

Renal osteodystrophy: definition, nomenclature and classification
Disturbances of bone and mineral metabolism are a hallmark of chronic kidney disease (CKD). Renal osteodystrophy (ROD) is the traditional term for bone lesions in conjunction with CKD and is now considered a part of the ‘chronic kidney disease—mineral and bone disorder’ (CKD-MBD) [1]. ROD comprises various subtypes with substantial differences in aetiology and fundamental differences in treatment strategies. In long-term dialysis patients the prevalence of some types of ROD is virtually 100% [2].
A simple, easy to apply but still sophisticated and comprehensive descriptive system of ROD is the TMV system [1]. The TMV system comprises bone turnover (T), bone mineralization (M) as well as bone volume (V). Bone turnover and bone volume may both be classified as high, normal or low. Bone mineralization may be categorized as normal or abnormal. As an alternative to volume, the bone balance may be considered [3,4].
Based on the above system, the NKF/KDOQI guidelines distinguish six types of bone pathology in CKD-MBD (Table 1) (Figures 1 and 2). The focus on cancellous bone parameters in this classification system has been questioned regarding the importance of cortical bone quality for structural integrity [5]. Moreover, bone histomorphometric parameters comprise a continuum, and categorization may be an oversimplified approach [5]. Nevertheless, categorical CKD-MBD classification is helpful for clinical practice and widely used as the basis for therapeutic decision making. In this review we will focus particularly on adynamic bone disease (ABD), which is increasing in prevalence and, in many CKD populations, now represents the most frequent type of bone lesion [6].
Table 1.
NKF/KDOQI guidelinesa and renal osteodystrophy classification
| Hyperparathyroid (high turnover) bone disease | |
|---|---|
| Mixed (high turnover with mineralization defect) bone disease | |
| Osteomalacia | |
| Adynamic bone disease (ABD) | |
| Additionally, two distinct causing agents for ROD are explicitly mentioned: amyloid bone disease and aluminium bone disease |
ahttp://www.kidney.org/professionals/KDOQI/guidelines_bone/index.htm.
Fig. 1.
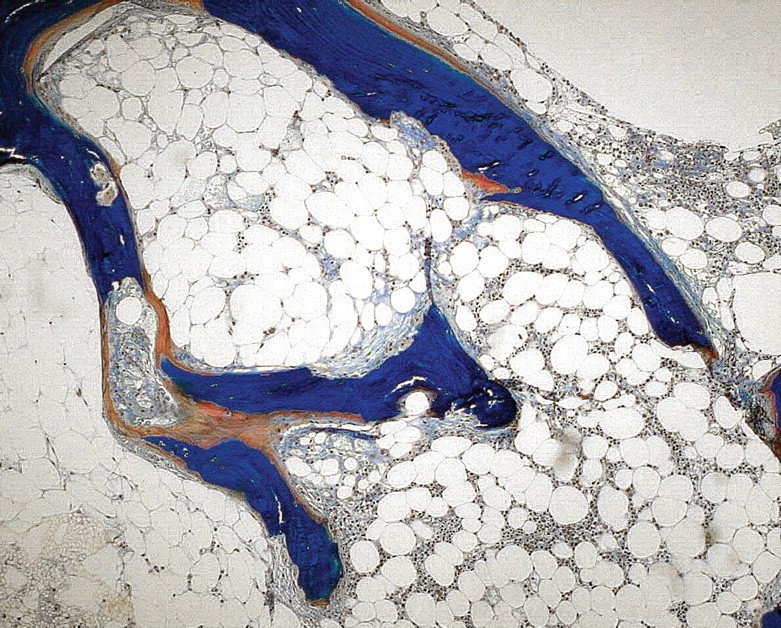
Mixed uraemic osteodystrophy: high resorptive activity, osteoid accumulation, peritrabecular fibrosis (Goldner stain) (courtesy of Dr. G. Lehmann, Jena).
Fig. 2.

Adynamic renal osteodystrophy: absence of cellular activity and osteoid, low cancellous bone volume (osteopenia) (Goldner stain) (courtesy of Dr. G. Lehmann, Jena).
What is ABD?
The term ‘aplastic’ or ‘adynamic’ bone disease was introduced in the early 1980s [7,8]. ABD is characterized by a low-bone turnover without osteoid accumulation, i.e. with a thin osteoid seam. Both the rate of collagen synthesis by osteoblasts and the subsequent mineralization of bone collagen are subnormal. The latter distinguishes ABD from the second low-turnover form, i.e. osteomalacia, where a mineralization defect exceeds the defects in bone formation, resulting in a relative osteoid excess [9,10]. In ABD, there are few or no osteoblasts, and minimal or no peritrabecular fibrosis or marrow fibrosis (in contrast to osteitis fibrosa). Especially the bone formation rate (BFR) is substantially diminished and the number of remodelling sites is low [9].
ABD in bone histomorphometry
The NKF-KDOQI guidelines suggest a number of histomorphometric parameters for the classification of ROD (Table 2).
Table 2.
Frequently applied histomorphometric parameters and normal levels according to K/DOQI
| Parameter | Normal level | |
|---|---|---|
| (1) | Bone volume relative to total tissue volume | 16–23% |
| (2) | Osteoid thickness | 4–20 μm |
| (3) | Osteoid surface relative to total bone surface | 1–39% |
| (4) | Osteoblast surface relative to total bone surface | 0.2–10% |
| (5) | Osteoclast surface relative to total bone surface | 0.15–1.2% |
| (6) | Activation frequencya | 0.49–0.72/year |
| (7) | Fibrosis volume relative to total tissue volume | Absent (%) |
| (8) | Mineralization lag time | <50 days |
ABD is diagnosed in the presence of subnormal values in criteria 2–6 in the absence of fibrosis.
aSee the text for comment.
Bone turnover may be assessed by the activation frequency or by BFR [3]. The activation frequency is defined as the reciprocal of the total remodelling time. The latter is the net result of bone resorption, reversal, formation and quiescent periods. Therefore, the activation frequency assesses both osteoclast (resorption) and osteoblast (formation) activity [11,12]. In contrast, BFR focuses only on osteoblast activity [11,13]. However, in ROD the correlation between these two parameters of bone turnover is excellent (r = 0.95 in dialysis and r = 0.97 in predialysis patients) [12] and both the activation frequency and BFR may be used for assessment of bone turnover.
Especially bone turnover, fibrosis quantification and bone mineralization assessment are required to differentiate between hyperparathyroid bone disease, osteomalacia and mixed and adynamic bone disease. Many previously published ROD studies thus rely on three histomorphometric parameters: BFR (μm2/mm2/day), osteoid accumulation (%) and the presence or absence of fibrosis [13–22]. For example, based on these three parameters, normal histology was defined as the absence of fibrosis, osteoid volume <12%, and BFR >97 but <613 μm2/mm2/day [16]. However, cut-off levels are inconstant. Concerning BFR, cut-off levels varying from 97 to 108 μm2/mm2/day have been applied to separate normal from low bone turnover. Other studies used one standard deviation below normal levels, or <5% of normal levels, to define adynamic BFR [13–23]. Similarly, cut-off levels for osteoid volume separating osteomalacia from ABD vary from 12% [16,21] to 15%, [13–15,17–20,23] (Figure 3).
Fig. 3.

Commonly applied definition criteria for ABD.
Parfitt has challenged such a high threshold for osteoid accumulation, since 5% is already ‘generous’, assuming that normal osteoid volume is 1.5 ± 1.2% [5]. If the lower 5% cut-off is used, more patients will be diagnosed with osteomalacia than with ABD [24]. In contrast, a fibrous tissue volume of <0.5% is a non-disputed criterion for ABD [13–21,23].
Within ABD, it is of major therapeutic importance to distinguish aluminium-induced and non-aluminium-induced forms [10,25,26]. Recently, a variant of ABD has been described (so-called ABD-V) [23], which is characterized by high osteoclastic resorption (osteoclast surface/bone surface more than two standard deviations higher than in controls). Its clinical relevance remains to be defined.
How to diagnose the subtype of renal osteodystrophy
The gold standard for the diagnosis and classification of ROD is histomorphometric analysis of an undecalcified bone sample [1]. Pre-biopsy in vivo tetracycline labelling as well as amyloid and aluminium stains are required for complete diagnostic work-up. A combination of dynamic and static bone parameters, both of cortical and trabecular bone, gives a complete overview upon bone metabolism [3,5]. The preferred site of biopsy is 2 cm posterior and 2 cm inferior to the anterior iliac crest using an instrument designed to obtain a core of bone of at least 4–5 mm diameter [9] (e.g. Meunier® bone biopsy device).
KDIGO and NKF-KDOQI guidelines recommend a bone biopsy in the following cases (Table 3).
Table 3.
Possible indications for an iliac crest bone biopsy in renal osteodystrophya
| If a CKD patient with serum levels of intact PTH (iPTH) between 100 and 500 pg/mL (11.0–55.0 pmol/L) develops unexplained hypercalcaemia, bone pain or an increase in bone alkaline phosphatase activity | |
| Inconsistencies among biochemical parameters that do not allow a definitive interpretation of bone metabolism | |
| Unexplained skeletal fracture or bone pain | |
| In the absence of other known causes of a bone fracture (e.g. malignancy); in the case of low trauma, unexplained fracture | |
| Severe progressive vascular calcification | |
| Unexplained hypercalcaemia | |
| Suspicion of aluminium overload or toxicity (or possibly other metals like strontium), especially before chelation treatment due to possible side effects of DFO | |
| Before parathyroidectomy if there has been significant exposure to aluminium in the past or if the results of biochemical determinations are not consistent with advanced secondary or tertiary hyperparathyroidism | |
| Consider a biopsy before beginning treatment with bisphosphonates |
aModified after [1] and http://www.kidney.org/professionals/KDOQI/guidelines_bone/index.htm.
