

Abstract
Background
RNA secondary structure prediction methods based on probabilistic modeling can be developed using stochastic context-free grammars (SCFGs). Such methods can readily combine different sources of information that can be expressed probabilistically, such as an evolutionary model of comparative RNA sequence analysis and a biophysical model of structure plausibility. However, the number of free parameters in an integrated model for consensus RNA structure prediction can become untenable if the underlying SCFG design is too complex. Thus a key question is, what small, simple SCFG designs perform best for RNA secondary structure prediction?
Results
Nine different small SCFGs were implemented to explore the tradeoffs between model complexity and prediction accuracy. Each model was tested for single sequence structure prediction accuracy on a benchmark set of RNA secondary structures.
Conclusions
Four SCFG designs had prediction accuracies near the performance of current energy minimization programs. One of these designs, introduced by Knudsen and Hein in their PFOLD algorithm, has only 21 free parameters and is significantly simpler than the others.
Background
Many RNAs conserve a base-paired secondary structure that is important to their function [1,2]. Accurate RNA secondary structure predictions help in understanding an RNA's function, in identifying novel functional RNAs in genome sequences, and in recognizing evolutionarily related RNAs in other organisms. Most RNA secondary structure prediction algorithms are based on energy minimization [2-7]. Alternatively, probabilistic modeling approaches using stochastic context-free grammars (SCFGs) can be used [8-10]. A potential advantage of a probabilistic modeling approach is that it is more readily extended to include other sources of statistical information that constrain a structure prediction.
For example, an outstanding problem is consensus RNA secondary structure prediction for a small number of structurally homologous RNA sequences. Comparative sequence analysis is probably the most powerful source of information for RNA structure prediction [11-14]. Homologous RNAs tend to conserve a common base-paired secondary structure, and conserved base-pairing interactions are revealed by compensatory mutations in multiple RNA sequence alignments [12,15-19]. Comparative sequence analysis is extremely reliable, and has produced strikingly accurate RNA structure predictions [14,20], but one is usually not blessed with the large number of sequences (nor the time and human expertise) that a purely comparative approach requires. There is a need for automated approaches that combine evolutionary information from comparative sequence analysis with biophysical knowledge of what structures are most plausible.
However, it is not clear how best to combine probabilistic evolutionary information with the thermodynamic parameters of the standard energy minimization model into a meaningful, mathematically defensible objective function. Clearly one can use the Gibbs-Boltzmann equation to convert an overall ΔG for a structure into a probability of that structure in an ensemble of all possible structures [21,22], but it is not possible to interpret individual energy parameters in the thermodynamic model as log probabilities. Consequently, nearly all consensus RNA structure prediction methods that have been introduced optimize an ad hoc weighted combination of thermodynamic parameters and comparative sequence analysis terms, either for consensus structure prediction from predetermined multiple RNA sequence alignments [18,23-26], or for the harder problem of simultaneous folding and alignment of initially unaligned RNAs [27-34]. A notable exception is the approach described by Knudsen and Hein, who developed a full probabilistic model that combines an explicit stochastic evolutionary model with an SCFG-based probabilistic model of structure plausibility, so they can find a consensus structure that optimizes a joint probability of that structure and multiple aligned homologous sequences [18,26].
In addition to the Knudsen and Hein approach, at least three other SCFG-based approaches to RNA secondary structure prediction have been described. These include an SCFG-based mirror of the standard Zuker algorithm for single-sequence structure prediction [35], and two "pair-SCFG" approaches for simultaneous folding and alignment of two homologous RNAs [31,36]. All four papers use different underlying SCFG designs. No group appears to have explored different possible SCFG designs before settling on the one they used. Only Knudsen and Hein reported any benchmark results for the accuracy of their secondary structure predictions [18,26]. It is not known how different designs affect the accuracy of SCFG-based secondary structure prediction. Flexibility in model design comes from the fact that SCFG probability parameter estimation can be done by counting frequencies in databases of trusted RNA secondary structures, so it is easy to parameterize different models that vary in complexity and capture different features of RNA structure. In contrast, energy minimization algorithms are based on a standard set of thermodynamic parameters, most of which are determined experimentally [2,7], so it would take substantial effort to develop a radically new thermodynamic model.
Design decisions are likely to be particularly important in consensus structure prediction applications, because a natural trade-off arises. A complex RNA folding SCFG might predict structures for single sequences better than a simpler model, but extending a complex RNA folding SCFG to deal with multiple evolutionarily correlated sequences can easily result in a combinatorial explosion of parameters, making the model impractical. One wants to build consensus prediction models on top of small, simple (i.e. "lightweight") SCFG designs that sacrifice as little RNA structure prediction accuracy as possible, relative to state-of-the-art energy minimization approaches.
