

Abstract
Background
An acute surplus of carbohydrates, and other substrates, can be converted and safely stored as lipids in adipocytes via de novo lipogenesis (DNL). However, in obesity, a condition characterized by chronic positive energy balance, DNL in non-adipose tissues may lead to ectopic lipid accumulation leading to lipotoxicity and metabolic stress. Indeed, DNL is dynamically recruited in liver during the development of fatty liver disease, where DNL is an important source of lipids. Nonetheless, a number of evidences indicates that DNL is an inefficient road for calorie to lipid conversion and that DNL may play an important role in sustaining metabolic homeostasis.
Scope of review
In this manuscript, we discuss the role of DNL as source of lipids during obesity, the energetic efficiency of this pathway in converting extra calories to lipids, and the function of DNL as a pathway supporting metabolic homeostasis.
Major conclusion
We conclude that inhibition of DNL in obese subjects, unless coupled with a correction of the chronic positive energy balance, may further promote lipotoxicity and metabolic stress. On the contrary, strategies aimed at specifically activating DNL in adipose tissue could support metabolic homeostasis in obese subjects by a number of mechanisms, which are discussed in this manuscript.
Keywords: Ectopic lipids, Glucose disposal, Thermogenesis, Lipokines, Obesity, Metabolic flexibility
1. Introduction
Metabolic homeostasis is an essential condition to sustain life, and metabolic derangements are at the origin of several debilitating diseases. Obesity, a pathological condition characterized by excessive accumulation of body lipids, is by far the most prevalent metabolic disease affecting hundreds of millions of people worldwide. The leading cause of this excessive lipid accumulation is a chronic positive energy balance combined with energy partitioning toward lipids. Lifestyle interventions are in principle ideal solutions to fight the obesity pandemics, although in practice the implementation of such measures has largely failed so far [1]. Hence, it is important to better understand the mechanisms driving excessive lipid deposition and the role of such mechanisms in impaired metabolic homeostasis. In this context, it has been proposed that lipids stored in non-adipose tissues (i.e., ectopic lipid accumulation) play a major role in the pathogenesis of obesity-related diseases [2,3]. It is also established that de-novo lipogenesis (DNL) in humans can be, under specific conditions, an important source of adipose tissue lipids and ectopically stored lipids. Indeed, DNL was shown to be an important source of intrahepatocellular lipids in the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Here we review the current literature on the role of DNL in metabolic homeostasis. We conclude that although DNL significantly contributes to ectopic lipid accumulation, the specific recruitment of this pathway may in fact limit the effects of excessive calories on ectopic lipid deposition and glucose intolerance by a number of mechanisms. We also propose that reactivation of DNL in adipose tissue of obese people could be a promising strategy for the treatment of obesity-driven diseases.
2. The DNL road to ectopic lipids
2.1. Contribution of DNL to fatty liver disease
Intracellular lipids can accumulate from dietary lipid influx or can be synthesized de-novo from Acetyl-CoA and Malonyl-CoA produced from the catabolism of different substrates (Figure 1A). For conditions in which lean body mass is not increasing, excess calories from positive energy balance are stored as triglycerides in white adipocytes. Triglycerides are energy dense and chemical stable compounds, and the adipocyte is a specialized cell type, which can safely store large amounts of triglycerides in a monolocular lipid droplet [4]. However, this system has a saturation limit, and excessive calories over a long period may lead to the deposition of triglycerides and other more toxic lipids in non-adipose tissues, leading to lipotoxicity and inflammation [2,3,5–7]. Hence, ectopic lipid deposition is considered a major source of metabolic stress and a major pathogenic factor in obesity-associated diseases. In humans, DNL can be an important contributor to adipose tissue and ectopic lipid accumulation [8,9], and several cell types express the enzymes implicated in this pathway. Because the hepatocyte is the non-adipose cell type displaying the highest lipogenic capacity, NAFLD is an ideal model to investigate the role of DNL in ectopic lipid deposition. The contribution of DNL to fatty acids stored in the liver of patients with NAFLD has been quantified in an elegant study using multiple stable isotopes to trace the different sources of liver fat [10]. DNL contributed about 26% of fatty acids in livers of NAFLD patients, free fatty acids from adipose tissue and from the diet contributed 59% of liver lipids, and dietary triglycerides associated with chylomicrons contributed 15% of liver fat. Hence, DNL can contribute about a quarter of the liver lipids in patients with NAFLD. A more recent study from the same laboratory investigated the contribution of the different lipid sources above to liver fat of human subjects with either low or high liver lipid content but with similar adiposity [11]. The results show that DNL was the only source of lipids displaying a statistically significant increase in subjects with high liver lipid content, compared to those with lower liver lipid content but comparable adiposity [11]. From these results it can be concluded that DNL is specifically recruited during the development of fatty liver. These findings led the authors to suggest that DNL plays a causative role in the pathogenesis of NAFLD and that suppression of DNL by carbohydrate restriction may be a valuable approach for the treatment of NAFLD [11]. In support of the latter concept, it has been reported that carbohydrate restriction clears liver lipids faster than lipid restriction in NAFLD patients [12]. However, no difference in liver lipids was observed between carbohydrate-restricted or fat-restricted patients after a weight loss of about 7% of the starting body weight [12,13]. Furthermore, it is not known whether the faster clearance of hepatic lipids observed during carbohydrate restriction, compared to fat restriction, depends on differences in DNL. Indeed, it is likely that carbohydrate restriction, more than fat restriction, will lead to increased hepatic glucose production, an important caloric output from the liver, which could further promote hepatocyte negative energy balance. Additional evidence to support a role for carbohydrate and DNL in human NAFLD comes from fructose overfeeding studies. Healthy men fed a 35% caloric surplus as fructose for seven days showed significantly elevated intrahepatocellular lipids compared to control subjects not receiving the fructose caloric surplus [14]. Furthermore, extra calories from fructose synergize with extra calories from fat to promote fatty liver [14]. Importantly, it was shown that an extra 25% calories in the form of fructose fed to healthy men for six days is sufficient to cause a marked increase of fractional DNL [15]. Taken together, these studies demonstrate that DNL is a significant source of intrahepatocellular lipid in fatty liver disease, and many have proposed that DNL is a major pathway in the pathogenesis of NAFLD. However, It is important to note that these studies are correlative and do not establish whether the specific recruitment of DNL in people with high-liver lipids is a promoting or a protecting factor in the progression of NAFLD.
Figure 1.
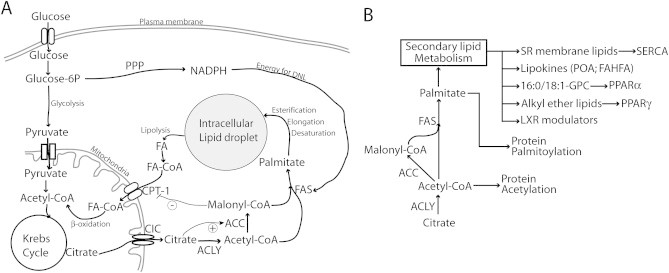
Energetic and biosynthetic functions of de novo lipogenesis (DNL) in the control of metabolic homeostasis. (A) DNL produces lipids by disposing of glucose and calories. Whereas lipids can be directly incorporated into intracellular stores in an energetically efficient manner, the conversion of glucose into intracellular lipids via DNL is a costly process. Glucose enters the cell via specific glucose transporters. It is converted to pyruvate via glycolysis and, in this form, enters the mitochondria where it is converted to acetyl-CoA in order to enter the Krebs cycle. In the presence of excessive glucose and calories, citrate from the Krebs cycle is exported to the cytoplasm via the citrate carrier (CIC). The latter is the first committed step of DNL. Indeed, citrate is a powerful inducer of acetyl-CoA carboxylase (ACC) activity, which produces malonyl-CoA, a major intermediate of fatty acid synthesis and an inhibitor of the fatty acid transporter CPT-1. However, it is important to consider that fatty acid synthase (FAS) consumes malonyl-CoA, limiting its accumulation and consequent inhibition of CPT-1. Because DNL and β-oxydation of fatty acids are distinct pathways, fatty acid synthesis and β-oxidation can occur simultaneously, creating futile cycles as described during brown adipose tissue activation and browning of white adipose tissue. The pentose phosphate pathway (PPP) is an important intracellular source of NADPH, which provides energy for DNL. Altogether DNL is an energetically inefficient way to form intracellular lipids and, in specific circumstances, can act as a considerable sink for calories and glucose. (B) DNL was also proposed to support the synthesis of several signaling molecules implicated in the control of metabolic homeostasis. These include specific lipids of the sarcoplasmic reticulum (SR) membrane controlling SERCA function and intracellular calcium; secreted lipids with cytokine-like activity supporting metabolic homeostasis “lipokines”, such as palmitoleic acid (PAO) and branched fatty acid esters of hydroxy fatty acids (FAHFA); endogenous ligands of nuclear receptors including PPARα (16:0/18:1-GPC), PPARγ (alkyl ether lipids), and possibly LXR. DNL was also implicated in the palmitoylation and acetylation of specific proteins.
