibasaki T., Takahashi H., Miki T., Sunaga Y., Matsumura K., Yamanaka M. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jewell J.L., Oh E., Thurmond D.C. Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. The American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2010;298(3):R517–R531. doi: 10.1152/ajpregu.00597.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jewell J.L., Oh E., Ramalingam L., Kalwat M.A., Tagliabracci V.S., Tackett L. Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. The Journal of Cell Biology. 2011;193(1):185–199. doi: 10.1083/jcb.201007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramalingam L., Yoder S.M., Oh E., Thurmond D.C. Munc18c: a controversial regulator of peripheral insulin action. Trends in Endocrinology & Metabolism. 2014;25(11):601–608. doi: 10.1016/j.tem.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spurlin B.A., Thomas R.M., Nevins A.K., Kim H.J., Kim Y.J., Noh H.L. Insulin resistance in tetracycline-repressible Munc18c transgenic mice. Diabetes. 2003;52(8):1910–1917. doi: 10.2337/diabetes.52.8.1910. [DOI] [PubMed] [Google Scholar]
- 22.Oh E., Thurmond D.C. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes. 2009;58(5):1165–1174. doi: 10.2337/db08-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torres J., Funk H.M., Zegers M.M., ter Beest M.B. The syntaxin 4 N terminus regulates its basolateral targeting by munc18c-dependent and -independent mechanisms. The Journal of Biological Chemistry. 2011;286(12):10834–10846. doi: 10.1074/jbc.M110.186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.James D.E. MUNC-ing around with insulin action. The Journal of Clinical Investigation. 2005;115(2):219–221. doi: 10.1172/JCI24158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H., Rathore S.S., Lopez J.A., Davis E.M., James D.E., Martin J.L. Comparative studies of Munc18c and Munc18-1 reveal conserved and divergent mechanisms of Sec1/Munc18 proteins. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(35):E3271–E3280. doi: 10.1073/pnas.1311232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie L., Zhu D., Kang Y., Liang T., He Y., Gaisano H.Y. Exocyst sec5 regulates exocytosis of newcomer insulin granules underlying biphasic insulin secretion. PLoS One. 2013;8(7):e67561. doi: 10.1371/journal.pone.0067561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie L., Zhu D., Gaisano H.Y. Role of mammalian homologue of Caenorhabditis elegans unc-13-1 (Munc13-1) in the recruitment of newcomer insulin granules in both first and second phases of glucose-stimulated insulin secretion in mouse islets. Diabetologia. 2012;55(10):2693–2702. doi: 10.1007/s00125-012-2640-z. [DOI] [PubMed] [Google Scholar]
- 28.Rorsman P., Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46(8):1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 29.Braun M., Ramracheya R., Bengtsson M., Zhang Q., Karanauskaite J., Partridge C. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57(6):1618–1628. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- 30.Gillis K.D., Mossner R., Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16(6):1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 31.Yasuda T., Shibasaki T., Minami K., Takahashi H., Mizoguchi A., Uriu Y. Rim2alpha determines docking and priming states in insulin granule exocytosis. Cell Metabolism. 2010;12(2):117–129. doi: 10.1016/j.cmet.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 32.Olofsson C.S., Gopel S.O., Barg S., Galvanovskis J., Ma X., Salehi A. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Archiv. 2002;444(1–2):43–51. doi: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- 33.Spurlin B.A., Park S.Y., Nevins A.K., Kim J.K., Thurmond D.C. Syntaxin 4 transgenic mice exhibit enhanced insulin-mediated glucose uptake in skeletal muscle. Diabetes. 2004;53(9):2223–2231. doi: 10.2337/diabetes.53.9.2223. [DOI] [PubMed] [Google Scholar]
- 34.Spurlin B.A., Thurmond D.C. Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic beta-cells. Mol Endocrinology. 2006;20(1):183–193. doi: 10.1210/me.2005-0157. [DOI] [PubMed] [Google Scholar]
- 35.Yang C., Coker K.J., Kim J.K., Mora S., Thurmond D.C., Davis A.C. Syntaxin 4 heterozygous knockout mice develop muscle insulin resistance. The Journal of Clinical Investigation. 2001;107(10):1311–1318. doi: 10.1172/JCI12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwan E.P., Gaisano H.Y. Glucagon-like peptide 1 regulates sequential and compound exocytosis in pancreatic islet beta-cells. Diabetes. 2005;54(9):2734–2743. doi: 10.2337/diabetes.54.9.2734. [DOI] [PubMed] [Google Scholar]
- 37.Seino S., Shibasaki T., Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. The Journal of Clinical Investigation. 2011;121(6):2118–2125. doi: 10.1172/JCI45680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergman B.C., Cornier M.A., Horton T.J., Bessesen D.H., Eckel R.H. Skeletal muscle munc18c and syntaxin 4 in human obesity. Nutrition & Metabolism (London) 2008;5:21. doi: 10.1186/1743-7075-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ostenson C.G., Gaisano H., Sheu L., Tibell A., Bartfai T. Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes. 2006;55(2):435–440. doi: 10.2337/diabetes.55.02.06.db04-1575. [DOI] [PubMed] [Google Scholar]
- 40.Oh E., Stull N.D., Mirmira R.G., Thurmond D.C. Syntaxin 4 up-regulation increases efficiency of insulin release in pancreatic islets from humans with and without type 2 diabetes mellitus. The Journal of Clinical Endocrinology & Metabolism. 2014;99(5):E866–E870. doi: 10.1210/jc.2013-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cosen-Binker L.I., Binker M.G., Wang C.C., Hong W., Gaisano H.Y. VAMP8 is the v-SNARE that mediates basolateral exocytosis in a mouse model of alcoholic pancreatitis. The Journal of Clinical Investigation. 2008;118(7):2535–2551. doi: 10.1172/JCI34672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramalingam L., Lu J., Hudmon A., Thurmond D.C. Doc2b serves as a scaffolding platform for concurrent binding of multiple Munc18 isoforms in pancreatic islet beta-cells. Biochemical Journal. 2014;464(2):251–258. doi: 10.1042/BJ20140845. [DOI] [PMC free article] [PubMed] [Google Scholar]