Here we explore the impact of different SCFG designs on single-sequence RNA secondary structure prediction accuracy. Our goal is to identify lightweight SCFG model designs that can serve as cores underlying more complex integrated approaches. We have implemented nine different lightweight SCFGs, estimated their parameters from rRNA structure data, evaluated their prediction accuracy on a benchmark of trusted RNA structures, and compared these results to the accuracy of energy minimization methods.
Algorithms
Dynamic programming algorithms for non-pseudoknotted RNA secondary structure prediction work by calculating scores for optimal foldings for all subsequences xi...xj, starting with subsequences of zero length and working outwards recursively on increasingly longer sequences [2]. For example, an example of an RNA folding algorithm [3] is:
Initialization:
γ(i, i - 1) = 0
γ(i, i) = 0
Iteration:
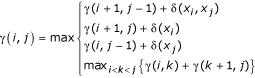
δ(xi) and δ(xj) are scores for single-stranded nucleotides, and δ(xi, xj) are scores for base pairs. For a sequence of length L, the score calculation terminates when γ(1, L) is calculated, the score of the best structure on the complete sequence. The optimal structure itself is retrieved by a traceback of the dynamic programming matrix.
When δ(xi, xj) = 1 for base pairs and all other scores are zero, this algorithm finds the structure that maximizes the number of base pairs with the final score γ(1, L) as the number of base pairs in the optimal structure [3]. In the more complicated loop-dependent algorithms (e.g. the Zuker algorithm), the algorithm is fundamentally the same (though with more terms, more matrices, and minimization instead of maximization); but the scores are energy parameters, and the final score is ΔG, the calculated thermodynamic stability of the optimal structure [2].
In a probabilistic approach, the algorithm again remains fundamentally the same, but the parameters are log probabilities (we will call the parameter set Θ) and the final score is the log probability of the optimal structure ( ) and the sequence (x) given the model parameters, log P(, x|Θ). This is the quantity we need for statistical inference approaches [37,38]. Probabilities must sum to one, so we have to be sure that for our model,
) and the sequence (x) given the model parameters, log P(, x|Θ). This is the quantity we need for statistical inference approaches [37,38]. Probabilities must sum to one, so we have to be sure that for our model, 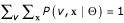 over all possible sequences x and all possible structures v for those sequences. But there are an infinite number of possible sequences and a combinatorial explosion of possible structures, so we can't enumerate all these possibilities; we need a formal system to be confident that we can form a correct probabilistic model.
over all possible sequences x and all possible structures v for those sequences. But there are an infinite number of possible sequences and a combinatorial explosion of possible structures, so we can't enumerate all these possibilities; we need a formal system to be confident that we can form a correct probabilistic model.
Stochastic context-free grammars (SCFGs) are that formal system. SCFGs are probabilistic models capable of capturing the long range, nested, pairwise correlations, such as those induced by base pairing in non-pseudoknotted RNA secondary structures [8-10]. Here we give a somewhat informal introduction to SCFGs, specifically as they apply to RNA folding, starting with (nonstochastic) context-free grammars. For a review of the use of SCFGs for RNA folding, see [10]. For more formal descriptions of CFGs, see [39-41].
Context-free grammars
A context-free grammar 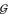 can be defined by = (V, T, P, S) where:
can be defined by = (V, T, P, S) where:
• V is a finite set of nonterminal symbols ("states"),
• T is a finite set of terminal symbols (for RNA: {a, c, g, u}),
• P is a finite set of production rules (described below), and
• S is the initial (start) nonterminal (S ∈ V).
Production rules describe how the grammar generates an observed symbol string in steps, starting from the initial start nonterminal S. Production rules take the form A → α where A ∈ V and α is any combination of nonterminals T and/or terminals V. By convention, capital letters denote nonterminal symbols and lower-case letters are terminals. S → gSc is an example of a CFG production rule; it generates a 'g' and a 'c' terminal symbol in one correlated step. The ability to generate or score two or more correlated symbols in a single step is what gives CFGs the power to deal with base pairing.
At each step of a production from the grammar, one has a current string of terminals and/or nonterminals; one chooses a nonterminal, and transforms that nonterminal into a new substring using a valid production rule. This process starts with the initial string S, iterates until one arrives at a string consisting solely of terminal symbols.
For example, consider a "palindrome grammar" V = {S}, T = {a, b}, and P = {S → aSa, S → bSb, S → ε}, where ε is a null string used as an ending production. This little grammar generates only strings consisting of palindromes of a and b symbols. RNA base pairing is essentially palindromic, except pairings are complements rather than identities. An example production is S ⇒ aSa ⇒ abSba ⇒ abbSbba ⇒ abbbba, resulting in the string abbbba. Note that the grammar produces this string in an nested fashion (as opposed to a left-to-right fashion, the way simpler string alignment algorithms work), and that this CFG efficiently describes the set of all palindromes over the alphabet {a, b}.