2.2. DNL is the most “energetically costly” road for extra calories to ectopic lipid deposition
The studies described above demonstrate that DNL is specifically recruited during the development of NAFLD in humans, and that it significantly contributes to ectopic lipid accumulation. However, it could be argued that the specific recruitment of DNL in people accumulating liver lipids may offer some protection from NAFLD (and ectopic lipid deposition) progression, by minimizing the conversion efficiency of extra calories from positive energy balance to lipids. Indeed, among all the possible sources of ectopic lipid deposition DNL is by far the one that costs the most calories (Figure 1A). Based on stoichiometry, the synthesis of one molecule of palmitate from acetyl-CoA costs 7 molecules of ATP and requires the conversion of 14 high-energy NADPH molecules to NADP+. Different studies evaluating feeding efficiency in pigs consistently indicate an energetic cost for storing fat via DNL of 20%–25% of metabolizable energy intake [16,17]. In humans the energy expenditure for DNL has not been evaluated to the same extent as in pigs, but a study comparing the effects of overfeeding healthy humans with fat or carbohydrates on energy storage reported consistent results with the studies above [18]. Interestingly, a study reported that feeding healthy humans a 35% surplus of calories in the form of fructose for seven days caused an increase of 16% of intrahepatocellular lipids relative to control subjects, whereas a 30% caloric surplus in the form of dietary fat for only four days led to an 86% increase of intrahepatocellular lipids relative to control subjects [14]. It must be mentioned that in this latter, study the authors do not measure DNL, which is limiting data interpretation. Nonetheless, these data indicate that a caloric surplus of fructose (a highly lipogenic nutrient) is much less efficient than a similar caloric surplus of dietary lipids in promoting hepatic lipid deposition [14]. Consistently, in humans DNL has been shown to be positively correlated with post-prandial energy expenditure [19], and carbohydrate overfeeding (leading to induction of DNL gene-expression), but not fat overfeeding, significantly elevated energy expenditure [20,21]. Collectively, these data indicate that carbohydrate conversion to lipids via DNL may lead to significantly less ectopic lipid deposition for the same caloric surplus than what dietary lipids or lipids mobilized from adipose tissue can yield.
In addition, it is also important to consider that, in humans, adipose tissue is considered to be a major site for DNL, significantly contributing to body lipid reserves [22–25]. Thus, it is remarkable that hepatic DNL is positively correlated with insulin resistance and fatty liver, while the opposite correlation is observed between adipose tissue DNL, fatty liver, and insulin resistance. Indeed, in humans, large adipocyte size is correlated with insulin resistance and decreased expression of DNL genes in adipose tissue [24,26–28]. Furthermore, studies simultaneously investigating liver and adipose tissue DNL in lean and obese men show that insulin resistance and steatosis are positively correlated with DNL in liver and negatively correlated with DNL in adipose tissue [29,30].
Overall, current human studies indicate that not only is hepatic DNL specifically recruited during ectopic lipid deposition but also DNL is an energy-inefficient source of lipids. Indeed, by consuming a larger share of the caloric surplus driving ectopic lipid accumulation, DNL yields fewer lipids per calorie than what direct ectopic deposition of lipids derived from diet or adipose tissue would yield (Figure 1A). Finally, DNL in adipose tissue is negatively correlated with fatty liver and insulin resistance. Hence, reactivation of DNL in adipose tissue may be a promising strategy for the treatment of lipotoxicity-related diseases.
3. DNL, a biosynthetic pathway sustaining homeostasis
More mechanistic insights about the role of DNL in ectopic lipid deposition and metabolic homeostasis can be derived from mouse genetic studies indicating a specific role for DNL in the production of a number of bioactive molecules that contribute to metabolic homeostasis in a number of different cell types (Figure 1B). Below, we review some basic literature from mouse genetic studies targeting essential enzymes along the DNL pathway and we discuss the role of DNL in the control of fatty acid oxidation and cell signaling.
3.1. ATP-citrate lyase and acetyl-CoA carboxylase
Malonyl-CoA, an essential substrate for DNL, also acts as potent inhibitor of mitochondrial fatty acid oxidation via carnitine palmitoyltransferase-1 (CPT-1), such that elevated cytoplasmic levels of malonyl-CoA may promote cellular lipid accumulation (Figure 1A). As ATP-citrate lyase (Acly) and acetyl-CoA carboxylase (ACC) are both upstream of the synthesis of malonyl-CoA the roles of these two DNL enzymes in ectopic lipid deposition and insulin resistance are discussed together below.
Targeted genetic inactivation of the Acly locus leads to early embryonic lethality as no homozygote embryos for Acly inactivation were found from 8.5 days post-coitum [31]. Hence Acly produces lipids necessary for the early phase of embryonic development, which cannot be functionally provided by the mother. More information comes from experiments where Acly expression is suppressed in livers of mice by adenoviral-mediated delivery of small interfering RNA (siRNA). Genetically obese mice lacking a functional leptin receptor (db/db) were infected with either an adenovirus expressing siRNA against Acly or a control virus [32]. db/db mice infected with Acly-siRNA adenovirus showed marked suppression of liver Acly expression, were protected from NAFLD, and displayed improved glucose tolerance compared to db/db mice infected with a control virus [32]. These results suggest that inhibition of DNL in hepatocytes can protect against ectopic lipid storage. However, mice infected with Acly-siRNA virus displayed a marked reduction of malonyl-CoA [32] and thus the protective effects of Acly silencing are at least in part the consequence of increased fatty acid oxidation due to reduced CPT-1 inhibition from malonyl-CoA (Figure 1A). It is also important to consider that genetically obese mice lacking leptin function, such as db/db mice, display exaggerated hepatic DNL [33] and defective lipolysis in white adipocytes [34,35]. Thus the contribution of DNL to liver lipids in db/db mice on a standard chow (low fat) diet is most likely significantly higher than the value of 26% measured in humans [10]. Hence Acly inhibition may not protect or may even promote NAFLD in humans, for whom most liver lipids come from the diet and adipose tissue. Indeed, suppression of Acly in wild-type mice by infection with Acly-siRNA virus did not protect from NAFLD but promoted the development of fatty liver, in particular when mice were placed on high-fat diet for 25 days [36]. Hence, under conditions of physiological DNL and adipose tissue lipolysis, as it occurs in wild type mice, liver Acly activity protects from accumulation of liver lipids.
Downstream to Acly, the enzymes acetyl-CoA carboxylase (ACC) 1 and 2 directly catalyze the reaction leading to the formation of malonyl-CoA (Figure 1A). Genetic deletion of the ACC1 gene in mice leads to early embryonic lethality [37], indicating a fundamental role for DNL in early embryonic development. More information about a role for ACC1 in metabolism comes from a study using mice with a liver-specific conditional deletion of ACC1. Mice lacking ACC1 in hepatocytes maintained on a chow diet displayed a reduction of about one third of liver lipid content compared to control mice [38]. Importantly, the difference in lipid content between hepatocyte-specific ACC1 knockout mice and control mice was exacerbated by feeding the mice a fat-free diet and blunted by feeding the mice a high-fat diet [38]. The latter results are consistent with what was discussed above on liver Acly-inactivation as ACC1 inactivation in hepatocytes caused an important reduction of liver lipids only in lean mice on extreme lipogenic conditions (fat-free diet) and no protection was observed when dietary lipids are abundant (high-fat diet).
ACC2 knockout mice are viable [39–42], and it has been reported that mice lacking ACC2 display a leaner phenotype and reduced hepatic lipid content [39,40]. However, reduced liver lipids were not due to reduced hepatic DNL, as malonyl-CoA levels in liver of ACC2 knockout mice were comparable to those found in control mice [39]. Hence, reduced liver lipids reported in ACC2 knockout mice are most likely explained by the leaner phenotype of these mice. It must be also noted that other laboratories which generated mice lacking ACC2 could not observe a leaner phenotype for ACC2 knockout mice, thereby weakening the link between ACC2, adiposity, and NAFLD [41,42]. Further information on the role of ACC on NAFLD can be derived from experiments where ACC1 and ACC2 are both inactivated in liver. In one study liver expression of ACC1 and ACC2 was markedly reduced (by about 80%) in rats, using modified antisense oligonucleotides against ACC1 and ACC2 [43]. Rats receiving the ACC1 and ACC2 antisense oligonucleotides displayed reduced liver lipids and improved insulin sensitivity. However, it is important to note that in rats receiving the ACC1-2 antisense oligonucleotides, ACC1 and ACC2 activities were only partially reduced, as indicated by a 50% decrease in malonyl-CoA levels [43]. Furthermore, analysis of DNL and fatty acid oxidation showed that ACC1-2 antisense oligonucleotides caused a rather modest reduction of DNL (about one third) and a marked increase in fatty acid oxidation (about four times) [43]. The findings that hepatocytes treated with ACC1-2 antisense oligonucleotides display a marked four-fold elevation of fatty acid oxidation and close to control levels of DNL (about two thirds) suggest the possibility that liver lipids may be largely disposed by a futile cycle between DNL and fatty acid oxidation (Figure 1A). Hence, DNL and fatty acid oxidation could be coordinated in a way to form, under specific conditions, a futile cycle protecting cells from ectopic lipid deposition by dissipating extra calories in the form of heat [44–49].