The indications for a bone biopsy after renal transplantation are less clear and not explicitly discussed in current guidelines. Basically, the above-mentioned indications also apply for the posttransplant situation.
Bone biopsy and the role of tetracycline labelling
It is mandatory to distinguish between static and dynamic bone parameters in histomorphometry. Static histomorphometric parameters include bone volume/tissue volume, osteoid thickness, osteoid surface/bone surface, osteoblast surface/bone surface, osteoclast surface/bone surface, and fibrosis volume/tissue volume. In contrast, BFR, activation frequency and mineralization lag time are dynamic bone parameters.
In order to evaluate the underlying dynamics of bone morphology, in vivo tetracycline labelling is necessary [4,25]. Tetracyclines show fluorescence in ultraviolet light and bind to actively forming bone areas. Calcium-containing phosphate binders should not be given in parallel to tetracycline. In patients with severely impaired renal function, one possible scheme for tetracycline labelling is shown in Table 4 [27]. Information is enhanced by using two different tetracylines with different fluorescence [27]. After the second labelling period, 4–6 days should elapse to give the second tetracycline line sufficient time to get buried by osteoid, in order to protect it from washout during in vitro staining. A modified, short-term ‘emergency’ labelling scheme is possible [9,27]. The most appropriate labelling scheme should be chosen in agreement with the local bone pathologist.
Table 4.
Example of tetracycline labelling in patients with suspected renal osteodystrophy
| First label: doxycyclin 100 mg SID for 3 days | |
| wait 14 days (8–15 days) | |
| Second label: minocyclin 50 mg BID for 3 days (2–4 days) | |
| perform a biopsy 4–6 days later |
Several alternatives from the tetracycline family are available, e.g. tetracycline hydrochloride (250 mg TID/BID depending on renal function on days 25–23 before biopsy) followed by demeclocycline (days 4–2 before biopsy) (note: demeclocycline capsules or tablets are not available in parts of the European Community).
Alternatively, two doses of tetracycline hydrochloride 10 days apart may be used.
Assessment of renal osteodystrophy without a bone biopsy
Measurements of bone mineral density or plain bone radiographs are not suitable for a diagnosis of ROD [25], although the latter may identify Looser's zones. None of the known biochemical markers for parathyroid status, bone formation and bone resorption have reached a sufficient level of diagnostic accuracy (reviews in [1,25,28]), and none so far can replace the diagnostic power of a bone biopsy.
Whereas plasma iPTH levels at the extremes, i.e. <50 pg/ml and >800 pg/ml, are usually associated with ABD and high-turnover bone disease, respectively, in particular levels between about 100 and 500 pg/ml exhibit variable associations with types of bone lesions. This diagnostic uncertainty of intermediate, K/DOQI target-compliant PTH levels has recently been confirmed by bone biopsy studies from Brazil [29] and Portugal [30]. The situation is complicated further by wide variations in iPTH results if different test assays are employed [31,32] and by potentially variable ratios of agonistic (PTH1–84) and antagonistic (PTH7–84) PTH forms [33].
Bone alkaline phosphatase (BAP) is probably the single most useful biochemical parameter for the assessment of bone formation. Elevated levels of bone alkaline phosphatase virtually exclude an adynamic renal bone disease [25,28]; however, elevations of BAP along with total AP may be seen in cases of severe osteomalacia. Combinations of biochemical markers hold promise [22], at least for the differentiation for high-turnover versus adynamic forms. Such combinations could be, for example, iPTH plus osteoprotegerin [2] or iPTH plus bone-specific alkaline phosphatase [28]. Another approach is to measure the ratio of PTH(1–84) to PTH(7–84) [33].
Currently, the domain of biochemical markers is the long-term monitoring of ROD evolution. Changes of bone markers, such as bone-specific alkaline phosphatase, over time, may be suitable indicators for the assessment of therapeutic effects.
Aluminium bone disease
Hyperaluminaemia in end-stage renal disease (ESRD) patients was reported as early as 1970 [34]. The true dimension of aluminium-related complications in dialysis patients, including bone disease, emerged in the 1980s [35,36]. In the 1980s, aluminium overload was the predominant cause for the development of low-turnover bone disease in dialysis patients [35,37]. In aluminium-treated dialysis patients with osteitis fibrosa, the distribution of aluminium in bone is diffuse, whereas in aluminium-induced osteomalacia, or ABD, there is a predominant localization along the mineralization front [35]. Aluminium causes mineralization defects, and markedly reduces both osteoclast resorption and osteoblast surface [10]. It profoundly decreases PTH synthesis and release [38,39] even in the presence of excessive hyperphosphataemia [40]. A chronic low-dose exposure with concomitant high dosages of vitamin D may preferentially lead to ABD [10] rather than osteomalacia. Clinically, the aluminium-induced ABD forms appear particularly prone to causing bone pain, hypercalcaemia and fractures [10,26]. A current example that even recent trial findings have to take into account the underlying aluminium exposure is the study by Barreto et al. from Brazil [29]. They recorded a high proportion of low-turnover bone disease (∼2/3) in their entire cohort. Of these patients ∼60% had substantial aluminium staining (>25% aluminium bone staining) in contrast to about a third of the patients with high-turnover bone disease.
Sources of aluminium
Prior to widespread usage of reverse osmosis, water contamination used to be a major source of aluminium for dialysis patients [35]. Aluminium toxicity has also been described in CKD patients ingesting aluminium hydroxide who had never been treated with dialysis [41]. Although aluminium-containing phosphate binder usage has substantially declined, in 1995 about a quarter of patients exhibited positive aluminium bone staining, and in a study published in 2004, 57% of the dialysis patients had been treated with aluminium ‘in the past’ [42]. Thus, aluminium overload or intoxication continues to be a clinical concern. There is certainly a difference in the clinical relevance of aluminium-induced bone disease between ‘developing’ and ‘developed’ countries simply due to the different prescription patterns. Especially in emerging countries aluminium is used more generously, and as a consequence in the year 2000, 95% of the bone biopsies contained Al in Uruguay compared to 19% in Spain [43].
How to diagnose aluminium bone disease
Serum aluminium levels do not correctly reflect body aluminium stores and do not correlate well with signs of aluminium toxicity. A desferrioxamine (DFO) test increases the diagnostic accuracy (Table 5).
Table 5.
How to perform a DFO test
| Stop iron supplements 5 days before DFO testing | |
| Serum aluminium measurements need to be performed using an aluminium-free blood collection tube | |
| Measure aluminium in serum before dialysis session; choose dialysis session before long dialysis interval | |
| Administer DFO 1 h before end of dialysis session by i.v‥ DFO tests may be performed with high-dose (40 mg/kg) [42] or low-dose DFO (5–10 mg/kg) [44,45] | |
| Measure aluminium again in serum after long interval before next dialysis |
Depending on the dosage of DFO administered, the aluminium increase in serum regarded as diagnostic varies from exceeding 50 μg/L [44] to 200 μg/L [45]. The NKF-K/DOQI guidelines recommend performing the low-dose test because of possible DFO side effects (ophthalmologic damage and mucormycosis). The sensitivity and specificity of the low-dose DFO test to diagnose Al-bone disease are 87% and 95%, respectively, if at the same time iPTH levels are <150 pg/mL [46]. Candidates for DFO testing are patients with elevated serum aluminium levels (between 60 and 200 μg/L) and clinical symptoms and/or signs suggestive of aluminium toxicity. Patients exposed to significant amounts of aluminium in the past and who are scheduled for parathyroidectomy should also be tested, since aluminium-related bone disease can worsen after parathyroidectomy. The latter concern may also apply if calcimimetics are used in such patients, but this is not proven yet.
Upon bone biopsy staining using the aluminon reagent or the acid solochrome azurine (ASA) stain is required in order to detect aluminium [47,48]. Aluminium-positive surfaces <5% are usually not considered to be significant, while those >25% are considered to be strongly positive [49].
When does ABD occur in the course of CKD?
ABD frequently occurs before ESRD is reached [21,50,51]. Bone biopsies in patients new on dialysis or with advanced CKD (mean age 54 ± 12 years) revealed ABD in 23% of the patients [21]. None of these patients had received calcitriol or aluminium during the course of CKD. An even higher ABD prevalence of 49% in predialysis CKD stage 5 patients was reported [19]. The prevalence of ABD was 13% in patients with a creatinine clearance of 20 ± 12 ml/ min [51]. No data are available on the evolution of ABD in patients who progress from CKD stages 3 to 5.
Evolution of ABD prevalence over the last decades
The prevalence of ABD has increased over the last 15– 20 years, despite the fact that aluminium-induced low-turnover bone disease has become more and more infrequent [6,18] (Figure 4). Non-aluminium-induced ABD has now emerged as the dominant lesion in a mixed cohort of adult haemodialysis and peritoneal dialysis patients [18], and in particular in diabetic ESRD patients, prevalences up to 67% have been observed [21]. In parallel, the former predominance of hyperparathyroid bone disease has diminished [6,52]. The increase in ABD prevalence parallels two major developments in dialysis patients. First, the proportion of elderly and diabetic patients is steadily growing. Second, many patients were exposed to relatively high vitamin D and oral calcium dosages. It is currently impossible to quantify the relative impact of these two potentially causative factors.