Now consider an example CFG that generates RNA structures: V = {S}, T = {a, c, g, u}, and P =

where '|' represents 'or' between production rules. The productions can be rewritten in a shorthand as:
S → aS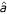 | aS | Sa | SS | ε
| aS | Sa | SS | ε
where we are now using a generically to represent any single terminal symbol in T, and the rule S → aS implies a basepairs with . (We could also allow GU pairs here. In SCFGs, below, we will have probability scores for all 16 base pairs including noncanonical ones, or in the case of grammars that take base-pair stacking into account, the full 16 × 16 matrix of possibilities.)
A CFG derivation has an elegant representation known as a parse tree, π. Figure 1 shows an example RNA structure and two example parse trees for the RNA CFG. Given appropriate production rules, a parse tree has a natural correspondence with an RNA secondary structure.
Figure 1.

Examples of CFG parse trees for an example RNA structure Left: an RNA secondary structure with two stems. Middle: a parse tree for that structure using the grammar S → aS | aS | Sa | SS | ε, with nonterminals in red and terminals in black. Note the correspondence between the RNA structure and the structure of the parse tree; that the individual steps in the grammar correspond to base pairs and single nucleotides (that is, the grammar is used to factor the structure down into individually scored steps); and that the RNA sequence can be read off the parse tree by following the margin of the tree counterclockwise. Right: an alternative parse tree for the same structure, demonstrating that this grammar is structurally ambiguous.
Nonstochastic CFGs are used in pattern search applications, where one represents an RNA structural consensus as a CFG and ask if a particular sequence matches or doesn't match that query. They are not useful for structure prediction. For the CFG above, for example, for any RNA sequence there will be a huge number of valid parse trees, each of which corresponds to a possible RNA secondary structure. However, our problem in structure prediction is not to determine whether an RNA sequence has at least one possible structure. Given a sequence, we want to score and rank the possible parse trees for that sequence to infer the optimal one. To score and rank parse trees, we need to use stochastic context free grammars. In addition, we need efficient algorithms for finding the optimal SCFG parse tree for a given sequence.
Stochastic context-free grammars
In an SCFG , we associate a probability with each CFG production rule. The probabilities of the set of productions from any given nonterminal must sum to one. We refer to the full set of probabilities (the parameters of a model) as Θ. The probability P(x, π|, Θ) is the product of all the probabilities of the production rules used in a parse tree π for sequence x. An SCFG is a probabilistic model that describes a joint probability distribution P(x, π|, Θ) over all RNA sequences x and all possible parse trees π.
Given a parameterized SCFG (, Θ) and a sequence x, the Cocke-Younger-Kasami (CYK) dynamic programming algorithm finds an optimal (maximum probability) parse tree 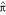 for a sequence x,
for a sequence x,
= argmaxπ P(π, x|, Θ).
For notational and formal reasons, the CYK algorithm is usually described for SCFGs in a special "Chomsky normal form" with only two kinds of production rules, A → AA and A → a; any SCFG can provably be converted to a set of production rules of this form [10]. But in applications, it is convenient to avoid this conversion and express a CYK algorithm directly in terms of the grammar's own production rules. For an RNA SCFG based on the example grammar in the previous section, the CYK algorithm is:
Initialization:
γ(i, i - 1) = log p(S → ε)
Iteration:
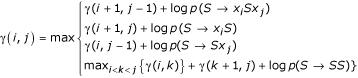
When the algorithm terminates, γ(1, L) is logP(x, |, Θ), the log probability of the most probable parse tree for the sequence x given the grammar and parameters Θ. A traceback recovers this optimal parse tree.
The near-exact correspondence between the CYK algorithm and standard dynamic programming algorithms for RNA folding should be clear. SCFG algorithms are essentially the same as existing RNA folding algorithms, but the scoring system is probabilistic, based on factoring the score for a structure down into a sum of log probability terms, rather than factoring the structure into a sum of energy terms or arbitrary base-pair scores. The thermodynamic scoring parameters for energy minimization are largely derived by experimental melting studies of small model structures [7]; in contrast, SCFG log probability parameters are derived from frequencies observed in training sets of known RNA secondary structures. That is, instead of scoring a G-C pair stacked on a C-G base pair by adding a term for the free energy contribution of the GC/CG stack, an SCFG would add a log probability that GC/CG stacks are observed in known RNA structures.
A related SCFG algorithm, the Inside algorithm, is used to obtain the total probability of the sequence given the model summed over all parse trees,
![]()
The Inside algorithm replaces the max operations in CYK with sums and additions of terms with multiplications. It is analogous to the McCaskill algorithm for calculating partition functions using the thermodynamic model of RNA folding [10,21]. Suboptimal parse trees can be sampled from their posterior probability distribution by a probabilistic traceback of the Inside matrix, analogous to techniques used for energy minimization algorithms [10,22,42].
Grammar ambiguity
A grammar is said to be ambiguous when there is more than one possible parse tree for some sequence [39-41]. Any SCFG that will be useful for RNA secondary structure prediction must be ambiguous in this sense. We are interested in looking at all possible base paired structures for the sequence, represented as a set of possible parse trees, and finding the optimal (highest probability) one.