Strong support in favor of a role for DNL in the protection from NAFLD can also be derived from a study of mice with hepatocyte-specific double knockout for ACC1 and ACC2 [50]. Compared to wild type controls, mice lacking both ACC1 and ACC2 in hepatocytes displayed increased liver mass, increased liver triglyceride content, and more hepatocyte microvescicular fat. Importantly DNL, assessed by acetate incorporation in fatty acids, was blunted in primary hepatocytes from liver-specific ACC1-2 knockout mice [50]. Surprisingly, the authors also observed that fatty acid oxidation was reduced by 50% in primary hepatocytes lacking ACC1 and ACC2 [50], suggesting that basal levels of DNL may be required to sustain efficient fatty acid oxidation. Finally, the authors observed that ablation of ACC1 and ACC2 in hepatocytes leads to altered acetylation of hundreds of proteins, revealing an important role for liver ACC activity in sustaining physiological protein acetylation [50].
Altogether, studies targeting the key lipogenic enzymes Acly and ACC1-2 indicate that DNL plays an important role in embryonic development and, in adult mice, in the coordination of fatty acid oxidation by malonyl-CoA dependent and independent mechanisms. Furthermore, ACC activity in hepatocytes is required to sustain physiological protein acetylation. Although under specific conditions, ACC and Acly suppression can protect from the accumulation of intrahepatocelullar lipids, this effect seems to be more relevant to models in which DNL is the major source of hepatic lipids and cannot be easily compensated by other lipid sources (e.g. in db/db mice and in mice fed a fat-free diet). In wild type mice on chow diet (shown for Acly and ACC), or on high-fat diet (shown for Acly), targeting these enzymes was reported to promote lipid accumulation in hepatocytes, indicating a protective role for these enzymes against fatty liver. An explanation for the latter could be the high energetic cost of DNL and the possibility to form futile substrate cycling between DNL and fatty acid oxidation (Figure 1A) [44–49,51].
3.2. Fatty acid synthase inactivation indicates a role for DNL in signaling
In contrast to Acly and ACC1-2, fatty acid synthase (FAS) consumes malonyl-CoA instead of producing it. Hence, inhibition of FAS is expected to increase intracellular concentrations of malonyl-CoA (Figure 1A).
A small-molecule FAS inhibitor, platensimycin, was shown to effectively inhibit FAS activity in mice [52]. Platensimycin protected mice from liver triglyceride accumulation induced by high-fructose diet but did not significantly protect db/db mice from fatty liver [52]. Thus, acute systemic FAS inhibition was shown to protect mice from liver triglyceride accumulation only when the mice are kept on high-fructose diet, an experimental model in which the unique road for extra calories to ectopic lipid is DNL.
In line with what was observed for Acly and ACC1 knockout mice, targeted genetic deletion of the Fas gene in the mouse causes a lethal phenotype during embryonic development [53], and feeding the mothers with a fat-rich diet did not rescue lethality of Fas knockout embryos. Hence FAS may produce lipids with essential structural or signaling function for embryonic development. Indeed, subsequent studies of mouse tissue-specific conditional gene targeting showed that FAS is required to sustain the levels of specific signaling lipids (Figure 1B). When the Fas gene is specifically targeted in mature hepatocytes, mice display suppressed liver FAS activity and higher levels of liver malonyl-CoA and develop more steatosis when placed on a fat-free diet [54]. The latter results demonstrate that hepatic DNL is not required for fat-free diet-induced steatosis, and show that inhibition of hepatic DNL downstream malonyl-CoA (which inhibits fatty acid oxidation) exacerbates the steatotic effects of a fat-free diet (Figure 1A). Interestingly, hepatocyte-specific FAS knockout mice kept on a fat-free diet display reduced expression of PPARα target genes in liver and developed hypoglycemia compared to control mice, a phenotype consistent with decreased PPARα activity [54]. Administration of a PPARα agonist to these mice rescued steatosis and hypoglycemia, suggesting that FAS activity may be required to sustain functional concentrations of a physiological PPARα agonist. Indeed, the same laboratory reported that FAS activity in the hepatocyte is necessary to sustain the concentration of the phospholipid 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC), which binds and activates PPARα [55]. FAS was also implicated in the synthesis of PPARα agonists in non-typical lipogenic cells such as neurons [56]. Mice lacking FAS in the hypothalamus and pancreatic β-cells display normal β-cell function but are leaner than control mice due to reduced food intake [56]. Loss of FAS activity in the hypothalamus correlated with decreased expression of PPARα-regulated genes, and intrahypothalamic, but not systemic, administration of a low dose of a PPARα agonist normalized food intake in these mice.
Further support for a role of FAS in the control of nuclear receptor function comes from conditional gene inactivation of FAS in myeloid cells [57]. Mice lacking FAS in myeloid cells are protected in a model of atherosclerosis and display decreased PPARα-mediated gene expression. The authors reported that macrophages lacking FAS were less prone to transform to foam cells because of decreased cholesterol uptake and increased cholesterol export. Interestingly, macrophages lacking FAS activity display elevated levels of the nuclear receptor LXR and of its target genes ABCA1 and SREBP1. Overall, the authors concluded that FAS metabolites in macrophages modulate the activity of the nuclear receptors PPARα and LXR to orchestrate cellular cholesterol traffic [57]. Hence, the action of FAS as synthetic pathway for nuclear receptor ligands is not limited to PPARα but also involves other nuclear receptors in a tissue-specific manner (e.g. LXR in macrophage). Consistently with the latter concept, ablation of FAS activity in white and brown adipocytes leads to a leaner phenotype and browning of subcutaneous adipose tissue independently from the generation of PPARα agonists in adipocytes [58]. Indeed, the authors report increased PPARα-driven gene expression in FAS-deficient adipocytes and show evidence that FAS activity in adipocytes is necessary to sustain the production of specific alkyl ether lipids, which act as PPARγ agonists.
Collectively, these studies indicate that FAS generates specific metabolites controlling the activity of different nuclear receptors, including PPARα; LXR; and PPARγ, with specific patterns for specific cell types (Figure 1B).
In addition to sustaining physiological levels of nuclear receptor ligands the main product of FAS is palmitate, the substrate of protein palmitoylation, an important post-translational modification affecting activity and cellular localization of proteins (Figure 1B). Mice lacking FAS in endothelial cells display defective angiogenesis and are sensitized to the effects of LPS, a phenotype that was explained by defective palmitoylation of nitric oxide synthase (eNOS) [59]. Conditional deletion of FAS in the intestinal epithelium of mice causes defective mucous barrier consequent to defective palmitoylation and secretion of mucin-2, an essential constituent of the intestinal mucus barrier [60]. These last two studies indicate that FAS activity is required for efficient functional palmitoylation of some proteins implicated in different physiological functions.
Another mechanism by which FAS activity was reported to control cellular function is through the modulation of physiological calcium signaling. Mice lacking FAS specifically in the myocardium and in skeletal muscle display increased calcium signaling [61,62], which was explained by defective activity of the sarcoplasmic calcium transporter SERCA [62]. The action of FAS on SERCA activity did not appear to be direct but through modulation of the composition of the sarcoplasmic reticulum membrane [62]. Thus, this study indicates that FAS activity in skeletal muscle is required to sustain sarcoplasmic reticulum membrane lipid composition, and consequently calcium homeostasis (Figure 1B). Furthermore, the fact that FAS expression in skeletal muscle was elevated in response to feeding a high fat diet [62] suggests a possible role for FAS in sustaining myocyte physiological lipid composition in response to a dietary lipid load. However, to date, it is not clear whether FAS is important for sustaining the physiological composition of membrane compartments other than that of the sarcoplasmic reticulum.
From the mouse genetic studies discussed it can be concluded that FAS activity is implicated in the synthesis of lipids required to maintain important cellular signaling and regulatory functions. More specifically, FAS activity is required to sustain physiological levels of specific nuclear receptor ligands, palmitoylation of specific target proteins, lipid composition of the sarcoplasmic reticulum membrane, SERCA function, and calcium homeostasis (Figure 1B). Importantly, FAS deletion in liver did not protect but exacerbated steatosis in mice kept on fat-free diet. The latter indicates that the protective effects of ACC1 liver inactivation on intrahepatocellular lipid accumulation observed in mice kept on a fat free diet are better explained by reduced levels of malonyl-CoA rather than decreased DNL [38] (Figure 1A).
3.3. Emerging roles for DNL in the immune system
Several lines of evidence suggest a close relationship between the molecular machinery controlling immune functions and the control of lipid handling.