Fig. 4.
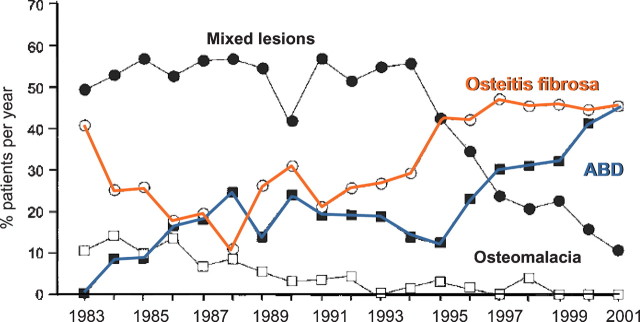
Evolution of ROD distribution pattern over time (modified after [6]).
Of note, not all studies confirm a high ABD prevalence. For example, Lehmann et al. [20] used the static histological parameters, osteoclast-covered surface/bone surface (OcS/BS <1%) and osteoblast-covered surface/bone surface (ObS/BS <1%), to stratify patients into low- versus high-turnover osteopathy. With this classification, only ∼7% of both pre- and dialysis patients suffered from low-turnover osteopathy.
What are risk factors for the development of ABD?
Besides aluminium, several other factors or conditions decrease bone turnover and bone remodelling activity (Table 6). Low bone turnover is not limited to advanced CKD, but also occurs in other conditions that are frequent in dialysis populations such as advanced age, glucocorticoid-induced osteoporosis, diabetes and hypoparathyroidism. A relative ‘hypoparathyroidism’ is regarded as an important risk factor for ABD [14,18,50,53]. It may be due to low iPTH(1–84) levels or to a relative excess of antagonistic PTH fragments (e.g. PTH(7–84)) that negatively affect bone metabolism [54]. In a bone biopsy study in dialysis patients, iPTH plasma levels determined by immunoradiometric assay (Nichols Allegro) were highly predictive of ABD if <120 pg/mL, while levels >450 pg/mL virtually excluded ABD [19]. When considering the optimal osteoblast surface (1.5%) and the absence of fibrosis, the authors defined an iPTH range between 120 and 250 pg/mL as desirable (in patients not treated with calcitriol). In agreement with this report, bone biopsies in parathyroidectomized dialysis patients with a persistent iPTH plasma level <70 pg/mL uniformly revealed low turnover or ABD at 1 year after the operation [55].
Table 6.
Factors associated with a high prevalence of ABD
Apart from absolute or relative hypoparathyroidism, ABD is frequently characterized by skeletal resistance to bone-anabolic PTH actions, presumably via a down-regulation of the PTH/PTHrp receptor on osteoblasts [56,57]. In patients with ABD, the parathyroid gland responsiveness to hypocalcaemia is diminished. As a consequence, PTH pulsatility, an important parameter accounting for PTH anabolic bone actions, is impaired in ABD.
Diabetes mellitus negatively affects bone metabolism. In type 1 diabetics with ESRD bone biopsies exhibited reduced trabecular and osteoid bone volumes and marked reductions in indices of bone formation and resorption [58]. Diabetic dialysis patients are also particularly prone to aluminium accumulation and PTH resistance [59].
Calcium administration and vitamin D as triggers for ABD will be discussed in the ‘treatment’ section.
It is clear from the above that the pathophysiology of ABD is certainly multifactorial. Further mediators may include uraemic toxins as well as derangements in cytokines and growth factors [10, 60]. In summary, on the background of relative PTH resistance in a uraemic milieu and presumably several other factors, there is only a thin line in CKD between allowing sufficient hyperparathyroidism to maintain sufficient bone metabolism versus oversuppression of PTH leading to low-turnover bone disease [61].
Low-turnover bone disease and clinical symptoms
Skeletal pain may occur in all subtypes of ROD, but is especially common in patients with (aluminium-induced) osteomalacia [10]. Proximal muscle weakness together with axial skeletal pain and fractures of the ribs, vertebral bodies, pelvis and hips has been described as common features of aluminium-induced osteomalacia [35]. However, these signs and symptoms may also occur in the absence of aluminium overload in patients with osteomalacic bone lesions [68] (Figure 5). The classical triad [35] of dialysis encephalopathy [69], microcytic anaemia and osteopathy suggesting aluminium toxicity is very rare nowadays. It has been claimed that ‘aluminium-induced bone disease is the only form of low-turnover producing symptoms and ultimately death’ [10]. This idea came from observation that side effects of low-turnover bone disease (pain, hypercalcaemia, fractures) were associated with aluminium covering >20% of the bone surface [10]. However, non-aluminium induced ABD also carries significant morbidity and mortality [42] (see below) and it is now clear that any type of ABD may cause bone pain. However, there is no pathognomonic clinical sign of ABD.
Fig. 5.
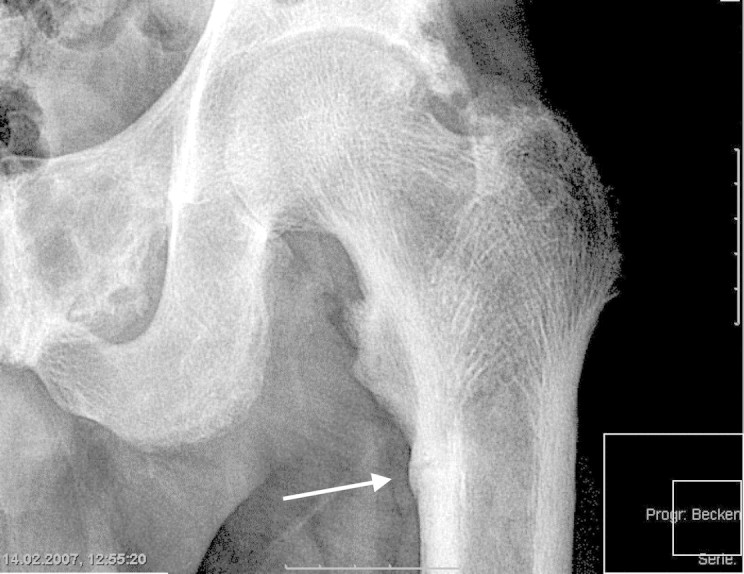
Painful bone lesions in a patient with low-turnover osteodystrophy and Looser zones (arrow) (courtesy of Professor R. Guenther, Aachen).
ABD and calcium metabolism
ABD is characterized by a reduced ability to incorporate serum calcium into the bone compartment [70]. In calcium isotope experiments in dialysis patients with biopsy-proven ROD, calcium accretion in bone was significantly lower in ABD compared to hyperparathyroid bone lesions [70]. Patients with reduced bone turnover exhibited a higher systemic calcium exposure while enteral calcium absorption did not differ between high- and low-turnover bone lesions [70]. In agreement with this, low biochemical markers of bone turnover predicted the development of hypercalcaemia after the initiation of calcium carbonate [71]. The reduced bone capacity to buffer calcium loads in ABD has now been widely confirmed [72].
ABD and ectopic calcification
Cardiovascular calcifications and associated mortality are prominent clinical problems in patients with ESRD [73–75]. Several studies noted a relation between bone metabolism and such calcifications. In 224 prevalent Turkish haemodialysis patients, low turnover was detected in 75% of the bone biopsies [76]. Patients with the lowest bone activation frequencies, i.e. the lowest bone turnover, exhibited the most pronounced coronary artery calcification (CAC) scores. Similar findings were obtained in 101 Brazilian haemodialysis patients [15]. London et al. [42] quantified vascular calcifications of the common carotid arteries, the abdominal aorta, iliofemoral axis as well as legs. Increasing calcification score levels were associated with decreasing mean iPTH, tetracycline double-labelled surface and osteoblast surface, while the aluminium-stained surface predicted the calcification score in a multiple stepwise regression analysis [42]. All these findings point to an association of low-bone turnover with cardiovascular calcifications (Figures 6 and 7).
Fig. 6.
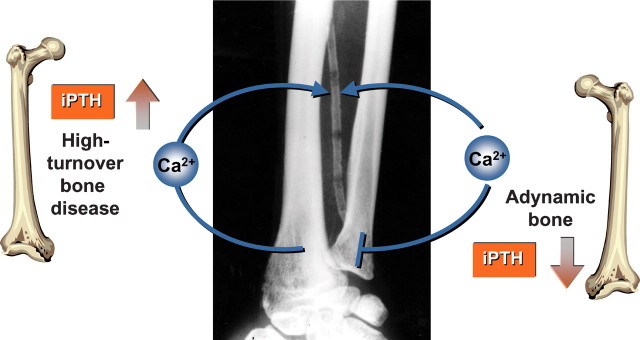
Impaired bone buffering capacity in both high-turnover osteopathy and adynamic bone turnover.
Fig. 7.
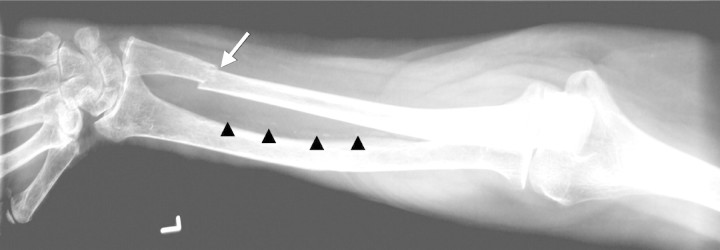
Coexistence of bone (arrow) and vascular (arrowheads) disease in uraemia.