A more subtle form of grammar ambiguity concerns us here. The CYK algorithm finds the mathematically optimal parse tree ; we want the optimal secondary structure . The optimal parse tree gives us the optimal structure if and only if there is a one to one correspondence between parse trees and secondary structures. However, a given secondary structure does not necessarily have a unique parse tree. For instance, consider the two possible parse trees for the example in Figure 1, both of which express the same set of base pairs but use different series of production rules. (We consider two structures to be identical if they have the same set of base pairs.) When multiple valid parse trees describe the same secondary structure, we call the grammar structurally ambiguous. If a grammar is structurally ambiguous, then we cannot equate the probability of a parse tree with the probability of its structure [43]. The probability of a structure is a sum over the probabilities of all parse trees consistent with that structure. This summation is not reconcilable with the CYK algorithm; an optimal structure cannot be calculated efficiently if we need to do the summation over multiple possible parse trees for each structure. Thus, we will either have to use grammars that are structurally unambiguous, or we will have to assume that it is a valid approximation to assume an optimal parse tree gives us the optimal structure. We explore this issue in the results.
It can be tricky to write grammars that are structurally unambiguous, and it appears to be difficult to prove that they are so, except in simple cases. Determining grammar ambiguity in the usual formal language sense is undecidable [40,41]. We can, however, test empirically whether a particular grammar is unambiguous for a given sequence x and structure v. We do this with a conditional Inside algorithm that calculates:
![]()
by limiting the Inside calculation to  (v), those parse trees consistent with the given structure v. For example, the conditional Inside algorithm for the RNA grammar described above is:
(v), those parse trees consistent with the given structure v. For example, the conditional Inside algorithm for the RNA grammar described above is:
Initialization:
γ(i, i - 1) = p(S → ε)
Iteration:
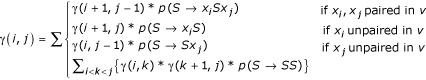
If P(x, |, Θ) calculated by CYK equals P(x, v|, Θ) calculated by conditional Inside then there is only one parse tree, namely , of nonzero probability for the structure v. Operationally, we consider a grammar to be structurally unambiguous if this condition holds for a large sample of different sequences.
An interesting difference between the thermodynamic and probabilistic approaches with respect to ambiguity is worth noting. The thermodynamic scoring scheme is not normalized, so structural ambiguity is not an issue for finding optimal structures; regardless of how many different ways there are of scoring the energy of a structure, the lowest energy structure still wins. However, ambiguity becomes a painstaking issue for calculating the equilibrium partition function [21], where one must be careful not to count any structure more than once. For SCFG-based methods, with normalized probabilities as scores, exactly the opposite is the case. Ambiguity is an issue for optimal structure prediction, but the summed Inside calculation (the analog of the summed partition function calculation) gives the correct result even for ambiguous grammars.
SCFG designs
The RNA SCFG shown above factors a secondary structure into scoring terms for each individual base pair and each individual unpaired residue. In this paper we will examine four additional grammars of this type. However, state of the art thermodynamic models use a loop-dependent thermodynamic model that factors a structure in a more complex way, into nearest-neighbor base stacking terms (as opposed to individual base pairs) and tables of penalties for different lengths of different kinds of loops (bulge, interior, hairpin, and multifurcation). SCFG methods can also capture more sophisticated folding features.
Base pair stacking is a first order Markov dependence. It can be modeled in a grammar by capturing a limited amount of context dependency, a technique called lexicalization in natural language processing. One uses production rules of the type, 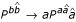 , where the probability of emitting a new pair a, is dependent on the previous base pair b,
, where the probability of emitting a new pair a, is dependent on the previous base pair b, 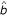 . The notation
. The notation 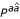 indicates that the P nonterminal will "remember" that it just generated an a, pair. Formally, in the production rules, this means 16 nonterminals, one for each pair of nucleotides, each one of which can generate 16 possible pairs: thus, we have the desired 16 × 16 table for base stacking parameters in terms of our production rules. However, lexicalized nonterminals do not incur any extra storage nor additional computational cost when parsing, because the lexicalization depends completely on the local sequence context; the 16 nonterminals can just be treated as one "meta" P nonterminal, with scores conditioned on the local context around the base pair xi, xj.
indicates that the P nonterminal will "remember" that it just generated an a, pair. Formally, in the production rules, this means 16 nonterminals, one for each pair of nucleotides, each one of which can generate 16 possible pairs: thus, we have the desired 16 × 16 table for base stacking parameters in terms of our production rules. However, lexicalized nonterminals do not incur any extra storage nor additional computational cost when parsing, because the lexicalization depends completely on the local sequence context; the 16 nonterminals can just be treated as one "meta" P nonterminal, with scores conditioned on the local context around the base pair xi, xj.