Most notably, the transcription factor interferon regulatory factor 4 (IRF4) was reported to be a major transcriptional regulator of the gene expression program for lipolysis [63,64], whereas the pro-inflammatory class-1 phosphoinositide 3 kinase γ (PI3Kγ) emerged as a potent post-translational regulator of lipolysis [65,66]. Conversely, lipids released from lipolysis may play an important role in the control of immune functions [67,68]. IRF4 ablation in fibroblasts resulted in elevated DNL, indicating a crosstalk between IRF4, lipolysis, and DNL [64], suggesting that lipolysis and DNL may be sources of immunomodulatory lipids. As discussed above, FAS in macrophages modulates the activity of the nuclear receptors PPARα and LXR [57]. Mice lacking FAS in the hypothalamus display reduced levels of inflammatory cytokines in response to bacterial lipopolysaccharides (LPS) [69]. Mice lacking FAS activity in endothelial cells display defective angiogenesis and are sensitized to the effects of LPS, a phenotype that was explained by defective palmitoylation of nitric oxide synthase eNOS [59]. Hence, DNL activity may be implicated in the response to bacterial LPS. Consistently, macrophages undergoing classical LPS-mediated “M1” activation display high-rates of glucose utilization compared to monocytes and “M2” activated macrophages [70]. Treatment of macrophages with either LPS or the M1 cytokines TNFα or INFγ causes a marked elevation of the expression of the key DNL proteins mitochondrial citrate carrier and citrate lyase [71–73]. Furthermore, mitochondrial citrate carrier and citrate lyase activities in macrophage have been proposed to play a role in the production of reactive oxygen species, NO and the prostaglandin PGE2 [71–73]. Thus, increased glucose utilization for DNL is a distinct characteristic of M1-activated macrophages, which may play a role in sustaining innate immune functions. LPS induces citrate lyase expression and DNL also in B-cells, and inhibition of citrate lyase causes proliferation arrest, decreased survival, and defective expansion of the endoplasmic reticulum of B-cell following LPS treatment [74]. Hence, DNL may be implicated in proliferation, survival, and activation of B-cells in response to LPS. T-cell metabolism also depends on the specific T-cell type and on the activation state [75]. Activated proliferating T-cells display elevated glucose uptake, but the general role of DNL in T-cell function is not known. However, it was recently reported that memory CD8+ T-cells utilize glucose to support oxidative phosphorylation of fatty acids via DNL, implying an energy demanding futile cycle between fatty acids synthesis and oxidation [51].
DNL is also important in maintaining the integrity of intestinal mucous barrier, as FAS ablation in the intestinal epithelium of mice causes a defective mucous barrier, increased intestinal permeability, and predisposition to intestinal inflammation [60]. Nonetheless, it was also reported that acute FAS inhibition protects mice from the dextran sulfate induced colitis, indicating a complex action for FAS in mucosal immunity [76].
Altogether, emerging evidence indicates that DNL is dynamically recruited in specific leukocytes during differentiation or activation. DNL action in immunity is in part metabolic, providing structural lipids necessary for expansion of cell membranes or components of the intestinal mucous barrier. Furthermore, several lines of evidence suggest that DNL may also be required for the synthesis of specific lipids implicated in signal transduction. However, our understanding of the role of DNL in immunity is partial, and further studies are needed to clarify the role of DNL in specific immune reactions.
3.4. DNL is an important mechanism for glucose disposal
In the adipocyte, as in the hepatocyte, DNL and the pentose phosphate pathway generating NADPH (the energy source for DNL) are important pathways for glucose utilization. Mice with adipose tissue specific ablation of GLUT-4 display reduced glucose utilization in adipose tissue, glucose intolerance, and decreased insulin signaling in liver and skeletal muscle relative to control mice [77]. Hence, glucose utilization in adipose tissue is required to support glucose homeostasis in the mouse through both local and systemic effects. Interestingly, GLUT-4 ablation in either muscle or adipose tissue resulted in similar glucose intolerance, and GLUT-4 ablation in both tissues did not lead to additive effects on glucose intolerance, which was explained by a compensatory increase in hepatic DNL [78]. Thus, liver DNL is an important pathway for glucose disposal, which can in part compensate for a severe impairment of muscle and adipose tissue glucose utilization to sustain blood glucose homeostasis. Furthermore, another study reported that the glucose intolerance observed in mice lacking GLUT-4 in muscle can be completely corrected by over-expression of GLUT-4 in adipose tissue [79]. Hence, a severe defect in muscle glucose uptake can be corrected by increasing glucose uptake in adipose tissue. More recently, it was shown that the beneficial effects of adipose tissue GLUT-4 over-expression are blunted in mice lacking the transcription factor carbohydrate-responsive element-binding protein (ChREBP), which mediates glucose-dependent induction of adipose tissue DNL gene expression [80]. Hence, the beneficial effects of increased adipose tissue glucose uptake on blood glucose homeostasis depend on adipose tissue DNL.
Further evidence for a role of adipose tissue DNL in glucose homeostasis comes from mice lacking essential regulators of the expression of DNL enzymes in liver. The transcription factor sterol regulatory element-binding protein (SREBP) 1c plays an important role in the activation of the DNL gene-expression program in the hepatocyte in response to insulin. SREBP1c forms a complex with the protein SREBP-activating protein (SCAP), which is important for SREBP1 translocation to Golgi apparatus and subsequent activation. Mice lacking SCAP in the liver (L-SCAP) display reduced DNL in liver, which was compensated by increased DNL in adipose tissue, so that total DNL was unchanged [81]. Interestingly, mice lacking L-SCAP display improved fasting blood glucose, and improved glucose and insulin tolerance, indicating that shifting DNL from liver to adipose tissue could promote blood glucose homeostasis [81].
A shift of glucose utilization via DNL from liver to adipose tissue also has been observed in mice lacking the transcription factors liver-X-receptors (LXR) α and β in ob/ob mice (LOKO mice) [82]. LXRα and LXRβ control the expression of several genes including SREBP1c and specific genes in the DNL program.
Compared to control ob/ob mice, LOKO mice show reduced DNL gene expression program in liver but elevated expression in adipose tissue. LOKO mice are protected from hepatic steatosis and display improved insulin sensitivity and adipose tissue glucose uptake despite elevated expression of inflammatory markers in adipose tissue [82]. These latter results indicate that enhanced adipose tissue DNL promotes adipocyte glucose utilization even in the context of marked adipose tissue inflammation. However, it must be noted that LOKO mice were glucose intolerant because of a severe defect in circulating insulin levels consequent to reduced pancreatic β-cell mass. The cause of this loss of β-cell mass is not clear [82], but it is possible that the observed elevated adipose tissue inflammation may promote β-cell loss. Indeed, elevated adipose tissue DNL is expected to promote adipocyte hypertrophic growth, which in the context of morbid obesity, as in the case for ob/ob mice, may exacerbate adipose tissue inflammation.
Finally, recent studies indicate that adipose tissue DNL supports metabolic homoeostasis by producing cytokine-like lipids “lipokines” with antidiabetogenic and anti-inflammatory activity, such as C16:1n17 palmitoleic acid (PAO), and branched fatty acid esters of hydroxy fatty acid (FAHFA) [83,84] (Figure 1B).
Altogether, the studies above indicate that DNL in liver and, more notably, in adipose tissue is an important mechanism for glucose disposal, playing important roles in sustaining blood glucose homeostasis by mechanisms involving direct glucose utilization (liver and adipose tissue) and synthesis of lipokines (adipose tissue).