Calcific uraemic arteriolopathy (CUA), formerly called calciphylaxis, has also been linked to ABD [77]: five out of seven patients with CUA had biopsy-confirmed ABD (Figure 8).
Fig. 8.
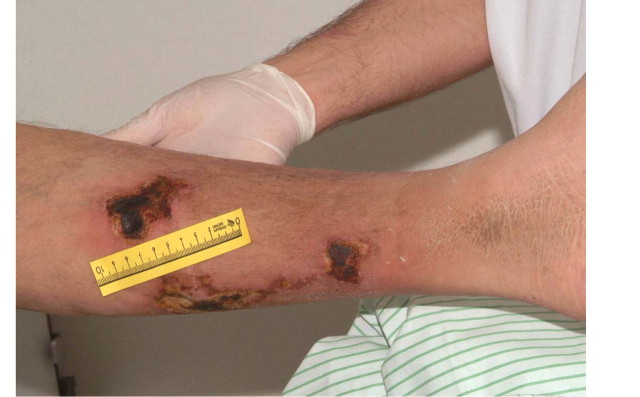
Painful, cutaneous lesions in a patient with calcific uraemic arteriolopathy, CUA (calciphylaxis).
The above human data are supported by animal studies performed in LDL receptor knock-out mice (LDL-R−/−). These mice, when fed a high-fat or diabetogenic diet, also exhibit the combination of low-turnover osteodystrophy and vascular calcifications [78,79]. This is accelerated by superimposed experimental CKD [78,79]. Administration of anabolic bone stimulating agents such as bone morphogenic protein 7 (BMP-7) [78] or synthetic PTH(1–34) [79] improved bone turnover and skeletal mineralization and decreased calcium deposition in the aorta.
Low iPTH and increased mortality
The causal relation between ABD and vascular disease may at least in part explain why iPTH plasma levels <150 pg/mL led to a significant, 1.4-fold increase in mortality in 58 000 ESRD patients after extensive multivariate adjustments [80]. Ganesh et al. confirmed a U-curve relationship in their 2-year follow-up study in 12 800 dialysis patients: Both very low (<32 pg/ml) and high iPTH levels (>496 pg/ml) increased the risk for sudden death [81]. Similar findings were reported in other smaller studies [82;83]. In particular the combination of low iPTH and high serum calcium levels (plus high serum phosphate), a combination typical for ABD, was associated with substantial mortality [84]. However, such a U-curve-shaped relationship between PTH and mortality has not been uniformly confirmed. After multiple adjustments, Block et al. revealed a linear association of the two parameters [85].
ABD and bone stability
ABD is associated with a diminished ability to repair microdamage [5]. Accumulated microdamage may result in an increased fracture risk [53,86]. In a retrospective study in 9000 haemodialysis patients, a U-curve relationship between fracture risk and plasma iPTH levels was indeed detectable [87]. Fracture risk was comparable for hip, vertebrae and pelvis in patients with iPTH levels <150 pg/mL and those with iPTH exceeding 800 pg/mL and was lowest around ∼300 pg/mL [87]. Another study determined that, compared to the normal population, hip fracture incidence was 17 times higher in ESRD patients [88]. One of the significant predictors of fracture risk was an iPTH level <195 pg/mL. Atsumi et al. [86] retrospectively showed that the lowest tertile of iPTH, in particular in men, was associated with a 22% increase in the risk of vertebral fractures. However, all these studies have significant limitations and may only serve to create a hypothesis rather than to establish evidence, since none assessed bone histologies or parathyroidectomy rates and they were retrospective and uncontrolled.
In prepubertal children, ABD was associated with decreased linear growth and worsened growth retardation [89].
Management of the patient with ABD
General considerations
In contrast to high-turnover bone disease, the management of ABD is not well investigated and large-scale prospective randomized trials are absent [90]. The treatment currently follows two principles: first, to reduce calcium and vitamin D load and second, to restore PTH activity (Table 7). Using these approaches, ABD is reversible in a substantial number of patients [13,91]. However, while the approaches mentioned above appear intuitive, the situation clearly is more complex given data from large databases indicating that treatment with active vitamin D is associated with a survival benefit even in patients with very low PTH levels [92]. Thus, potential bone benefits of avoiding active vitamin D in ABD patients may be offset by the resulting lack of other beneficial actions of a pleiotropic compound such as vitamin D, emphasizing the need for large controlled prospective trials in this area.
Table 7.
Therapeutic strategies in ABD
| Stop calcium-containing phosphate binders and replace with non-calcium-, non-aluminium-containing phosphate binders | |
| Assess oral dietary calcium intake and reduce to <2000 mg/day | |
| Reduce or stop active vitamin D compounds | |
| Lower dialysate calcium to 1.25 mmol/L or below | |
| In selected cases consider a biopsy to confirm diagnosis and to assess bone aluminium content and distribution | |
| Stop aluminium exposition; consider aluminium mobilisation and removal (DFO treatment) | |
| Consider PTH(1–34) in ABD plus severe fracturing osteoporosis | |
| Calcimimetics and calcilytics currently of unknown value | |
| Avoid bisphosphonates, strontium and fluoride administration |
Aluminium removal in cases of significant exposure
DFO mobilizes aluminium from bone and decreases the proportion of protein-bound aluminium in plasma, thereby facilitating removal by dialysis. Discontinuation of aluminium and administration of DFO improved signs of aluminium-induced bone lesions in vivo [93,94]. Human data with serial biopsies after DFO treatment have shown marked declines in stainable bone-surface aluminium that were associated with increases in BFR [17]. Long-term application of DFO (11 ± 4 months, dosage 42 ± 17 mg/kg administered once weekly) also improved signs of dementia and increased erythrocyte mean corpuscular volume, but side effects were common [95]. Polysulfone dialyzers offer maximum clearance of DFO-aluminium complexes [96]. Parathyroidectomy should be avoided in patients with aluminium-induced bone disease, since the decrease in bone turnover after surgery may be associated with an accelerated accumulation of aluminium in bone [97]. A repeat bone biopsy with quantification of stainable aluminium on the trabecular surface may help to guide the duration of chelation therapy [27].
Reduction of intradialytic calcium loading
Serum ionized calcium levels are probably the most powerful regulator of PTH synthesis and excretion. Especially in conjunction with vitamin D treatment a positive calcium balance depresses bone turnover [63]. For both haemodialysis and CAPD patients there are convincing laboratory data and first histomorphometry results showing that lowering dialysate calcium concentration improves ABD [13,98–100]. Reducing the dialysate calcium concentration from 1.75 or 1.5 mmol/L to 1.25 mmol/L reduced serum ionized calcium, diminished episodes of hypercalacemia and increased iPTH (fourfold), bone-specific alkaline phosphatase and TRAP-5b levels within 3–6 months [100]. In a prospective trial in 51 CAPD patients with biopsy-proven ABD, two batch calcium concentrations (1.62 mM or 1.0 mM) were compared [13]. Repeat bone biopsies after 16 months showed that the low-calcium batch led to a normalization of BFR, which increased from 18.1 ± 5.6 to 159 ± 59 μm2/mm2/day. The low-calcium group experienced a decrease in serum ionized calcium levels resulting in a 300% increase in serum iPTH values (from 57 ± 15 to 237 ± 34 pg/mL). In 40% of the patients, ABD had resolved after 16 months.
The current NKF K/DOQI guidelines recommend limiting daily oral calcium intake (dietary calcium plus phosphate binder) to <2000 mg. Dialysate calcium concentrations of 1.75 mmol/L should not be used routinely. In cases of ABD, reduction of dialysate calcium to 1.25 or 1.00 mmol/L is advisable and usually tolerated well clinically.
Usage of calcium-free phosphate binders
Oral calcium-containing phosphate binders are the other major source of calcium. Recently developed calcium- and aluminium-free phosphate binders now offer alternatives. Two prospective bone biopsy studies have compared the effects of calcium-free versus calcium-containing phosphate binders on bone metabolism and histology in dialysis patients [30,91]. D'Haese et al. [91] compared the bone effects of lanthanum carbonate versus calcium carbonate in 63 dialysis patients. The median intake of calcium carbonate and lanthanum carbonate was 2000 (n = 30) and 1250 mg/day (n = 33), respectively. After 1 year of treatment, the number of patients increasing their bone turnover after an initial diagnosis of ABD was similar in both groups (3/6 calcium carbonate versus 4/6 lanthanum). However, during the follow-up, bone turnover decreased to ABD after an initial diagnosis of high-turnover ROD in six patients of the calcium carbonate group versus one patient in the lanthanum group. Regarding sevelamer, Ferreira et al. analysed repetitive bone biopsies in 68 patients after 1 year of treatment with either sevelamer (dosage increased from 3.3 ± 2.0 to 5.0 ± 2.7 g/day) or calcium carbonate (dosage increased from 3.8 ± 2.2 to 4.0 ± 2.5 g/day) [30]. Only the sevelamer group exhibited a significant increase in BFR per bone surface. At the end of the study three patients (9%) had developed de novo ABD in the sevelamer group compared to six (17%) in the calcium group. However, the comparability between these two bone biopsy studies is limited due to different histomorphometric criteria of ABD [30,91]. Several additional lines of evidence also point towards an improved bone turnover following a switch from calcium-containing phosphate binders to sevelamer [101,102]. In the Treat-to-Goal study, 200 haemodialysis patients were randomized either to 6.5 g/day sevelamer or 4.6 g/day calcium acetate or 3.9/day g calcium carbonate (mean intake) over 53 weeks. Mean iPTH remained stable in the sevelamer group (∼220 pg/mL), whereas it dropped significantly from 200 to 138 pg/ml in the calcium group. In a post hoc analysis of this study, it was shown that calcium-treated subjects showed a decrease in thoracic vertebral trabecular bone attenuation, a surrogate marker of bone density, whereas sevelamer-treated subjects exhibited stable values [102]. Similar data were obtained in a 2-year prospective study that also compared calcium carbonate-treated (4.3 ± 1.7 g/day) with sevelamer-treated (6.9 ± 2.6 g/day) haemodialysis patients [103]. The calcium carbonate group in comparison to the sevelamer group exhibited decreasing iPTH levels, significantly more hypercalcaemic episodes, and a loss of trabecular bone density [103] (Figure 9).
