

Abstract
IgG4-related systemic disease, including autoimmune pancreatitis, is a multi-organ disorder characterized by elevated serum immunoglobulin G4 (IgG4) concentration and IgG4-positive plasma cell infiltration. We report the case of a 67-year-old man with IgG4-related tubulointerstitial nephritis, presenting with markedly enlarged kidneys and renal dysfunction. The serum IgG4 level was elevated with 4200 mg/dl and pathological examination revealed patchy, clearly fringed areas of IgG4-positive plasma cell infiltration and advanced fibrosis in the renal parenchyma, perirenal tissue and lymph nodes. With oral prednisolone at a dose of 60 mg daily, a contraction of the kidneys and an improvement of renal function were observed. No recurrence of the disease was observed during the reduction of prednisolone to 2 mg daily over 4 years.
Keywords: IgG4, renal dysfunction, retroperitoneal fibrosis, tubulointerstitial nephritis
Introduction
IgG4-related systemic disease, a recently proposed clinical entity that is characterized by high serum immunoglobulin G4 (IgG4) level and massive tissue infiltration of IgG4 positive plasma cells [1,2], has been shown to affect several organs. Autoimmune pancreatitis (AIP) is the best-documented cause of this clinical entity [1,3]. AIP has been reported to cause multiple extra-pancreatic lesions [4], sometimes with renal involvement [5–8]. Recently, nephropathies without AIP have also been reported [2,9].
Case report
In January 2004, a 67-year-old man with type 2 diabetes mellitus was referred to our department due to bulky tumour-like appearance of both kidneys detected by ultrasound. He had been found to have an asymptomatic polyclonal hypergammaglobulinaemia 7 years earlier. CT scan demonstrated markedly enlarged kidneys with irregular contrast staining, retroperitoneal fibrosis and cervical, mediastinal and paraaortic lymph node swelling (Figure 1). There was no morphological abnormality in the pancreas. A subsequent MRI confirmed the CT findings. Cervical lymph node and open renal biopsies were performed.
Fig. 1.
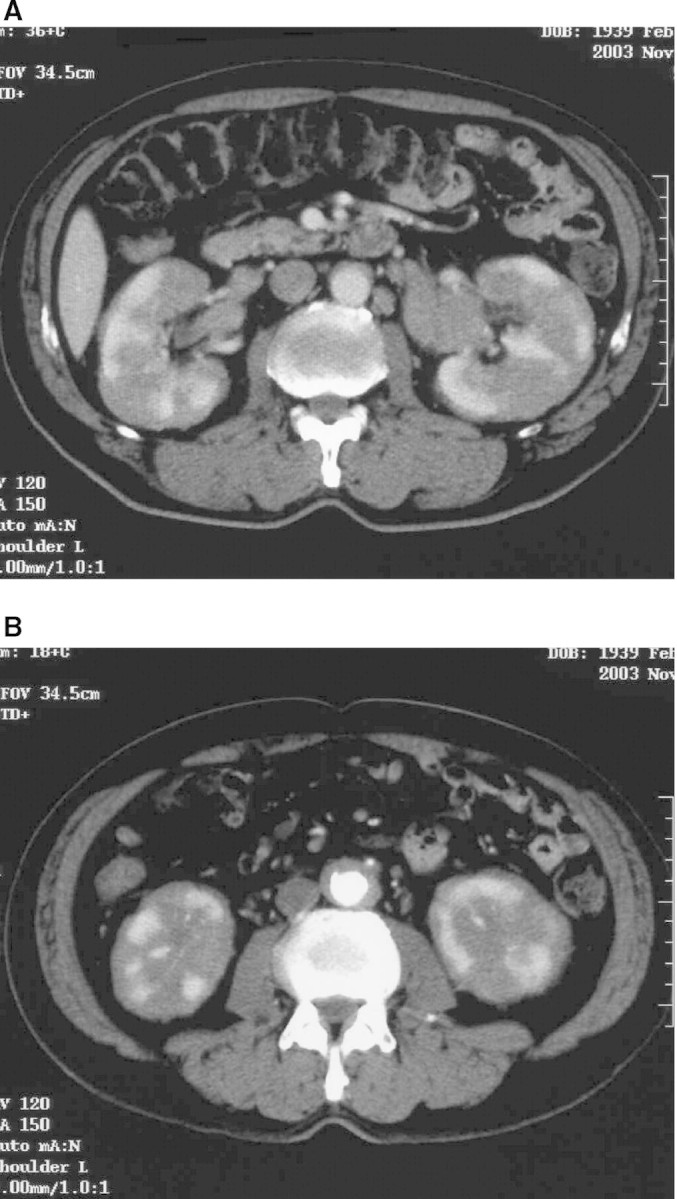
CT imaging at referral. (A) Both kidneys were markedly enlarged and showed irregular contrast staining. The pancreas was normal morphologically. (B) A paraaortic low-density area was considered to be retroperitoneal fibrosis.