The simplest (and most common) model of loop lengths in a grammar uses recursive rules like S → aS, which imply a geometric length distribution. Explicit loop length distributions can be modeled by enumerating a set of production rules, one for each possible length. A set of such loop length probabilities is no different in effect than the lookup table of loop length penalties in the thermodynamic parameters.
Base stacking and explicit loop lengths are the most important two features of SCFGs that would closely mirror the current thermodynamic model. Most other desirable features, such as parameterizing stacked terminal mismatches for hairpin and internal loops, dangled bases, and special stable tetraloops, can also be dealt with using techniques like the above. Coaxial stacking [44,45] can also be accommodated [46], but adds to complexity. In this paper, four grammars model base stacking, but we do not explore the use of explicit loop lengths or other more complex features included in the thermodynamic model.
Parameterization
The parameters of each SCFG were estimated from frequencies observed in annotated secondary structures. The training data were large and small subunit rRNAs, obtained from the European Ribosomal Database [47,48]. Sequences containing more than 5% ambiguous bases and with less than 40% base pairing are discarded. The resulting data set was then filtered to remove sequences with greater than 80% identity. The final training set contains randomly chosen equal numbers of LSU and SSU sequences from the filtered data, totaling 278 sequences, 586,293 nucleotides, and 146,759 base pairs.
For a given grammar, a parse tree for each structure is determined from the secondary structure annotation, and the number of occurrences of each production type is counted. Production probabilities are then estimated from these counts using a Laplace (plus-one) prior [10].
When determining a parse tree for an ambiguous grammar, one possible parse tree was arbitrarily chosen. A more sophisticated approach is possible (for example, expectation maximization using a conditional Inside/Outside algorithm, to estimate the expected number of occurrences of productions in all possible parse trees given a structure), but we did not explore this.
We separated each production rule into a product of a transition probability (to go from one nonterminal to the new one) and an emission probability (for generating zero or more terminal symbols). This is the standard treatment in hidden Markov models.
Testing
A benchmark data set was built from the Ribonuclease P database [49], the Signal Recognition Particle database [50] and the tmRNA database [51]. Each of these databases provides high quality individual sequence structure information and curated structural alignments in a readily parsable form. Each structural alignment was filtered to remove sequences with greater than 80% identity. The structure for each member of the filtered families were then retrieved from the databases. Any sequence containing ambiguous bases was removed. The resulting test set contains 403 total sequences, consisting of 225 RNase P's, 81 SRP's, and 97 tmRNA's.
We count the number of base pairs that are correctly predicted, given a predicted structure and the trusted benchmark structure. That is, if we predict a base pair i, j, it is correct if and only if base pair i, j is present in the trusted structure. No helix offsetting/sliding is allowed, so our accuracy percentages are systematically lower than other published benchmarks that count base pairs as correct if they are within one base of a trusted base pair.
Predicted base pairs that are in the trusted structure are true positives (TP); predicted base-pairs not in the trusted structure are false positives (FP); base-pairs in the trusted structure but not predicted by the algorithm are false negatives (FN).
Sensitivity is the fraction of base pairs in the trusted structures that are predicted correctly: TP / (TP + FN). Positive predictive value (PPV; often called specificity) is the number of predicted base pairs that are in the trusted structure: TP / (TP + FP).
Rfam v5.0 [52] was utilized to build a second, larger test set for grammar ambiguity experiments, from a wide variety of different RNA structures. This set was constructed by filtering the seed alignments at 80% identity and then imposing the family's consensus structure onto each sequence to infer individual secondary structures. Any sequence containing ambiguous bases was removed. The resulting dataset contains 2455 sequences from 174 different RNA families.
Implementations
Training (parameter estimation), CYK parsing (structure prediction), and conditional Inside (ambiguity testing) code was written for each of the nine grammars. Inside with stochastic traceback (suboptimal sampling of parse trees) was implemented for the ambiguous grammars. The ANSI C source code for these implementations is freely available under the GNU General Public License (GPL) from http://www.genetics.wustl.edu/eddy/publications/#DowellEddy04. The data sets (training, testing, and benchmark) are freely available from the same URL.
For benchmarking experiments, we used mfold v3.1.2, Pfold (Oct 2003), PKNOTS v 1.01, RNAstructure v4.0, and the Vienna RNA package vl.4. The mfold software was obtained from Michael Zuker at http://www.bioinfo.rpi.edu/~zukerm/rna/mfold-3.0.html. An early release of RNAstructure v4.0 [53] was graciously provided by Dave Mathews. Both mfold and RNAstructure was run with MAX = 750 P = 20 W = 0 and efn2 rearrangement as recommended by [7]. The Vienna RNA package was obtained at http://www.tbi.univie.ac.at/~ivo/RNA/ and was run with default parameters. Bjarne Knudsen provided Pfold executables and guidance on how to run the package (personal communication). The PKNOTS program was run with default parameters, which does not attempt to predict pseudoknots. All benchmarks were conducted on Intel-based servers running a GNU/Linux operating system, with the exception of RNAstructure which was run in batch mode under Windows XP Pro 2002.