Fig. 9.
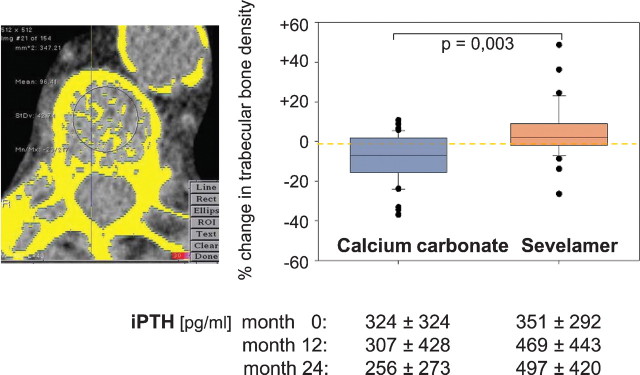
Change in trabecular bone density: comparison between sevelamer and calcium carbonate treatment (modified after [103]).
These human data are in line with experimental results indicating that high dosages of calcium supplementation in uraemic rats suppress osteoclastic and chondroclastic activity [104].
Avoidance of vitamin D over-treatment
The administration of active vitamin D compounds reduces bone turnover in CKD patients. One hundred and seventy-six CKD patients (GFR 15–50 ml/min) were randomized to alphacalcidol (0.25 μg every other day to 1.0 μg/day) or placebo treatment over 2 years [49]. Bone biopsies were performed at the study entry and end. In patients with ROD at baseline (75%), alphacalcidol treatment significantly reduced osteoblast surface, number of osteoblasts, eroded surface and BFR while these parameters changed insignificantly with placebo. Biopsy studies indicate that high dosages of active vitamin D (calcitriol) in patients with ESRD may eventually lead to the development of ABD. In a prospective 12-month study with serial bone biopsies in 14 children on peritoneal dialysis, all exhibited hyperparathyroidism-associated bone lesions at baseline and 11 overt osteitis fibrosa [63]. Intermittent oral or intraperitoneal calcitriol decreased BFR by ∼60% and six children developed ABD (43%) [63]. Similar results emerged from another 12-month repeat biopsy study in 16 peritoneal dialysis children, who, after an initial diagnosis of osteitis fibrosa (n = 9) or mild lesions of secondary HPT (n = 7), developed ABD under calcitriol in 25% of the cases [89]. However, in all these studies high-dosage active vitamin D treatment was associated with higher incidences of hypercalcaemia and higher mean serum calcium levels. Therefore, it is difficult to assess the particular impact of non-calcaemic versus calcaemic vitamin D actions upon bone metabolism. Moreover, the high dialysate calcium content of 1.75 mmol/L certainly contributed to ABD development in the two studies.
Preliminary in vitro data point towards a lower osteoblast activity suppression of novel vitamin D receptor (VDR) agonists (paricalcitol) [105]. Additionally, paricalcitol increased while calcitriol decreased the PTH(1–84)/PTH C-fragment ratio in haemodialysis patients indicating a positive effect by paricalcitol on skeletal PTH resistance [106]. However, no human bone biopsy data are available to verify whether newer VDR agonists indeed affect bone turnover in a better way than calcitriol.
Teriparatide as a bone-stimulating agent
The daily subcutaneous application of PTH(1–34), teriparatide, is a powerful anti-osteoporotic treatment. In theory, teriparatide offers the chance to restore bone metabolism in patients with ABD ([79] see above). The administration of PTH(1–34) in patients with ‘non-renal’ hypoparathyroidism (mostly post-surgical or with gain-of-function mutations in the calcium-sensing receptor) over 3 years led to significant elevations of bone turnover markers [107]. However, controlled human trials in CKD have not been performed so far. Nevertheless, in anecdotal reports, teriparatide (e.g. 20 μg s.c. three times per week after haemodialysis) has been used in bone biopsy-confirmed ABD patients with severe fracturing osteoporosis. Reductions of bone pain and transient increases of bone-specific alkaline phosphatase have been reported.
Restoring the pulsatile PTH secretion pattern
The biological action of PTH on bone largely depends on pulsatile PTH secretion [108]. This may explain the risk for ABD in patients receiving active vitamin D or peritoneal dialysis, since in the former case vitamin D activity builds up over days and then continuously suppresses PTH release, whereas in PD patients there is often a constant exposure to high calcium dialysate levels, in contrast to the fluctuating calcium level in HD patients.
Two classes of compounds may help re-establish a pulsatile, oscillatory secretion pattern of PTH in patients with ABD: the calcimimetics and the calcilytics. The calcimimetic agent cinacalcet has a half-life of <24 h and initially reduces iPTH levels markedly, but this is followed by a strong iPTH rebound in plasma so that circadian swings of plasma iPTH increase [109]. In vivo experiments already showed a bone protective, bone anabolic effect of calcimimetics [110]. Untreated rats with adriamycin-induced CKD developed a low-turnover bone disease resembling osteomalacia [110]. Two treatment arms with NPS-568, a short-acting calcimimetic agent, were tested: one with daily oral gavage, the other with a continuous subcutaneous infusion. While the continuous infusion normalized PTH-levels in the previously hyperparathyroid CKD animals, large fluctuations of PTH were detectable in the gavage group: at 1 h after gavage, PTH decreased by 78%, while levels had returned to baseline after 14 h. After 57 days, several parameters of bone formation were significantly improved in the daily gavage arm compared to the animals treated continuously.
Finally, calcilytic agents, which temporarily block the calcium sensing receptor at the parathyroid gland and thereby promote PTH secretion, may also help to stimulate bone turnover by increasing the pulsatile PTH secretion pattern. The oral calcilytic agent NPS 2143 has been applied to a model of bone loss and osteopenia (ovarectomized rats) [111] and compared with the action of s.c. PTH(1–34). Increases of plasma PTH after the administration of NPS 2143 were prolonged (>4 h) in contrast to short increases with s.c. PTH(1–34). Indeed, both agents stimulated bone turnover. However, NPS 2143 resulted in a dramatic increase in both bone formation and resorption, with no net effect on bone mass. In contrast, PTH(1–34) also increased both resorption and formation, but formation exceeded resorption, resulting in increased bone mass. Only the coapplication of the calcilytic agent plus estradiol led to an increase in bone mass, presumably due to the hormonal antiresorptive effect in this experiment. Calcilytic agents therefore need further proof of bone protective properties.
ABD: closing remarks
ABD is not an innocent bystander in CKD [65]. It is possibly the most prevalent bone lesion in advanced CKD, is associated with impaired calcium metabolism and linked to cardiovascular disease and mortality in CKD patients. ABD is, at least in part, often iatrogenic and it is this part in particular, which lends itself to prevention or therapeutic intervention. Reducing the calcium load is the best investigated preventive or therapeutic option in non-aluminium induced ABD.
Acknowledgments
Conflict of interest statement. The results presented in this paper have not been published previously in whole or part. J.F. is a regular consultant for Amgen and Genzyme and has received honoraria for lectures and panels from the following companies: Abbott, Amgen, Genzyme and Shire. V.B. has received travel grants and honoraria for lectures from Genzyme, Abbott and Shire.