In the renal tissue, light microscopic examination showed massive infiltration of lymphocytes and plasma cells with extensive fibrosis and tubular atrophy in the interstitium (Figure 2). Interestingly, the cellular infiltration was observed in a patchy form, and the affected area and normal parenchyma were clearly separated. Glomeruli were almost normal. There were no findings of diabetic nephropathy. Marked lymphoplasmacytic infiltration with extensive fibrosis was also observed in the lymph node and the perirenal tissue. The infiltrated cells showed no dysplastic changes, and Castleman's disease was clinically excluded.
Fig. 2.
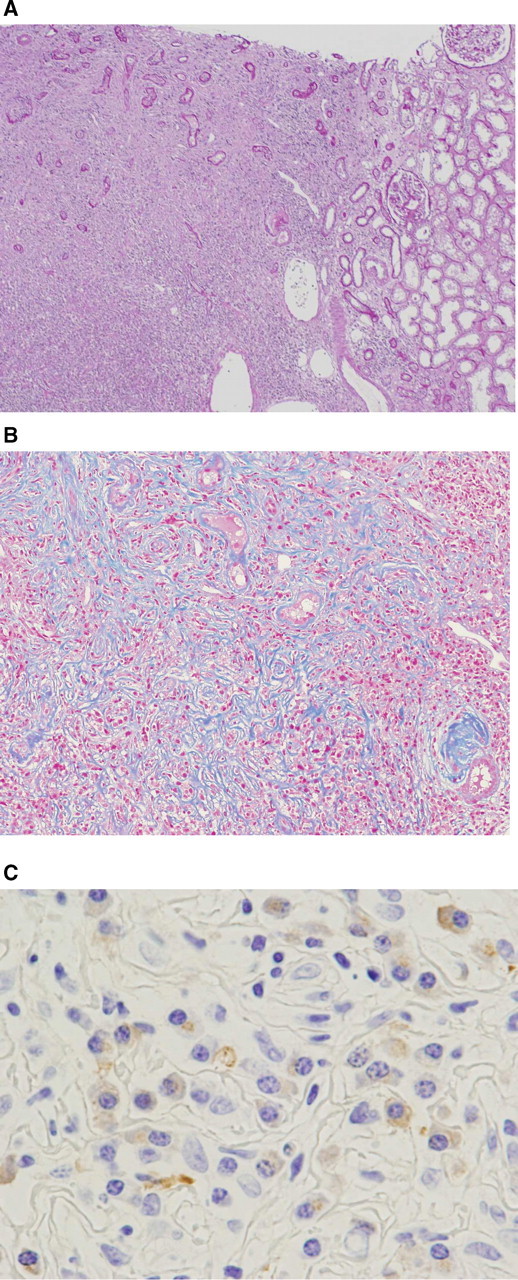
Microscopic findings of renal biopsy. (A) The affected area had a patchy distribution and was clearly separated from normal renal parenchyma (periodic acid Schiff staining, original magnification × 40). (B) Extensive fibrosis with marked tubular atrophy and lymphoplasmacytic infiltration (Masson Trichrome staining, original magnification × 200). (C) IgG4 positive plasma cell infiltration (immunohistochemistry, original magnification × 1200).