For all the grammars in this paper, the CYK and Inside algorithms are O(ML2) and O(ML3) in memory and time respectively, for M nonterminals in the grammar and a sequence of length L.
Results and discussion
Two ambiguous grammars
We first implemented two structurally ambiguous grammars:
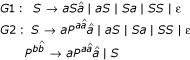
G1 is the simple grammar we used as an example in Methods. G2 extends it to include base pair stacking parameters.
It seemed possible that structural ambiguity might be only a formal concern. This would be the case if, in a set of possible parse trees for any particular structure, one parse tree has a probability that dominates the others, and approximates the summed probability of the ensemble. For example, many of the alternative parse trees are pathological cases that invoke fruitless cycles of bifurcation and destruction; any S ⇒ ε derivation in a parse tree could also be S ⇒ SS ⇒ εS ⇒ εε, but these more elaborate trees have probabilities that asymptote towards zero. Additionally, our parameterization of ambiguous grammars will tend to be biased towards concentrating probability in a single possible parse tree, because we nonrandomly choose only one possible parse tree for each training structure, favoring smaller, more sensible parse trees where ambiguous choices were possible. And finally, if all structures have about the same amount of probability diffusion to ambiguous parses, the rank order of the best parse trees might still reflect the rank order of the best structures, so that a CYK algorithm that finds the best parse tree would still recover the best structure.
To test whether structural ambiguity made a practical impact on structure prediction, we performed the following "reordering" experiment. For a given RNA sequence, we use CYK to find the optimal parse tree . We sample N more suboptimal parse trees from the posterior distribution (using Inside with stochastic traceback). We rank the N + 1 trees by their probabilities P(x, π|, Θ), where the optimal CYK parse is ranked first by definition. Then for each parse tree π, we get the base-paired structure v and use conditional Inside to calculate the P(x, v|, Θ) summed over all parse trees for that structure. We then ask two questions: first, if the rank order of the N + 1 parse trees is the same as the rank order of their N + 1 structures; second, if the structure of the optimal CYK parse tree becomes suboptimal in the ranked list of structure probabilities P(x, v|, Θ). If ambiguity does not matter in practice, we should not see many differences in rank order, and we hope to never see the top-ranked structure differ from the structure implied by the top-ranked CYK parse. We did the reordering experiment on all 2455 sequences in the Rfam5-based test set, for N varying from 10 to 3000 suboptimal parse trees.
The results showed that even in a small number of samples, the rank order of the parse tree probabilities and the structural probabilities were nearly always significantly different. For N = 10 suboptimals, for grammar G1 a better structure than the CYK parse was found for 2% of the sequences (41/2455), and for G2, a better structure was found for 69% (1704/2455). Sampling deeper into the posterior probability distribution for parse trees increases the chance of finding a more optimal structure; for N = 1000 suboptimals, better structures than the CYK parse were found for about 20% of the sequences for G1 (495/2455), and 98% of the sequences for G2 (2417/2455).
Thus these results showed that the CYK algorithm often does not give the optimal secondary structure if one is using a structurally ambiguous grammar. Structural ambiguity appears to be a practical concern. We therefore focused on developing several lightweight unambiguous grammars.
Four unambiguous grammars
It is something of a challenge to make unambiguous grammars with a small number of productions. We do not know of a way to systematically generate and explore the space of unambiguous grammars. There appear to be many different ways to make unambiguous RNA grammars that have the same simple emission parameterization (4 probabilities for unpaired residues, 16 probabilities for base pairs). We used the following four grammars:

G3 was developed by RDD. We challenged Graeme Mitchison to make a smaller one, and he produced G4 (G. Mitchison, personal communication). The ultracompact G5 grammar is from Ivo Hofacker (personal communication). G6 is the Knudsen/Hein grammar utilized in the Pfold package [18,26]. Each of the four grammars was conjectured to be unambiguous by inspection. Each one also passed the empirical test for ambiguity described in Methods, using the test set of 2455 Rfam sequences.
Each grammar imposes slightly different restrictions on the "language" of possible structures. G3 imposes a minimum hairpin loop length of one nucleotide, G6 has a minimum of two, and G4 and G5 do not impose minimum hairpin loop lengths. Also, G3 and G6 can not emit an empty string, ε, whereas G4 and G5 can. Figure 2 shows parse trees for an example RNA structure using these four grammars G3-G6. The figure shows how each grammar factors a structure into elementary scorable steps in a different manner.
Figure 2.