References
- 1.Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2006;69:1945–1953. doi: 10.1038/sj.ki.5000414. [DOI] [PubMed] [Google Scholar]
- 2.Haas M, Leko-Mohr Z, Roschger P, et al. Osteoprotegerin and parathyroid hormone as markers of high-turnover osteodystrophy and decreased bone mineralization in hemodialysis patients. Am J Kidney Dis. 2002;39:580–586. doi: 10.1053/ajkd.2002.31409. [DOI] [PubMed] [Google Scholar]
- 3.Malluche HH, Monier-Faugere MC. Renal osteodystrophy: what's in a name? Presentation of a clinically useful new model to interpret bone histologic findings. Clin Nephrol. 2006;65:235–242. doi: 10.5414/cnp65235. [DOI] [PubMed] [Google Scholar]
- 4.Ferreira A. Development of renal bone disease. Eur J Clin Invest. 2006;36(Suppl 2):2–12. doi: 10.1111/j.1365-2362.2006.01661.x. [DOI] [PubMed] [Google Scholar]
- 5.Parfitt AM. Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens. 2003;12:387–403. doi: 10.1097/00041552-200307000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Malluche HH, Mawad H, Monier-Faugere MC. The importance of bone health in end-stage renal disease: out of the frying pan, into the fire. Nephrol Dial Transplant. 2004;19(Suppl1):i9–i13. doi: 10.1093/ndt/gfh1002. [DOI] [PubMed] [Google Scholar]
- 7.Sherrard DJ, Ott S, Maloney NA. Uremic osteodystrophy, classification, cause, and treatment. In: Frame B, Potts JRJ, editors. Clinical Disorders of Bone and Mineral Metabolism. Amsterdam: Excerpta Medica; 1983. pp. 254–259. [Google Scholar]
- 8.Malluche HH, Faugere MC. Atlas of Mineralized Bone Histology. Basel: Karger; 1986. [Google Scholar]
- 9.Spasovski GB. Bone biopsy as a diagnostic tool in the assessment of renal osteodystrophy. Int J Artif Organs. 2004;27:918–923. doi: 10.1177/039139880402701103. [DOI] [PubMed] [Google Scholar]
- 10.Cannata-Andia JB. Hypokinetic azotemic osteodystrophy. Kidney Int. 1998;54:1000–1016. doi: 10.1046/j.1523-1755.1998.00080.x. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez CJ, Hazelwood SJ, Martin RB. The relationship between basic multicellular unit activation and origination in cancellous bone. Bone. 1999;25:585–587. doi: 10.1016/s8756-3282(99)00201-x. [DOI] [PubMed] [Google Scholar]
- 12.Ballanti P, Coen G, Mazzaferro S, et al. Histomorphometric assessment of bone turnover in uraemic patients: comparison between activation frequency and bone formation rate. Histopathology. 2001;38:571–583. doi: 10.1046/j.1365-2559.2001.01139.x. [DOI] [PubMed] [Google Scholar]
- 13.Haris A, Sherrard DJ, Hercz G. Reversal of adynamic bone disease by lowering of dialysate calcium. Kidney Int. 2006;70:931–937. doi: 10.1038/sj.ki.5001666. [DOI] [PubMed] [Google Scholar]
- 14.Monier-Faugere MC, Malluche HH. Trends in renal osteodystrophy: a survey from 1983 to 1995 in a total of 2248 patients. Nephrol Dial Transplant. 1996;11(Suppl 3):111–120. doi: 10.1093/ndt/11.supp3.111. [DOI] [PubMed] [Google Scholar]
- 15.Barreto DV, Barreto FC, Carvalho AB, et al. Coronary calcification in hemodialysis patients: the contribution of traditional and uremia-related risk factors. Kidney Int. 2005;67:1576–1582. doi: 10.1111/j.1523-1755.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 16.Salusky IB, Coburn JW, Brill J, et al. Bone disease in pediatric patients undergoing dialysis with CAPD or CCPD. Kidney Int. 1988;33:975–982. doi: 10.1038/ki.1988.96. [DOI] [PubMed] [Google Scholar]
- 17.Andress DL, Nebeker HG, Ott SM, et al. Bone histologic response to deferoxamine in aluminum-related bone disease. Kidney Int. 1987;31:1344–1350. doi: 10.1038/ki.1987.148. [DOI] [PubMed] [Google Scholar]
- 18.Sherrard DJ, Hercz G, Pei Y, et al. The spectrum of bone disease in end-stage renal failure—an evolving disorder. Kidney Int. 1993;43:436–442. doi: 10.1038/ki.1993.64. [DOI] [PubMed] [Google Scholar]
- 19.Torres A, Lorenzo V, Hernandez D, et al. Bone disease in predialysis, hemodialysis, and CAPD patients: evidence of a better bone response to PTH. Kidney Int. 1995;47:1434–1442. doi: 10.1038/ki.1995.201. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann G, Stein G, Huller M, et al. Specific measurement of PTH (1–84) in various forms of renal osteodystrophy (ROD) as assessed by bone histomorphometry. Kidney Int. 2005;68:1206–1214. doi: 10.1111/j.1523-1755.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- 21.Spasovski GB, Bervoets AR, Behets GJ, et al. Spectrum of renal bone disease in end-stage renal failure patients not yet on dialysis. Nephrol Dial Transplant. 2003;18:1159–1166. doi: 10.1093/ndt/gfg116. [DOI] [PubMed] [Google Scholar]
- 22.Bervoets AR, Spasovski GB, Behets GJ, et al. Useful biochemical markers for diagnosing renal osteodystrophy in predialysis end-stage renal failure patients. Am J Kidney Dis. 2003;41:997–1007. doi: 10.1016/s0272-6386(03)00197-5. [DOI] [PubMed] [Google Scholar]
- 23.Rocha LA, Higa A, Barreto FC, et al. Variant of adynamic bone disease in hemodialysis patients: fact or fiction. Am J Kidney Dis. 2006;48:430–436. doi: 10.1053/j.ajkd.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 24.Llach F, Felsenfeld AJ, Coleman MD, et al. The natural course of dialysis osteomalacia. Kidney Int Suppl. 1986;18:S74–S79. [PubMed] [Google Scholar]
- 25.Schwarz C, Sulzbacher I, Oberbauer R. Diagnosis of renal osteodystrophy. Eur J Clin Invest. 2006;36(Suppl 2):13–22. doi: 10.1111/j.1365-2362.2006.01666.x. [DOI] [PubMed] [Google Scholar]
- 26.Cannata-Andia JB. Pathogenesis, prevention and management of low-bone turnover. Nephrol Dial Transplant. 2000;15(Suppl 5):15–17. doi: 10.1093/ndt/15.suppl_5.15. [DOI] [PubMed] [Google Scholar]
- 27.Malluche HH, Monier-Faugere MC. The role of bone biopsy in the management of patients with renal osteodystrophy. J Am Soc Nephrol. 1994;4:1631–1642. doi: 10.1681/ASN.V491631. [DOI] [PubMed] [Google Scholar]
- 28.Urena P, De Vernejoul MC. Circulating biochemical markers of bone remodeling in uremic patients. Kidney Int. 1999;55:2141–2156. doi: 10.1046/j.1523-1755.1999.00461.x. [DOI] [PubMed] [Google Scholar]
- 29.Barreto FC, Barreto DV, Moyses RM, et al. K/DOQI-recommended intact PTH levels do not prevent low-turnover bone disease in hemodialysis patients. Kidney Int. 2008;73:771–777. doi: 10.1038/sj.ki.5002769. [DOI] [PubMed] [Google Scholar]
- 30.Ferreira A, Frazao JM, Monier-Faugere MC, et al. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J Am Soc Nephrol. 2008;19:405–412. doi: 10.1681/ASN.2006101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodman WG. Comments on plasma parathyroid hormone levels and their relationship to bone histopathology among patients undergoing dialysis. Semin Dial. 2007;20:1–4. doi: 10.1111/j.1525-139X.2007.00230.x. [DOI] [PubMed] [Google Scholar]
- 32.Goodman WG. The evolution of assays for parathyroid hormone. Semin Dial. 2005;18:296–301. doi: 10.1111/j.1525-139X.2005.18405.x. [DOI] [PubMed] [Google Scholar]
- 33.Monier-Faugere MC, Geng Z, Mawad H, et al. Improved assessment of bone turnover by the PTH-(1–84)/large C-PTH fragments ratio in ESRD patients. Kidney Int. 2001;60:1460–1468. doi: 10.1046/j.1523-1755.2001.00949.x. [DOI] [PubMed] [Google Scholar]
- 34.Berlyne GM, Ben Ari J, Pest D, et al. Hyperaluminaemia from aluminum resins in renal failure. Lancet. 1970;2:494–496. doi: 10.1016/s0140-6736(70)90113-3. [DOI] [PubMed] [Google Scholar]
- 35.Nebeker HG, Coburn JW. Aluminum and renal osteodystrophy. Annu Rev Med. 1986;37:79–95. doi: 10.1146/annurev.me.37.020186.000455. [DOI] [PubMed] [Google Scholar]
- 36.Savory J, Bertholf RL, Wills MR. Aluminium toxicity in chronic renal insufficiency. Clin Endocrinol Metab. 1985;14:681–702. doi: 10.1016/s0300-595x(85)80012-8. [DOI] [PubMed] [Google Scholar]
- 37.Ott SM, Maloney NA, Coburn JW, et al. The prevalence of bone aluminum deposition in renal osteodystrophy and its relation to the response to calcitriol therapy. N Engl J Med. 1982;307:709–713. doi: 10.1056/NEJM198209163071202. [DOI] [PubMed] [Google Scholar]
- 38.Morrissey J, Slatopolsky E. Effect of aluminum on parathyroid hormone secretion. Kidney Int Suppl. 1986;18:S41–S44. [PubMed] [Google Scholar]