At first, we did not initiate therapy, because the serum creatinine concentration (Cr) at referral was 1.3 mg/dl and remained stable for several months. However, renal function deteriorated gradually from Cr 1.3 mg/dl to 2.1 mg/dl over 6 months and he was re-admitted to our department. Urinary testing on admission showed only slight proteinuria without haematuria. Blood tests revealed elevated serum total protein of 11.2 g/dl and decreased albumin of 3.2 g/dl. HbA1c was 5.6%. Serum C3, C4 and total serum haemolytic activity (CH50) were decreased to 43 mg/dl, 2 mg/dl and 12 U/dl, respectively. ANA was positive with a titre of 1:40. Hypergammaglobulinaemia persisted with the elevated serum IgG level of 7380 mg/dl (IgG4 of 3160 mg/dl), IgA 145 mg/dl and IgM 72 mg/dl. With these pathological and serological findings, IgG4-related systemic disease was suspected, although there was no immunohistochemical confirmation. We therefore started prednisolone at a dose of 60 mg daily. This therapy effectively improved renal function with creatinine falling to 1.1 mg/dl, reduced the renal size, lymphadenopathy, retroperitoneal fibrosis and lowered the serum IgG level to the normal range. Prednisolone was gradually tapered to 2 mg daily over 4 years and no recurrence was observed during the observation period.
Subsequent immunohistochemical examination revealed abundant IgG4-positive plasma cells in the renal, perirenal and lymph tissue (Figure 2), which confirmed the diagnosis of an IgG4-related systemic disease.
Discussion
IgG4-related systemic disease is characterized by (1) elevated serum IgG4 concentration, (2) massive infiltration of IgG4-positive plasma cells and extensive fibrosis in affected organs and (3) favourable response to corticosteroid therapy [1,2]. As the nomenclature is not established, this disorder is also called IgG4-related disease, IgG4-related sclerosing disease, IgG4-related plasmocytic disease, autoimmune multi-organ lymphoproliferative syndrome or IgG4 positive plasma cell disease [2]. Recently, a new clinical entity of IgG4-positive multi-organ lymphoproliferative syndrome (IgG4+MOLPS) has been proposed [10]. The most well-documented form of this disorder is AIP [1–3], also called sclerosing pancreatitis. AIP was associated with several extra-pancreatic lesions, such as sclerosing cholangitis, retroperitoneal fibrosis, sclerosing sialadenitis, swelling of lacrimal glands and lymphadenopathy [4]. Recently, renal involvement of IgG4-related systemic disease was reported with or without AIP [2,5–9]. Mikulicz's disease, a sclerosing sialadenitis, has also been considered to be involved in this entity [9,10], and a case of Mikulicz's disease complicated by tubulointerstitial nephritis with IgG4-positive plasma cell infiltration has been reported [9].
Renal lesions in IgG4-related sclerosing disease exhibit several patterns. Two cases of tubulointerstitial nephritis with AIP were reported in 2004 [5,6]. On the other hand, tumour-like focal masses have been described [2,7]. Moreover, retroperitoneal fibrosis with hydronephrosis has also been considered as another renal manifestation in this disorder [2]. In microscopic examinations, the affected renal area has demonstrated focal lymphoplasmacytic sclerosing lesions with IgG4-positive plasma cells, similar to AIP [2,8], and often clearly separated from the normal parenchyma. This characteristic tendency of focal demarcation is observed at a microscopic level, even when the macroscopic examination shows the diffuse distribution [2,8]. Indeed, in the present case, the renal lesion was first recognized as tumour-like on ultrasound. Later, bulky enlargement of bilateral kidneys was observed by CT and MRI. Histologically, a lesion demonstrating patchy distribution and a clear demarcated area of infiltration with IgG4-positive plasma cells was observed. These findings suggest that focal multiple lymphoplasmacytic infiltrations might either unite or expand, and finally form a diffuse or tumour-like lesions during the progression of the disease. In addition, the understanding of these characteristics is helpful for the correct diagnosis of IgG4-related systemic disease and unnecessary nephrectomy can be avoided. Indeed, AIP is often misdiagnosed as pancreatic carcinoma, and one previous case has been reported of the renal lesion with AIP mimicking renal metastasis of pancreas carcinoma [7].