3.5. DNL in metabolic homeostasis: lessons from caloric restriction and “catch-up fat” during re-feeding
The transition between fasting and feeding is characterized by a remarkable metabolic flexibility consisting of a switch from glucose to lipid oxidation during fasting, and rapid induction of glucose utilization after re-feeding a high-carbohydrate diet [85]. The major modulators of this metabolic flexibility are insulin, dietary carbohydrates, and dietary lipids [85]. Insulin promotes glucose utilization by inducing the membrane translocation of GLUT-4 in muscle and adipose tissue, promoting glycogen deposition in muscle and liver, and inducing a DNL gene expression program in liver and adipose tissue [86]. Dietary carbohydrates also induce DNL in adipose tissue by a mechanism involving the transcription factor ChREBP. Hence the action of insulin in DNL is in part consequent to its promotion of glucose transport via GLUT-4 in adipose tissue [77,80], but not in liver, where glucose uptake is mediated by the constitutive transporter GLUT-2, and DNL is more dependent on the transcription factor SREBP1c [87]. Dietary lipids are known to antagonize glucose utilization since the time of the Randal cycle hypothesis [88,89], but our mechanistic understanding of such phenomenon is still partial. At the molecular level, it was proposed that during obesity ectopic deposition of lipids decreases glucose utilization by causing defective insulin-dependent PI3K-AKT signaling [7,90,91]. However, the role of these pathways in the acute effects of dietary lipids on glucose utilization (e.g. during re-feeding) is not clear. In recent years, it has emerged that DNL plays a major role in energy disposal and glucose utilization during the physiological metabolic response to caloric restriction and re-feeding. In a study investigating lipid metabolism during caloric restriction, DNL was measured in caloric restricted mice at different time-points during the day, together with 24 h indirect calorimetry and continuous food intake measurement [92]. Once a day, the calorie-restricted mice received 70% of the food consumed by ad-libitum control fed mice. Importantly, calorie-restricted mice consumed all the provided food within 1 h (i.e. gorging), whereas the food intake of control ad-libitum fed mice was distributed thoughout the day. Hence, calorie-restricted mice display a circadian pattern of 1 h gorging and 23 h fasting, and during the 1-h gorging period calorie restricted mice consume over ten times the amount of calories consumed by the ad-libitum fed mice. This gorging feeding is a remarkable allostatic load of calories for the metabolism of calorie-restricted mice, which exhibit markedly reduced energy expenditure before gorging [92]. Because caloric restriction increases life-span, mice must have developed an effective mechanism to sustain metabolic homeostasis during the high rate of calorie intake occurring during this gorging phase. Indeed, energy expenditure in calorie-restricted mice was rapidly elevated following gorging up to the levels of control fed mice for about 6 h and then decreased to pre-gorging levels by 10 h after food was provided. The increase in energy expenditure was paralleled by a marked change of respiratory quotient from values close to 0.7 (indicating a preference for fatty acid oxidation) to values above 1 (indicating high rates of DNL) during the 6 h following gorging [92]. Metabolic labeling of newly synthesized lipids demonstrated that DNL was rapidly and markedly elevated in both liver and adipose tissue following gorging, with rates of DNL peaking during the thermogenic response occurring after gorging. Hepatic gene expression of ACC1 and FAS was markedly reduced in the calorie-restricted group compared to control mice before gorging, but was rapidly induced above the level of control fed mice 3 h after gorging. Interestingly, adipose tissue expression of ACC1 and FAS was markedly elevated above control fed levels even before gorging [92], suggesting that during the gorging phase DNL is first recruited to adipose tissue and then to liver. Altogether, this study indicates that a two-step, fat first and liver second, lipogenic response could have evolved as a mechanism to safely dispose calories and glucose during gorging following periods of famine.
Further evidence indicating an important role for DNL as a homeostatic mechanism for glucose and energy disposal comes from a rat model of semi-starvation/re-feeding characterized by rapid restoration of body fat (or catch-up fat) even in the absence of hyperphagia [93–97]. In this model, growing rats are restricted to 50% of caloric intake for two weeks (leading to growth arrest and marked depletion of body fat stores) and are then re-fed the equivalent amount of food consumed by control ad-libitum fed rats, which were not previously food restricted. Under these conditions of isocaloric feeding on chow (low-fat) diet, the “re-fed” rats deposit fat at a rate that is more than twice as fast as in spontaneously growing control-fed rats. This catch-up fat phenomenon is explained by a 10% reduction in energy expenditure and associated with hyperinsulinemia after a glucose load but normal glucose tolerance [93,95]. Hyperinsulinemic euglycemic clamp experiments showed that, compared to control animals, rats re-fed on a low-fat diet display muscle insulin resistance largely compensated by a marked increase in insulin-driven glucose utilization in adipose tissue [94]. These high levels of adipose tissue glucose disposal are paralleled by elevated FAS and glucose 6 phosphate dehydrogenase (G6PDH) activities, indicating increased utilization of glucose for DNL and in the pentose phosphate pathway. Indeed, in-vivo measurements of DNL, using 3H labeled water and 14C labeled glucose as tracers, show that glucose utilization by DNL is drastically increased in adipose tissue and livers of rats re-fed on low-fat diet compared to control fed animals [97]. These data indicate that under conditions of rapid adipose tissue expansion DNL can act as an important glucose sink sustaining physiological glucose levels.
Further support for the role of DNL in glucose and energy homeostasis can be derived from experiments in the “catch-up fat” model above comparing rats re-fed a low fat diet with rats re-fed with an isocaloric high-fat diet. Adipose tissue glucose utilization and adipose tissue and liver DNL are dramatically reduced in rats re-fed with high-fat diet compared to rats re-fed a low-fat diet. Furthermore, rats re-fed a high-fat diet display a remarkable 12% reduction of energy expenditure compared to rats re-fed a low-fat diet (which already saves energy compared to control fed rats), gain 53% more fat, and are glucose intolerant [93,95,97]. These data indicate that in the situation of rapid adipose tissue expansion (catch-up fat), adipose tissue and liver DNL act as an important sink for calories and glucose to sustain blood glucose homeostasis.
Overall, the studies on the cyclic diurnal pattern of gorging and fasting during chronic calorie restriction and the studies on catch-up fat re-feeding on low-fat or high-fat diet following caloric restriction, indicate that DNL is dynamically recruited in liver and adipose tissue during re-feeding as a mechanism for glucose and calories disposal. Such mechanisms may have evolved to support metabolic homeostasis during episodes of gorging-feeding following periods of food scarcity.
3.6. DNL in cold-induced thermogenesis
Brown adipose tissue (BAT) metabolizes glucose and lipids at high rates to generate heat for thermal regulation by a mechanism involving uncoupling protein-1 (UCP-1) mediated dissipation of the mitochondrial proton gradient [98]. Brown-like adipocytes, so called beige or brite adipocytes, are found in cold-exposed adult humans and the extraordinary glucose-disposing ability of these cells is evident from PET-scan imaging data [99–102]. The glucose utilization in DNL within the BAT was measured in animal models. Mice display high basal rates of DNL in BAT, which are dramatically induced by cold exposure to levels that are higher than in any other tissue and one order of magnitude higher than in white adipose tissue or liver [44]. It was estimated that the brown adipocyte synthesizes about 30% of the fatty acids that it utilizes via DNL [44], and it should be considered that the other 70% of fatty acids, which are directly utilized in BAT, are in part synthesized via DNL in liver and adipose tissue. Thus, cold-induced thermogenesis operates in the context of a substrate cycle between DNL in BAT, liver, and white adipose tissue, and fatty acid oxidation in BAT [44,45,103]. This substrate cycle may support thermogenesis by generating heat and by providing fatty acid substrates that fuel uncoupled mitochondria respiration in BAT.
Strong evidence in support of a functional role for DNL in cold-induced thermogenesis can be derived from the study of mice that do not express a functional type-2 deiodinase (Dio2−/−) [104,105]. Type-2 deiodinase, the expression of which is induced about 50 fold in BAT following cold exposure, mediates the local conversion of the thyroid hormone triiodothyronine (T3) from its pro-hormone thyroxine (T4). Compared to control mice, Dio2−/− mice display hypothermia during cold exposure due to a severe defect in BAT thermogenesis, which is in part compensated for by shivering [104]. Remarkably, the defective cold-induced thermogenic response in BAT from Dio2−/− mice occurs despite a dramatic increase of local norepinephrine turnover and increased UCP-1 levels compared to control mice during cold exposure [105]. However, Dio2−/− mice could not induce BAT lipogenesis in response to cold, leading to a rapid depletion of intracellular brown adipocyte lipids stores during cold exposure [105]. To further investigate the role of DNL in cold-induced BAT thermogenesis WT mice were fed for five days with MEDICA-16, a potent pharmacological inhibitor of DNL, and then cold-induced thermogenesis was evaluated. Cold-exposed mice fed with MEDICA-16 displayed hypothermia and a marked reduction in cold-induced BAT thermogenesis despite that UCP-1 protein levels were similar to those of control mice [105].
Altogether, the studies above indicate that DNL in BAT is rapidly and potently induced in response to cold and that DNL is required to sustain cold-induced BAT thermogenesis and to sustain the intracellular lipid stores in brown adipocyte during cold activation.
4. Conclusion: DNL is probably more friend than foe
Ectopic lipid deposition driven by chronic positive energy balance is considered to be a major metabolic stressor linking obesity, type-2 diabetes, and the metabolic syndrome. Together with dietary lipids, and lipids released from adipose tissue, DNL significantly contributes to ectopic lipid deposition. In particular, DNL was shown to be specifically recruited in livers of NAFLD patients where it contributes to about 26% of liver lipids [10]. Yet, a number of evidences discussed in this review indicate that the specific recruitment of DNL during chronic positive energy balance may protect from ectopic lipid deposition and glucose intolerance. Indeed, DNL is an energetically inefficient way to store extra calories to lipids. Hence, the direct deposition of lipids from diet or from the circulating pool of fatty acids released from adipose tissue will lead to more ectopic lipid accumulation than DNL. Because DNL produces lipids with an extra caloric cost and by disposing glucose, it should be a safer road for excessive calories to lipid storage than the alternative pathways. Importantly, DNL and fatty acid oxidation are distinct pathways, which can operate simultaneously in the cell generating a futile cycle between fatty acid synthesis and oxidation dissipating extra calories and disposing glucose [44–49,51] (Figure 1A). Indeed, this futile cycling between DNL and fatty acid oxidation was shown to be specifically recruited in adipose tissues during cold exposure or stimulation with a β-adrenergic agonist [44,45,49].