- 39.Diaz-Corte C, Fernandez-Martin JL, Barreto S, et al. Effect of aluminium load on parathyroid hormone synthesis. Nephrol Dial Transplant. 2001;16:742–745. doi: 10.1093/ndt/16.4.742. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez-Suarez I, Naves M, Diaz-Corte C, et al. Effect of aluminium on calcium-sensing receptor expression, proliferation, and apoptosis of parathyroid glands from rats with chronic renal failure. Kidney Int Suppl. 2003;63:S39–S43. doi: 10.1046/j.1523-1755.63.s85.10.x. [DOI] [PubMed] [Google Scholar]
- 41.Felsenfeld AJ, Gutman RA, Llach F, et al. Harrelson JM. Osteomalacia in chronic renal failure: a syndrome previously reported only with maintenance dialysis. Am J Nephrol. 1982;2:147–154. doi: 10.1159/000166631. [DOI] [PubMed] [Google Scholar]
- 42.London GM, Marty C, Marchais SJ, et al. Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol. 2004;15:1943–1951. doi: 10.1097/01.asn.0000129337.50739.48. [DOI] [PubMed] [Google Scholar]
- 43.Jorgetti V, Lopez BD, Caorsi H, et al. Different patterns of renal osteodystrophy in Iberoamerica. Am J Med Sci. 2000;320:76–80. doi: 10.1097/00000441-200008000-00002. [DOI] [PubMed] [Google Scholar]
- 44.D’Haese PC, Couttenye MM, Goodman WG, et al. Use of the low-dose desferrioxamine test to diagnose and differentiate between patients with aluminium-related bone disease, increased risk for aluminium toxicity, or aluminium overload. Nephrol Dial Transplant. 1995;10:1874–1884. [PubMed] [Google Scholar]
- 45.Milliner DS, Nebeker HG, Ott SM, et al. Use of the deferoxamine infusion test in the diagnosis of aluminum-related osteodystrophy. Ann Intern Med. 1984;101:775–779. doi: 10.7326/0003-4819-101-6-775. [DOI] [PubMed] [Google Scholar]
- 46.Nebeker HG, Andress DL, Milliner DS, et al. Indirect methods for the diagnosis of aluminum bone disease: plasma aluminum, the desferrioxamine infusion test, and serum iPTH. Kidney Int Suppl. 1986;18:S96–S99. [PubMed] [Google Scholar]
- 47.Walton JR, Diamond TH, Kumar S, et al. A sensitive stain for aluminum in undecalcified cancellous bone. J Inorg Biochem. 2007;101:1285–1290. doi: 10.1016/j.jinorgbio.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Hodsman AB, Steer BM. Serum aluminum levels as a reflection of renal osteodystrophy status and bone surface aluminum staining. J Am Soc Nephrol. 1992;2:1318–1327. doi: 10.1681/ASN.V281318. [DOI] [PubMed] [Google Scholar]
- 49.Hamdy NA, Kanis JA, Beneton MN, et al. Effect of alfacalcidol on natural course of renal bone disease in mild to moderate renal failure. BMJ. 1995;310:358–363. doi: 10.1136/bmj.310.6976.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coen G, Mazzaferro S, Ballanti P, et al. Renal bone disease in 76 patients with varying degrees of predialysis chronic renal failure: a cross-sectional study. Nephrol Dial Transplant. 1996;11:813–819. doi: 10.1093/oxfordjournals.ndt.a027404. [DOI] [PubMed] [Google Scholar]
- 51.Coen G, Ballanti P, Bonucci E, et al. Renal osteodystrophy in predialysis and hemodialysis patients: comparison of histologic patterns and diagnostic predictivity of intact PTH. Nephron. 2002;91:103–111. doi: 10.1159/000057611. [DOI] [PubMed] [Google Scholar]
- 52.Martin KJ, Olgaard K, Coburn JW, et al. Diagnosis, assessment, and treatment of bone turnover abnormalities in renal osteodystrophy. Am J Kidney Dis. 2004;43:558–565. doi: 10.1053/j.ajkd.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 53.Hutchison AJ, Moore PR. Low turnover bone disease. Perit Dial Int. 1996;16(Suppl 1):S295–S299. [PubMed] [Google Scholar]
- 54.Langub MC, Monier-Faugere MC, Wang G, et al. Administration of PTH-(7–84) antagonizes the effects of PTH-(1–84) on bone in rats with moderate renal failure. Endocrinology. 2003;144:1135–1138. doi: 10.1210/en.2002-221026. [DOI] [PubMed] [Google Scholar]
- 55.Yajima A, Ogawa Y, Ikehara A, et al. Development of low-turnover bone diseases after parathyroidectomy and autotransplantation. Int J Urol. 2001;8:S76–S79. doi: 10.1046/j.1442-2042.2001.00340.x. [DOI] [PubMed] [Google Scholar]
- 56.Picton ML, Moore PR, Mawer EB, et al. Down-regulation of human osteoblast PTH/PTHrP receptor mRNA in end-stage renal failure. Kidney Int. 2000;58:1440–1449. doi: 10.1046/j.1523-1755.2000.00306.x. [DOI] [PubMed] [Google Scholar]
- 57.Iwasaki-Ishizuka Y, Yamato H, Nii-Kono T, et al. Downregulation of parathyroid hormone receptor gene expression and osteoblastic dysfunction associated with skeletal resistance to parathyroid hormone in a rat model of renal failure with low turnover bone. Nephrol Dial Transplant. 2005;20:1904–1911. doi: 10.1093/ndt/gfh876. [DOI] [PubMed] [Google Scholar]
- 58.Vincenti F, Arnaud SB, Recker R, et al. Parathyroid and bone response of the diabetic patient to uremia. Kidney Int. 1984;25:677–682. doi: 10.1038/ki.1984.73. [DOI] [PubMed] [Google Scholar]
- 59.Andress DL, Hercz G, Kopp JB, et al. Bone histomorphometry of renal osteodystrophy in diabetic patients. J Bone Miner Res. 1987;2:525–531. doi: 10.1002/jbmr.5650020609. [DOI] [PubMed] [Google Scholar]
- 60.Couttenye MM, D’Haese PC, Verschoren WJ, et al. Low bone turnover in patients with renal failure. Kidney Int Suppl. 1999;73:S70–S76. doi: 10.1046/j.1523-1755.1999.07308.x. [DOI] [PubMed] [Google Scholar]
- 61.Hendy GN, Hruska KA, Mathew S, et al. New insights into mineral and skeletal regulation by active forms of vitamin D. Kidney Int. 2006;69:218–223. doi: 10.1038/sj.ki.5000091. [DOI] [PubMed] [Google Scholar]
- 62.Baker LR, Abrams SM, Roe CJ, et al. Early therapy of renal bone disease with calcitriol: a prospective double-blind study. Kidney Int Suppl. 1989;27:S140–S142. [PubMed] [Google Scholar]
- 63.Goodman WG, Ramirez JA, Belin TR, et al. Development of adynamic bone in patients with secondary hyperparathyroidism after intermittent calcitriol therapy. Kidney Int. 1994;46:1160–1166. doi: 10.1038/ki.1994.380. [DOI] [PubMed] [Google Scholar]
- 64.Slatopolsky E, Weerts C, Thielan J, et al. Marked suppression of secondary hyperparathyroidism by intravenous administration of 1,25-dihydroxy-cholecalciferol in uremic patients. J Clin Invest. 1984;74:2136–2143. doi: 10.1172/JCI111639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malluche HH, Monier-Faugere MC. Risk of adynamic bone disease in dialyzed patients. Kidney Int Suppl. 1992;38:S62–S67. [PubMed] [Google Scholar]
- 66.Pei Y, Hercz G, Greenwood C, et al. Renal osteodystrophy in diabetic patients. Kidney Int. 1993;44:159–164. doi: 10.1038/ki.1993.226. [DOI] [PubMed] [Google Scholar]
- 67.Kurz P, Tsobanelis T, Roth P, et al. Differences in calcium kinetic pattern between CAPD and HD patients. Clin Nephrol. 1995;44:255–261. [PubMed] [Google Scholar]
- 68.Hernandez JD, Wesseling K, Boechat MI, et al. Osteomalacia in a hemodialysis patient receiving an active vitamin D sterol. Nat Clin Pract Nephrol. 2007;3:227–232. doi: 10.1038/ncpneph0443. [DOI] [PubMed] [Google Scholar]
- 69.Alfrey AC, LeGendre GR, Kaehny WD. The dialysis encephalopathy syndrome. Possible aluminum intoxication. N Engl J Med. 1976;294:184–188. doi: 10.1056/NEJM197601222940402. [DOI] [PubMed] [Google Scholar]
- 70.Kurz P, Monier-Faugere MC, Bognar B, et al. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int. 1994;46:855–861. doi: 10.1038/ki.1994.342. [DOI] [PubMed] [Google Scholar]
- 71.Meric F, Yap P, Bia MJ. Etiology of hypercalcemia in hemodialysis patients on calcium carbonate therapy. Am J Kidney Dis. 1990;16:459–464. doi: 10.1016/s0272-6386(12)80059-x. [DOI] [PubMed] [Google Scholar]
- 72.Hercz G, Pei Y, Greenwood C, et al. Aplastic osteodystrophy without aluminum: the role of “suppressed” parathyroid function. Kidney Int. 1993;44:860–866. doi: 10.1038/ki.1993.323. [DOI] [PubMed] [Google Scholar]
- 73.Kestenbaum B, Belozeroff V. Mineral metabolism disturbances in patients with chronic kidney disease. Eur J Clin Invest. 2007;37:607–622. doi: 10.1111/j.1365-2362.2007.01840.x. [DOI] [PubMed] [Google Scholar]
- 74.Westenfeld R, Jahnen-Dechent W, Ketteler M. Vascular calcification and fetuin-A deficiency in chronic kidney disease. Trends Cardiovasc Med. 2007;17:124–128. doi: 10.1016/j.tcm.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 75.Kalpakian MA, Mehrotra R. Vascular calcification and disordered mineral metabolism in dialysis patients. Semin Dial. 2007;20:139–143. doi: 10.1111/j.1525-139X.2007.00261.x. [DOI] [PubMed] [Google Scholar]
- 76.Asci G. The link between cardiovascular and bone disease in hemodialysis patients (abstract) Nephrol Dial.Transplant. 2007;22(S6):217. [Google Scholar]