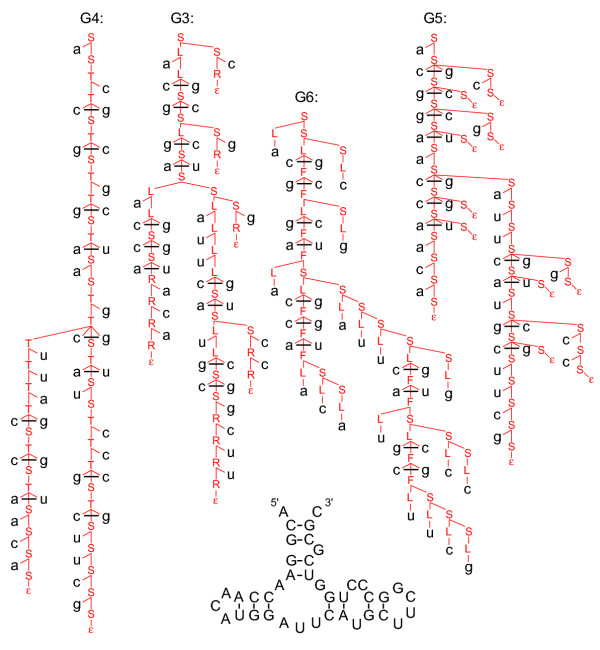
Example parse trees for four different unambiguous grammars, G3-G6. Each grammar's production rules defines how the example structure will be described. Some aspects seem artificial, having more to do with the constraints of being unambiguous rather than biologically meaningful features of RNA. G3 must bifurcate to accomodate a bulge on one side, but not the other. G4 shows a stutter-step behavior in stems, with a cycle of S ⇒ T ⇒ aS productions for each base pair. G5 (rightmost) uses a bifurcation and a null production for every base pair. G6 uses bifurcations at every single stranded residue.
Table 1 shows the resource requirements of these four grammars as well as the simple ambiguous grammar G1. Each non-terminal of a grammar requires storage of a dynamic programming matrix, so memory usage scales linearly with nonterminal number.
Table 1.
Simple grammar specifications.
| Parameters | |||||
| Grammar | NT | Total | Tied | C. elegans LSU Memory (MB) | Notes |
| G1 | 1 | 29 (25) | 25 (22) | 26.9 | ambiguous |
| G3 | 3 | 56 (47) | 28 (23) | 79.4 | min 1 nt loop; no ε string |
| G4 | 2 | 46 (40) | 26 (22) | 53.2 | none |
| G5 | 1 | 23 (20) | 23 (20) | 26.9 | none |
| G6 | 3 | 42 (36) | 26 (21) | 79.4 | min 2 nt loop; no ε string |
Each simple grammar requires a different number of nonterminals (NT) and parameters (with free parameters in parenthesis). Memory requirements were determined empirically by measuring the memory each grammar utilizes to fold a single C. elegans large subunit ribosomal RNA sequence (3662 nucleotides). Under "notes", we give some of the implications each grammar has on the language it describes.
Three unambiguous stacking grammars
The G6 grammar is readily extended to include stacking parameters, by changing the F nonterminal parameterization to a first order Markov chain,  . We call this grammar G6S. We also restructured two of the simple backbones (G3 and G4) to include base pair stacking parameters, resulting in grammars G7 and G8:
. We call this grammar G6S. We also restructured two of the simple backbones (G3 and G4) to include base pair stacking parameters, resulting in grammars G7 and G8:
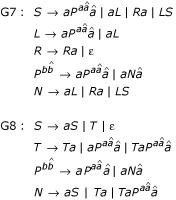
As written, these grammars do not permit lone pairs (a base-pair with no stacking partner). An alternative formulation (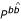 → N) that does allow lone pairs was found to give slightly poorer results (data not shown), even though roughly one quarter of the sequences in our benchmark set have annotated lone pairs.
→ N) that does allow lone pairs was found to give slightly poorer results (data not shown), even though roughly one quarter of the sequences in our benchmark set have annotated lone pairs.
Table 2 shows the general resource requirements for each of the stacking grammars. Each one is conjectured to be unambiguous. Each one passed the empirical ambiguity test for the set of 2455 Rfam sequences.
Table 2.
Stacking grammar specifications.
| Parameters | |||||
| Grammar | NT | Total | Tied | C. elegans LSU Memory (MB) | Notes |
| G2 | 2 | 287(281) | 283(278) | 53.2 | ambiguous |
| G7 | 5 | 341(326) | 289(281) | 128 | min 1 nt loop; no lone pairs; no ε string |
| G8 | 4 | 347(334) | 287(280) | 103 | min 1 nt loop; no lone pairs |
| G6S | 3 | 282(276) | 282(276) | 79.4 | min 2 nt loop; no ε string |
Stacking adds to grammars more nonterminals (NT) and total (free) parameters. This translates into increased memory requirements, as shown by the folding of C. elegans large subunit ribosomal RNA (3662 nucleotides). It also may introduce other restrictions such as not permitting lone pairs.
Prediction accuracy
The secondary structure prediction accuracy of each of the nine parameterized grammars was then measured, by calculating sensitivity and PPV on the benchmark set of 403 trusted secondary structures derived by comparative analysis. The results are shown in Table 3.