It has been reported that corticosteroid therapy is effective in IgG4-related systemic disease [1,2]. In treating the nephropathy, oral prednisolone at 40–60 mg daily has often been used as the initial therapy and achieved favourable responses [7–9]. In the present case, prednisolone was also effective, as in previous reports, and marked improvements of renal function and morphology were observed. There is no consensus as to the maintenance dose of steroid or long-term prognosis, but our case showed no recurrence of the disease with a staged reduction of prednisolone, from 60 mg to 2 mg daily over 4 years.
In summary, we describe a case of renal involvement in IgG4-related systemic disease with lymphadenopathy and retroperitoneal fibrosis, but without AIP. Patchy, clearly demarcated lymphoplasmacytic infiltrations with extensive fibrosis were observed on microscopic examination, although the radiographic imaging showed bulky enlargement of both kidneys. This case may be helpful in the diagnosis and the understanding of the pathophysiology of IgG4-related systemic disease.
Acknowledgments
We are grateful for the technical support in the immunohistochemical staining of IgG4 by Mr Shigeru Horita in the laboratory of pathology, Tokyo Women's Medical University.
Conflict of interest statement. None declared.
References
- 1.Kamisawa T. IgG4-related systemic disease. Intern Med. 2006;45:125–126. doi: 10.2169/internalmedicine.45.0137. [DOI] [PubMed] [Google Scholar]
- 2.Saeki T, Nishi S, Ito T, et al. Renal lesions in IgG4-related systemic disease. Intern Med. 2007;46:1365–1371. doi: 10.2169/internalmedicine.46.0183. [DOI] [PubMed] [Google Scholar]
- 3.Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 4.Kamisawa T, Egawa N, Nakajima H, et al. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol. 2005;39:904–907. doi: 10.1097/01.mcg.0000180629.77066.6c. [DOI] [PubMed] [Google Scholar]
- 5.Takeda S, Haratake J, Kasai T, et al. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant. 2004;19:474–476. doi: 10.1093/ndt/gfg477. [DOI] [PubMed] [Google Scholar]
- 6.Uchiyama-Tanaka Y, Mori Y, Kimura T, et al. Acute tubulointerstitial nephritis associated with autoimmune-related pancreatitis. Am J Kidney Dis. 2004;43:18–25. doi: 10.1053/j.ajkd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Rudmik L, Tropkox K, Nash C, et al. Autoimmune pancreatitis associated with renal lesions mimicking metastatic tumors. Can Med Assoc J. 2006;175:367–369. doi: 10.1503/cmaj.051668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson SJW, Jenkins DAS, Bellamy COS. Nephropathy in IgG4-related systemic disease. Am J Surg Pathol. 2006;30:1472–1477. doi: 10.1097/01.pas.0000213308.43929.97. [DOI] [PubMed] [Google Scholar]
- 9.Saeki T, Saito A, Yamazaki H, et al. Tubulointerstitial nephritis associated with IgG4-related systemic disease. Clin Exp Nephrol. 2007;11:168–173. doi: 10.1007/s10157-007-0464-9. [DOI] [PubMed] [Google Scholar]
- 10.Masaki Y, Dong L, Kurose N, et al. Proposal for a new clinical entity, IgG4-positive multi-organ lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis. 2008 doi: 10.1136/ard.2008.089169. doi: 10.1136/ard.2008.089169. [DOI] [PubMed] [Google Scholar]