Importantly, a relatively large body of evidence indicates that DNL is required for the synthesis of several signaling lipids implicated in different physiological functions, some of which may support metabolic homeostasis. Indeed malonyl-CoA and citrate coordinate fatty acid oxidation and synthesis by acting on CPT1 and ACC, respectively (Figure 1A). DNL was implicated in the synthesis of lipid ligands for nuclear receptors [54,57,58], lipids supporting sarcoplasmic reticulum membrane function and intracellular calcium homeostasis [61,62], physiological protein acetylation [50] and palmytoilation [59,60], and signaling lipids with systemic effects “lipokines” supporting metabolic homeostasis [83,84] (Figure 1B). Hence, it is possible that inhibition of DNL as a strategy to prevent ectopic lipid deposition (e.g. as a treatment for NAFLD) may be ineffective or even dangerous unless coupled to a reduction of the extra calories sustaining ectopic lipid accumulation. A low carbohydrate diet was shown to be effective in resolving hepatic lipids in NAFLD patients, but there are concerns about the long-term efficacy of dieting interventions. On the other hand, several lines of evidence indicate that reactivation of adipose tissue DNL in obesity could be a promising strategy to support metabolic homeostasis. A possible concern with the latter could be hypertrophic adipocyte growth promoting metabolic inflammation in overtly obese subjects [82]. However, an induction of DNL coupled with a simultaneous increase in fatty acid oxidation, as occurs during browning of white adipose tissue [49], may be an effective strategy to exploit the beneficial effects of DNL without promoting excessive adipose tissue hypertrophic growth and consequent inflammation.
Conflict of interest
The authors declare to have no conflict of interest in the topic covered by this manuscript.
Acknowledgments
The authors are grateful to Dr. Barbara Becattini for her feedback on this manuscript. This work is supported by a grant form Diabetesfonden to G.S.
References
- 1.Dombrowski S.U., Knittle K., Avenell A., Araujo-Soares V., Sniehotta F.F. Long term maintenance of weight loss with non-surgical interventions in obese adults: systematic review and meta-analyses of randomised controlled trials. British Medical Journal. 2014;348:g2646. doi: 10.1136/bmj.g2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unger R.H. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 3.Unger R.H., Clark G.O., Scherer P.E., Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochimica et Biophysica Acta. 2010;1801:209–214. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Thiam A.R., Farese R.V., Jr., Walther T.C. The biophysics and cell biology of lipid droplets. Nature Reviews Molecular Cell Biology. 2013;14:775–786. doi: 10.1038/nrm3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solinas G. Molecular pathways linking metabolic inflammation and thermogenesis. Obesity Reviews. 2012;13(Suppl. 2):69–82. doi: 10.1111/j.1467-789X.2012.01047.x. [DOI] [PubMed] [Google Scholar]
- 6.Virtue S., Vidal-Puig A. It's not how fat you are, it's what you do with it that counts. PLoS Biology. 2008;6:e237. doi: 10.1371/journal.pbio.0060237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solinas G., Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB Journal. 2010;24:2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 8.Jacome-Sosa M.M., Parks E.J. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Current Opinion In Lipidology. 2014;25:213–220. doi: 10.1097/MOL.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 9.Ameer F., Scandiuzzi L., Hasnain S., Kalbacher H., Zaidi N. De novo lipogenesis in health and disease. Metabolism. 2014;63:895–902. doi: 10.1016/j.metabol.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Donnelly K.L., Smith C.I., Schwarzenberg S.J., Jessurun J., Boldt M.D., Parks E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert J.E., Ramos-Roman M.A., Browning J.D., Parks E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirk E., Reeds D.N., Finck B.N., Mayurranjan S.M., Patterson B.W., Klein S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology. 2009;136:1552–1560. doi: 10.1053/j.gastro.2009.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haufe S., Engeli S., Kast P., Bohnke J., Utz W., Haas V. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology. 2011;53:1504–1514. doi: 10.1002/hep.24242. [DOI] [PubMed] [Google Scholar]
- 14.Sobrecases H., Le K.A., Bortolotti M., Schneiter P., Ith M., Kreis R. Effects of short-term overfeeding with fructose, fat and fructose plus fat on plasma and hepatic lipids in healthy men. Diabetes & Metabolism. 2010;36:244–246. doi: 10.1016/j.diabet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Faeh D., William J., Tappy L., Ravussin E., Bovet P. Prevalence, awareness and control of diabetes in the Seychelles and relationship with excess body weight. BMC Public Health. 2007;7:163. doi: 10.1186/1471-2458-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorbek G., Chwalibog A., Henckel S. Energetics of growth in pigs from 20 to 120 kg live weight. Z Tierphysiol Tierernahr Futtermittelkd. 1983;49:238–249. doi: 10.1111/j.1439-0396.1983.tb00805.x. [DOI] [PubMed] [Google Scholar]
- 17.Chwalibog A., Thorbek G. Energy expenditure by de novo lipogenesis. British Journal of Nutrition. 2001;86:309. doi: 10.1079/bjn2001401. [DOI] [PubMed] [Google Scholar]
- 18.Horton T.J., Drougas H., Brachey A., Reed G.W., Peters J.C., Hill J.O. Fat and carbohydrate overfeeding in humans: different effects on energy storage. American Journal of Clinical Nutrition. 1995;62:19–29. doi: 10.1093/ajcn/62.1.19. [DOI] [PubMed] [Google Scholar]
- 19.Marques-Lopes I., Ansorena D., Astiasaran I., Forga L., Martinez J.A. Postprandial de novo lipogenesis and metabolic changes induced by a high-carbohydrate, low-fat meal in lean and overweight men. American Journal of Clinical Nutrition. 2001;73:253–261. doi: 10.1093/ajcn/73.2.253. [DOI] [PubMed] [Google Scholar]
- 20.Minehira K., Bettschart V., Vidal H., Vega N., Di Vetta V., Rey V. Effect of carbohydrate overfeeding on whole body and adipose tissue metabolism in humans. Obesity Research. 2003;11:1096–1103. doi: 10.1038/oby.2003.150. [DOI] [PubMed] [Google Scholar]
- 21.Dirlewanger M., di Vetta V., Guenat E., Battilana P., Seematter G., Schneiter P. Effects of short-term carbohydrate or fat overfeeding on energy expenditure and plasma leptin concentrations in healthy female subjects. International Journal of Obesity and Related Metabolic Disorders. 2000;24:1413–1418. doi: 10.1038/sj.ijo.0801395. [DOI] [PubMed] [Google Scholar]
- 22.Letexier D., Pinteur C., Large V., Frering V., Beylot M. Comparison of the expression and activity of the lipogenic pathway in human and rat adipose tissue. Journal of Lipid Research. 2003;44:2127–2134. doi: 10.1194/jlr.M300235-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Strawford A., Antelo F., Christiansen M., Hellerstein M.K. Adipose tissue triglyceride turnover, de novo lipogenesis, and cell proliferation in humans measured with 2H2O. American Journal of Physiology. Endocrinology and Metabolism. 2004;286:E577–E588. doi: 10.1152/ajpendo.00093.2003. [DOI] [PubMed] [Google Scholar]
- 24.Minehira K., Vega N., Vidal H., Acheson K., Tappy L. Effect of carbohydrate overfeeding on whole body macronutrient metabolism and expression of lipogenic enzymes in adipose tissue of lean and overweight humans. International Journal of Obesity and Related Metabolic Disorders. 2004;28:1291–1298. doi: 10.1038/sj.ijo.0802760. [DOI] [PubMed] [Google Scholar]
- 25.Aarsland A., Chinkes D., Wolfe R.R. Hepatic and whole-body fat synthesis in humans during carbohydrate overfeeding. American Journal of Clinical Nutrition. 1997;65:1774–1782. doi: 10.1093/ajcn/65.6.1774. [DOI] [PubMed] [Google Scholar]
- 26.Salans L.B., Knittle J.L., Hirsch J. The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. Journal of Clinical Investigation. 1968;47:153–165. doi: 10.1172/JCI105705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weyer C., Foley J.E., Bogardus C., Tataranni P.A., Pratley R.E. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia. 2000;43:1498–1506. doi: 10.1007/s001250051560. [DOI] [PubMed] [Google Scholar]
- 28.Roberts R., Hodson L., Dennis A.L., Neville M.J., Humphreys S.M., Harnden K.E. Markers of de novo lipogenesis in adipose tissue: associations with small adipocytes and insulin sensitivity in humans. Diabetologia. 2009;52:882–890. doi: 10.1007/s00125-009-1300-4. [DOI] [PubMed] [Google Scholar]
- 29.Diraison F., Dusserre E., Vidal H., Sothier M., Beylot M. Increased hepatic lipogenesis but decreased expression of lipogenic gene in adipose tissue in human obesity. American Journal of Physiology. Endocrinology and Metabolism. 2002;282:E46–E51. doi: 10.1152/ajpendo.2002.282.1.E46. [DOI] [PubMed] [Google Scholar]