- 77.Mawad HW, Sawaya BP, Sarin R, et al. Calcific uremic arteriolopathy in association with low turnover uremic bone disease. Clin Nephrol. 1999;52:160–166. [PubMed] [Google Scholar]
- 78.Davies MR, Lund RJ, Mathew S, et al. Low turnover osteodystrophy and vascular calcification are amenable to skeletal anabolism in an animal model of chronic kidney disease and the metabolic syndrome. J Am Soc Nephrol. 2005;16:917–928. doi: 10.1681/ASN.2004100835. [DOI] [PubMed] [Google Scholar]
- 79.Shao JS, Cheng SL, Charlton-Kachigian N, et al. Teriparatide (human parathyroid hormone (1–34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. J Biol Chem. 2003;278:50195–50202. doi: 10.1074/jbc.M308825200. [DOI] [PubMed] [Google Scholar]
- 80.Kalantar-Zadeh K, Kuwae N, Regidor DL, et al. Survival predictability of time-varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int. 2006;70:771–780. doi: 10.1038/sj.ki.5001514. [DOI] [PubMed] [Google Scholar]
- 81.Ganesh SK, Stack AG, Levin NW, et al. Association of elevated serum PO(4), Ca × PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol. 2001;12:2131–2138. doi: 10.1681/ASN.V12102131. [DOI] [PubMed] [Google Scholar]
- 82.Avram MM, Mittman N, Myint MM, et al. Importance of low serum intact parathyroid hormone as a predictor of mortality in hemodialysis and peritoneal dialysis patients: 14 years of prospective observation. Am J Kidney Dis. 2001;38:1351–1357. doi: 10.1053/ajkd.2001.29254. [DOI] [PubMed] [Google Scholar]
- 83.Guh JY, Chen HC, Chuang HY, et al. Risk factors and risk for mortality of mild hypoparathyroidism in hemodialysis patients. Am J Kidney Dis. 2002;39:1245–1254. doi: 10.1053/ajkd.2002.33398. [DOI] [PubMed] [Google Scholar]
- 84.Stevens LA, Djurdjev O, Cardew S, et al. Calcium, phosphate, and parathyroid hormone levels in combination and as a function of dialysis duration predict mortality: evidence for the complexity of the association between mineral metabolism and outcomes. J Am Soc Nephrol. 2004;15:770–779. doi: 10.1097/01.asn.0000113243.24155.2f. [DOI] [PubMed] [Google Scholar]
- 85.Block GA, Klassen PS, Lazarus JM, et al. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15:2208–2218. doi: 10.1097/01.ASN.0000133041.27682.A2. [DOI] [PubMed] [Google Scholar]
- 86.Atsumi K, Kushida K, Yamazaki K, et al. Risk factors for vertebral fractures in renal osteodystrophy. Am J Kidney Dis. 1999;33:287–293. doi: 10.1016/s0272-6386(99)70302-1. [DOI] [PubMed] [Google Scholar]
- 87.Danese MD, Kim J, Doan QV, et al. PTH and the risks for hip, vertebral, and pelvic fractures among patients on dialysis. Am J Kidney Dis. 2006;47:149–156. doi: 10.1053/j.ajkd.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 88.Coco M, Rush H. Increased incidence of hip fractures in dialysis patients with low serum parathyroid hormone. Am J Kidney Dis. 2000;36:1115–1121. doi: 10.1053/ajkd.2000.19812. [DOI] [PubMed] [Google Scholar]
- 89.Kuizon BD, Goodman WG, Juppner H, et al. Diminished linear growth during intermittent calcitriol therapy in children undergoing CCPD. Kidney Int. 1998;53:205–211. doi: 10.1046/j.1523-1755.1998.00724.x. [DOI] [PubMed] [Google Scholar]
- 90.Martin KJ, Gonzalez EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol. 2007;18:875–885. doi: 10.1681/ASN.2006070771. [DOI] [PubMed] [Google Scholar]
- 91.D’Haese PC, Spasovski GB, Sikole A, et al. A multicenter study on the effects of lanthanum carbonate (Fosrenol) and calcium carbonate on renal bone disease in dialysis patients. Kidney Int Suppl. 2003;63:S73–S78. doi: 10.1046/j.1523-1755.63.s85.18.x. [DOI] [PubMed] [Google Scholar]
- 92.Teng M, Wolf M, Ofsthun MN, et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16:1115–1125. doi: 10.1681/ASN.2004070573. [DOI] [PubMed] [Google Scholar]
- 93.Finch JL, Bergfeld M, Martin KJ, et al. The effects of discontinuation of aluminum exposure on aluminum-induced osteomalacia. Kidney Int. 1986;30:318–324. doi: 10.1038/ki.1986.187. [DOI] [PubMed] [Google Scholar]
- 94.Jablonski G, Klem KH, Danielsen CC, et al. Aluminium-induced bone disease in uremic rats: effect of deferoxamine. Biosci Rep. 1996;16:49–63. doi: 10.1007/BF01201001. [DOI] [PubMed] [Google Scholar]
- 95.McCarthy JT, Milliner DS, Johnson WJ. Clinical experience with desferrioxamine in dialysis patients with aluminium toxicity. Q J Med. 1990;74:257–276. [PubMed] [Google Scholar]
- 96.Molitoris BA, Alfrey AC, Alfrey PS, et al. Rapid removal of DFO-chelated aluminum during hemodialysis using polysulfone dialyzers. Kidney Int. 1988;34:98–101. doi: 10.1038/ki.1988.150. [DOI] [PubMed] [Google Scholar]
- 97.Andress DL, Ott SM, Maloney NA, et al. Effect of parathyroidectomy on bone aluminum accumulation in chronic renal failure. N Engl J Med. 1985;312:468–473. doi: 10.1056/NEJM198502213120803. [DOI] [PubMed] [Google Scholar]
- 98.Spasovski G, Gelev S, Masin-Spasovska J, et al. Improvement of bone and mineral parameters related to adynamic bone disease by diminishing dialysate calcium. Bone. 2007;41:698–703. doi: 10.1016/j.bone.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 99.Lezaic V, Pejanovic S, Kostic S, et al. Effects of lowering dialysate calcium concentration on mineral metabolism and parathyroid hormone secretion: a multicentric study. Ther Apher Dial. 2007;11:121–130. doi: 10.1111/j.1744-9987.2007.00419.x. [DOI] [PubMed] [Google Scholar]
- 100.Fujimori A, Yorifuji M, Sakai M, et al. Low-calcium dialysate improves mineral metabolism in hemodialysis patients. Clin Nephrol. 2007;67:20–24. doi: 10.5414/cnp67020. [DOI] [PubMed] [Google Scholar]
- 101.Chertow GM, Burke SK, Raggi P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–252. doi: 10.1046/j.1523-1755.2002.00434.x. [DOI] [PubMed] [Google Scholar]
- 102.Raggi P, James G, Burke SK, et al. Decrease in thoracic vertebral bone attenuation with calcium-based phosphate binders in hemodialysis. J Bone Miner Res. 2005;20:764–772. doi: 10.1359/JBMR.041221. [DOI] [PubMed] [Google Scholar]
- 103.Asmus HG, Braun J, Krause R, et al. Two year comparison of sevelamer and calcium carbonate effects on cardiovascular calcification and bone density. Nephrol Dial Transplant. 2005;20:1653–1661. doi: 10.1093/ndt/gfh894. [DOI] [PubMed] [Google Scholar]
- 104.Sanchez CP, Kuizon BD, Abdella PA, et al. Impaired growth, delayed ossification, and reduced osteoclastic activity in the growth plate of calcium-supplemented rats with renal failure. Endocrinology. 2000;141:1536–1544. doi: 10.1210/endo.141.4.7436. [DOI] [PubMed] [Google Scholar]
- 105.Balint E, Marshall CF, Sprague SM. Effect of the vitamin D analogues paricalcitol and calcitriol on bone mineral in vitro. Am J Kidney Dis. 2000;36:789–796. doi: 10.1053/ajkd.2000.17667. [DOI] [PubMed] [Google Scholar]
- 106.Monier-Faugere MC, Mawad H, Malluche HH. Opposite effects of calcitriol and paricalcitol on the parathyroid hormone-(1–84)/large carboxy-terminal-parathyroid hormone fragments ratio in patients with stage 5 chronic kidney disease. Clin J Am Soc Nephrol. 2007;2:1255–1260. doi: 10.2215/CJN.03461006. [DOI] [PubMed] [Google Scholar]
- 107.Winer KK, Ko CW, Reynolds JC, et al. Long-term treatment of hypoparathyroidism: a randomized controlled study comparing parathyroid hormone-(1–34) versus calcitriol and calcium. J Clin Endocrinol Metab. 2003;88:4214–4220. doi: 10.1210/jc.2002-021736. [DOI] [PubMed] [Google Scholar]
- 108.Silver J, Bushinsky D. Harnessing the parathyroids to create stronger bones. Curr Opin Nephrol Hypertens. 2004;13:471–476. doi: 10.1097/01.mnh.0000133984.47806.00. [DOI] [PubMed] [Google Scholar]
- 109.Goodman WG, Hladik GA, Turner SA, et al. The Calcimimetic agent AMG 073 lowers plasma parathyroid hormone levels in hemodialysis patients with secondary hyperparathyroidism. J Am Soc Nephrol. 2002;13:1017–1024. doi: 10.1681/ASN.V1341017. [DOI] [PubMed] [Google Scholar]
- 110.Ishii H, Wada M, Furuya Y, et al. Daily intermittent decreases in serum levels of parathyroid hormone have an anabolic-like action on the bones of uremic rats with low-turnover bone and osteomalacia. Bone. 2000;26:175–182. doi: 10.1016/s8756-3282(99)00263-x. [DOI] [PubMed] [Google Scholar]
- 111.Gowen M, Stroup GB, Dodds RA, et al. Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats. J Clin Invest. 2000;105:1595–1604. doi: 10.1172/JCI9038. [DOI] [PMC free article] [PubMed] [Google Scholar]