- 30.Eissing L., Scherer T., Todter K., Knippschild U., Greve J.W., Buurman W.A. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nature Communications. 2013;4:1528. doi: 10.1038/ncomms2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beigneux A.P., Kosinski C., Gavino B., Horton J.D., Skarnes W.C., Young S.G. ATP-citrate lyase deficiency in the mouse. Journal of Biological Chemistry. 2004;279:9557–9564. doi: 10.1074/jbc.M310512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q., Jiang L., Wang J., Li S., Yu Y., You J. Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor-deficient mice. Hepatology. 2009;49:1166–1175. doi: 10.1002/hep.22774. [DOI] [PubMed] [Google Scholar]
- 33.Hems D.A., Rath E.A., Verrinder T.R. Fatty acid synthesis in liver and adipose tissue of normal and genetically obese (ob/ob) mice during the 24-hour cycle. Biochemical Journal. 1975;150:167–173. doi: 10.1042/bj1500167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fruhbeck G., Aguado M., Martinez J.A. In vitro lipolytic effect of leptin on mouse adipocytes: evidence for a possible autocrine/paracrine role of leptin. Biochemical and Biophysical Research. 1997;240:590–594. doi: 10.1006/bbrc.1997.7716. [DOI] [PubMed] [Google Scholar]
- 35.Fruhbeck G., Aguado M., Gomez-Ambrosi J., Martinez J.A. Lipolytic effect of in vivo leptin administration on adipocytes of lean and ob/ob mice, but not db/db mice. Biochemical and Biophysical Research. 1998;250:99–102. doi: 10.1006/bbrc.1998.9277. [DOI] [PubMed] [Google Scholar]
- 36.Wang Q., Li S., Jiang L., Zhou Y., Li Z., Shao M. Deficiency in hepatic ATP-citrate lyase affects VLDL-triglyceride mobilization and liver fatty acid composition in mice. Journal of Lipid Research. 2010;51:2516–2526. doi: 10.1194/jlr.M003335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abu-Elheiga L., Matzuk M.M., Kordari P., Oh W., Shaikenov T., Gu Z. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12011–12016. doi: 10.1073/pnas.0505714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao J., DeMayo F.J., Li H., Abu-Elheiga L., Gu Z., Shaikenov T.E. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8552–8557. doi: 10.1073/pnas.0603115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abu-Elheiga L., Matzuk M.M., Abo-Hashema K.A., Wakil S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 40.Abu-Elheiga L., Oh W., Kordari P., Wakil S.J. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10207–10212. doi: 10.1073/pnas.1733877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olson D.P., Pulinilkunnil T., Cline G.W., Shulman G.I., Lowell B.B. Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7598–7603. doi: 10.1073/pnas.0913492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoehn K.L., Turner N., Swarbrick M.M., Wilks D., Preston E., Phua Y. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metabolism. 2010;11:70–76. doi: 10.1016/j.cmet.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savage D.B., Choi C.S., Samuel V.T., Liu Z.X., Zhang D., Wang A. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. Journal of Clinical Investigation. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trayhurn P. Fatty acid synthesis in mouse brown adipose tissue. The influence of environmental temperature on the proportion of whole-body fatty acid synthesis in brown adipose tissue and the liver. Biochimica et Biophysica Acta. 1981;664:549–560. doi: 10.1016/0005-2760(81)90132-6. [DOI] [PubMed] [Google Scholar]
- 45.Yu X.X., Lewin D.A., Forrest W., Adams S.H. Cold elicits the simultaneous induction of fatty acid synthesis and beta-oxidation in murine brown adipose tissue: prediction from differential gene expression and confirmation in vivo. FASEB Journal. 2002;16:155–168. doi: 10.1096/fj.01-0568com. [DOI] [PubMed] [Google Scholar]
- 46.Solinas G., Summermatter S., Mainieri D., Gubler M., Montani J.P., Seydoux J. Corticotropin-releasing hormone directly stimulates thermogenesis in skeletal muscle possibly through substrate cycling between de novo lipogenesis and lipid oxidation. Endocrinology. 2006;147:31–38. doi: 10.1210/en.2005-1033. [DOI] [PubMed] [Google Scholar]
- 47.Dulloo A.G., Gubler M., Montani J.P., Seydoux J., Solinas G. Substrate cycling between de novo lipogenesis and lipid oxidation: a thermogenic mechanism against skeletal muscle lipotoxicity and glucolipotoxicity. International Journal of Obesity and Related Metabolic Disorders. 2004;28(Suppl 4):S29–S37. doi: 10.1038/sj.ijo.0802861. [DOI] [PubMed] [Google Scholar]
- 48.Solinas G., Summermatter S., Mainieri D., Gubler M., Pirola L., Wymann M.P. The direct effect of leptin on skeletal muscle thermogenesis is mediated by substrate cycling between de novo lipogenesis and lipid oxidation. FEBS Letters. 2004;577:539–544. doi: 10.1016/j.febslet.2004.10.066. [DOI] [PubMed] [Google Scholar]
- 49.Mottillo E.P., Balasubramanian P., Lee Y.H., Weng C., Kershaw E.E., Granneman J.G. Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic beta3-adrenergic receptor activation. Journal of Lipid Research. 2014;55:2276–2286. doi: 10.1194/jlr.M050005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chow J.D., Lawrence R.T., Healy M.E., Dominy J.E., Liao J.A., Breen D.S. Genetic inhibition of hepatic acetyl-CoA carboxylase activity increases liver fat and alters global protein acetylation. Molecular Metabolism. 2014;3:419–431. doi: 10.1016/j.molmet.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Sullivan D., van der Windt G.J., Huang S.C., Curtis J.D., Chang C.H., Buck M.D. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41:75–88. doi: 10.1016/j.immuni.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu M., Singh S.B., Wang J., Chung C.C., Salituro G., Karanam B.V. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5378–5383. doi: 10.1073/pnas.1002588108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chirala S.S., Chang H., Matzuk M., Abu-Elheiga L., Mao J., Mahon K. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6358–6363. doi: 10.1073/pnas.0931394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakravarthy M.V., Pan Z., Zhu Y., Tordjman K., Schneider J.G., Coleman T. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metabolism. 2005;1:309–322. doi: 10.1016/j.cmet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 55.Chakravarthy M.V., Lodhi I.J., Yin L., Malapaka R.R., Xu H.E., Turk J. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138:476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chakravarthy M.V., Zhu Y., Lopez M., Yin L., Wozniak D.F., Coleman T. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. Journal of Clinical Investigation. 2007;117:2539–2552. doi: 10.1172/JCI31183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider J.G., Yang Z., Chakravarthy M.V., Lodhi I.J., Wei X., Turk J. Macrophage fatty-acid synthase deficiency decreases diet-induced atherosclerosis. Journal of Biological Chemistry. 2010;285:23398–23409. doi: 10.1074/jbc.M110.100321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lodhi I.J., Yin L., Jensen-Urstad A.P., Funai K., Coleman T., Baird J.H. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARgamma activation to decrease diet-induced obesity. Cell Metabolism. 2012;16:189–201. doi: 10.1016/j.cmet.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei X., Schneider J.G., Shenouda S.M., Lee A., Towler D.A., Chakravarthy M.V. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. Journal of Biological Chemistry. 2011;286:2933–2945. doi: 10.1074/jbc.M110.193037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei X., Yang Z., Rey F.E., Ridaura V.K., Davidson N.O., Gordon J.I. Fatty acid synthase modulates intestinal barrier function through palmitoylation of mucin 2. Cell Host Microbe. 2012;11:140–152. doi: 10.1016/j.chom.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Funai K., Song H., Yin L., Lodhi I.J., Wei X., Yoshino J. Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. Journal of Clinical Investigation. 2013;123:1229–1240. doi: 10.1172/JCI65726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Razani B., Zhang H., Schulze P.C., Schilling J.D., Verbsky J., Lodhi I.J. Fatty acid synthase modulates homeostatic responses to myocardial stress. Journal of Biological Chemistry. 2011;286:30949–30961. doi: 10.1074/jbc.M111.230508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fabrizi M., Marchetti V., Mavilio M., Marino A., Casagrande V., Cavalera M. IL-21 is a major negative regulator of IRF4-dependent lipolysis affecting Tregs in adipose tissue and systemic insulin sensitivity. Diabetes. 2014;63:2086–2096. doi: 10.2337/db13-0939. [DOI] [PubMed] [Google Scholar]
- 64.Eguchi J., Wang X., Yu S., Kershaw E.E., Chiu P.C., Dushay J. Transcriptional control of adipose lipid handling by IRF4. Cell Metabolism. 2011;13:249–259. doi: 10.1016/j.cmet.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perino A., Beretta M., Kilic A., Ghigo A., Carnevale D., Repetto I.E. Combined inhibition of PI3Kbeta and PI3Kgamma reduces fat mass by enhancing alpha-MSH-dependent sympathetic drive. Science Signalling. 2014;7:ra110. doi: 10.1126/scisignal.2005485. [DOI] [PubMed] [Google Scholar]
- 66.Becattini B., Marone R., Zani F., Arsenijevic D., Seydoux J., Montani J.P. PI3Kgamma within a nonhematopoietic cell type negatively regulates diet-induced thermogenesis and promotes obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E854–E863. doi: 10.1073/pnas.1106698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schreiber R., Zechner R. Lipolysis meets inflammation: arachidonic acid mobilization from fat. Journal of Lipid Research. 2014;55:2447–2449. doi: 10.1194/jlr.C055673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dichlberger A., Schlager S., Maaninka K., Schneider W.J., Kovanen P.T. Adipose triglyceride lipase regulates eicosanoid production in activated human mast cells. Journal of Lipid Research. 2014;55:2471–2478. doi: 10.1194/jlr.M048553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chakravarthy M.V., Zhu Y., Yin L., Coleman T., Pappan K.L., Marshall C.A. Inactivation of hypothalamic FAS protects mice from diet-induced obesity and inflammation. Journal of Lipid Research. 2009;50:630–640. doi: 10.1194/jlr.M800379-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Galvan-Pena S., O'Neill L.A. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Infantino V., Iacobazzi V., Menga A., Avantaggiati M.L., Palmieri F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFalpha- and IFNgamma-triggered inflammation. Biochimica et Biophysica Acta. 2014;1839:1217–1225. doi: 10.1016/j.bbagrm.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Infantino V., Iacobazzi V., Palmieri F., Menga A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochemical and Biophysical Research. 2013;440:105–111. doi: 10.1016/j.bbrc.2013.09.037. [DOI] [PubMed] [Google Scholar]
- 73.Infantino V., Convertini P., Cucci L., Panaro M.A., Di Noia M.A., Calvello R. The mitochondrial citrate carrier: a new player in inflammation. Biochemical Journal. 2011;438:433–436. doi: 10.1042/BJ20111275. [DOI] [PubMed] [Google Scholar]
- 74.Dufort F.J., Gumina M.R., Ta N.L., Tao Y., Heyse S.A., Scott D.A. Glucose-dependent de novo lipogenesis in B lymphocytes: a requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. Journal of Biological Chemistry. 2014;289:7011–7024. doi: 10.1074/jbc.M114.551051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fox C.J., Hammerman P.S., Thompson C.B. Fuel feeds function: energy metabolism and the T-cell response. Nature Reviews Immunology. 2005;5:844–852. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 76.Matsuo S., Yang W.L., Aziz M., Kameoka S., Wang P. Fatty acid synthase inhibitor C75 ameliorates experimental colitis. Molecular Medicine. 2014;20:1–9. doi: 10.2119/molmed.2013.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abel E.D., Peroni O., Kim J.K., Kim Y.B., Boss O., Hadro E. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 78.Kotani K., Peroni O.D., Minokoshi Y., Boss O., Kahn B.B. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization. Journal of Clinical Investigation. 2004;114:1666–1675. doi: 10.1172/JCI21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carvalho E., Kotani K., Peroni O.D., Kahn B.B. Adipose-specific overexpression of GLUT4 reverses insulin resistance and diabetes in mice lacking GLUT4 selectively in muscle. American Journal of Physiology. Endocrinology and Metabolism. 2005;289:E551–E561. doi: 10.1152/ajpendo.00116.2005. [DOI] [PubMed] [Google Scholar]
- 80.Herman M.A., Peroni O.D., Villoria J., Schon M.R., Abumrad N.A., Bluher M. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuriyama H., Liang G., Engelking L.J., Horton J.D., Goldstein J.L., Brown M.S. Compensatory increase in fatty acid synthesis in adipose tissue of mice with conditional deficiency of SCAP in liver. Cell Metabolism. 2005;1:41–51. doi: 10.1016/j.cmet.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 82.Beaven S.W., Matveyenko A., Wroblewski K., Chao L., Wilpitz D., Hsu T.W. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metabolism. 2013;18:106–117. doi: 10.1016/j.cmet.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yore M.M., Syed I., Moraes-Vieira P.M., Zhang T., Herman M.A., Homan E.A. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014;159:318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cao H., Gerhold K., Mayers J.R., Wiest M.M., Watkins S.M., Hotamisligil G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galgani J.E., Moro C., Ravussin E. Metabolic flexibility and insulin resistance. American Journal of Physiology. Endocrinology and Metabolism. 2008;295:E1009–E1017. doi: 10.1152/ajpendo.90558.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saltiel A.R., Kahn C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 87.Shimano H., Shimomura I., Hammer R.E., Herz J., Goldstein J.L., Brown M.S. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. Journal of Clinical Investigation. 1997;100:2115–2124. doi: 10.1172/JCI119746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Randle P.J., Garland P.B., Hales C.N., Newsholme E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 89.Randle P.J., Garland P.B., Newsholme E.A., Hales C.N. The glucose fatty acid cycle in obesity and maturity onset diabetes mellitus. Annals of the New York Academy of Sciences. 1965;131:324–333. doi: 10.1111/j.1749-6632.1965.tb34800.x. [DOI] [PubMed] [Google Scholar]
- 90.Chavez J.A., Summers S.A. A ceramide-centric view of insulin resistance. Cell Metabolism. 2012;15:585–594. doi: 10.1016/j.cmet.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 91.Perry R.J., Samuel V.T., Petersen K.F., Shulman G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bruss M.D., Khambatta C.F., Ruby M.A., Aggarwal I., Hellerstein M.K. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. American Journal of Physiology. Endocrinology and Metabolism. 2010;298:E108–E116. doi: 10.1152/ajpendo.00524.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Crescenzo R., Samec S., Antic V., Rohner-Jeanrenaud F., Seydoux J., Montani J.P. A role for suppressed thermogenesis favoring catch-up fat in the pathophysiology of catch-up growth. Diabetes. 2003;52:1090–1097. doi: 10.2337/diabetes.52.5.1090. [DOI] [PubMed] [Google Scholar]
- 94.Cettour-Rose P., Samec S., Russell A.P., Summermatter S., Mainieri D., Carrillo-Theander C. Redistribution of glucose from skeletal muscle to adipose tissue during catch-up fat: a link between catch-up growth and later metabolic syndrome. Diabetes. 2005;54:751–756. doi: 10.2337/diabetes.54.3.751. [DOI] [PubMed] [Google Scholar]
- 95.Summermatter S., Mainieri D., Russell A.P., Seydoux J., Montani J.P., Buchala A. Thrifty metabolism that favors fat storage after caloric restriction: a role for skeletal muscle phosphatidylinositol-3-kinase activity and AMP-activated protein kinase. FASEB Journal. 2008;22:774–785. doi: 10.1096/fj.07-8972com. [DOI] [PubMed] [Google Scholar]
- 96.Summermatter S., Marcelino H., Arsenijevic D., Buchala A., Aprikian O., Assimacopoulos-Jeannet F. Adipose tissue plasticity during catch-up fat driven by thrifty metabolism: relevance for muscle-adipose glucose redistribution during catch-up growth. Diabetes. 2009;58:2228–2237. doi: 10.2337/db08-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marcelino H., Veyrat-Durebex C., Summermatter S., Sarafian D., Miles-Chan J., Arsenijevic D. A role for adipose tissue de novo lipogenesis in glucose homeostasis during catch-up growth: a Randle cycle favoring fat storage. Diabetes. 2013;62:362–372. doi: 10.2337/db12-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lowell B.B., Spiegelman B.M. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404:652–660. doi: 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- 99.Rothwell N.J., Stock M.J. A role for brown adipose tissue in diet-induced thermogenesis. Nature. 1979;281:31–35. doi: 10.1038/281031a0. [DOI] [PubMed] [Google Scholar]
- 100.Zingaretti M.C., Crosta F., Vitali A., Guerrieri M., Frontini A., Cannon B. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB Journal. 2009;23:3113–3120. doi: 10.1096/fj.09-133546. [DOI] [PubMed] [Google Scholar]
- 101.Vollmers C., Gill S., DiTacchio L., Pulivarthy S.R., Le H.D., Panda S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21453–21458. doi: 10.1073/pnas.0909591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van Marken Lichtenbelt W.D., Vanhommerig J.W., Smulders N.M., Drossaerts J.M., Kemerink G.J., Bouvy N.D. Cold-activated brown adipose tissue in healthy men. New England Journal of Medicine. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 103.Masoro E.J. Role of lipogenesis in nonshivering thermogenesis. Federation Proceedings. 1963;22:868–873. [PubMed] [Google Scholar]
- 104.de Jesus L.A., Carvalho S.D., Ribeiro M.O., Schneider M., Kim S.W., Harney J.W. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. Journal of Clinical Investigation. 2001;108:1379–1385. doi: 10.1172/JCI13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Christoffolete M.A., Linardi C.C., de Jesus L., Ebina K.N., Carvalho S.D., Ribeiro M.O. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. 2004;53:577–584. doi: 10.2337/diabetes.53.3.577. [DOI] [PubMed] [Google Scholar]