Table 3.
Grammar performance.
| Sensitivity % (PPV %) | ||||
| Grammar |
Full Benchmarking Set Sensitivity % (PPV %) |
RNase P | SRP | tmRNA |
| G1 | 17 (12) | 14 (11) | 37 (32) | 10(6) |
| G3 | 34 (31) | 37 (35) | 28 (28) | 31 (22) |
| G4 | 10 (8) | 10(8) | 19 (17) | 4(2) |
| G5 | 3(4) | 3(4) | 2(3) | 4(3) |
| G6 | 47 (45) | 49 (49) | 47 (49) | 44 (33) |
| G2 | 36 (25) | 31 (23) | 59 (48) | 33 (17) |
| G7 | 45 (43) | 46 (46) | 50 (52) | 40 (30) |
| G8 | 46 (44) | 46 (46) | 52 (53) | 44 (32) |
| G6S | 49 (45) | 50 (50) | 50 (51) | 44 (32) |
| mfold v3.1.2 | 56 (48) | 56 (51) | 70 (66) | 46 (30) |
| Vienna RNA | 55 (47) | 55 (51) | 67 (64) | 45 (30) |
| PKNOTS | 50 (41) | 53 (46) | 58 (55) | 38 (24) |
| RNAstructure | 56 (47) | 58 (52) | 62 (58) | 46 (30) |
| Pfold | 39 (69) | 42 (76) | 35 (64) | 33 (54) |
The first section contains simple grammars, the second contains stacking grammars, and the last section contains other available software packages that predict secondary structure. The nine grammars were trained on rRNA as described in the text. The second column gives the performance on the full benchmarking dataset, which is subdivided into each family in the subsequent columns. The metrics are calculated over base pairs as described in the text, evaluating the metrics over individual sequences shows similar results (data not shown).
In order to minimize performance differences due to differences in free parameter number, as opposed to grammar structure, we tied emission parameters together where possible. That is, each grammar uses only one emission distribution for 4 unpaired bases and 16 base pairs (and, in the case of the stacking grammars, 256 stacking parameters). Thus the effective (tied) number of parameters only differs because of the number of transitions in each grammar (Tables 1 and 2). To minimize differences caused by the grammars having slightly different languages, we also forced each grammar to have the same minimum hairpin loop size (two) by implementing the appropriate steps in dynamic programming parsers to ignore hairpin loops of length one or zero, even if the grammar permits them. Maximum a posteriori parameters were then determined for each grammar using a set of annotated ribosomal RNA secondary structures (see Methods).
For comparison, we also evaluated the performance of several implementations of energy minimization algorithms for predicting secondary structure on the same benchmark set: mfold v3.1.2, PKNOTS vl.0l, RNAstructure v4.0, and the Vienna RNA package vl.4. Vienna RNA and mfold use the thermodynamic parameters described in [7] but use slightly different implementations of the rules for exterior and multibranch loops. RNAstructure uses a recently revised set of parameters [53], and PKNOTS uses an earlier set of thermodynamic parameters [54].
We also benchmarked the Knudsen and Hein Pfold program, which independently implements the G6 grammar. The Pfold program was parameterized on a different training set composed of rRNAs and tRNAs, and has heuristics to call only confident base pairs, which increases PPV at the expense of sensitivity. Pfold is intended for consensus structure prediction, using an RNA evolutionary model to fold a given input RNA alignment, but we have used it here in single-sequence mode to compare to our implementation and parameterization of G6.
Among the four simple (nonstacking) unambiguous grammars, the simplest grammar, G5, with only three types of production rules, has an abysmal prediction performance. The grammar with the most rules, G3, did not give the best performance. The best simple SCFG design we tested is G6, the grammar used in Knudsen and Hein's Pfold. Though G6 does not perform quite as well as energy minimization methods, it does surprisingly well considering its simplicity. G6's performance is nearly comparable to the performance of PKNOTS, which uses the older (1995) thermodynamic energy rules. G6 has only 21 free probability parameters, compared to the several hundred thermodynamic parameters in energy minimization programs.
Performance for two of the grammars (G3 and G4) increased significantly when these grammars were extended to include stacking correlations (G7 and G8, respectively). Interestingly, G6 did not show any real performance gain when extended to include stacking correlations (G6S); this was unexpected. Once stacking correlations are included, four of the grammars (G6, G7, G8, and G6S) have comparably good performance, at the cost of increasing parameter number to include the 16 × 16 parameters set for the three with stacking correlations.
The reason for the strong differences between different designs is probably related to how naturally the production rules of the grammar correspond to the biological constraints of RNA structures. The compact G5 grammar, for instance, must invoke the same bifurcation rule S → aSS for every base pair and for every structural bifurcation, which are quite different structural features that occur with very different frequencies. The productions of G5 are thus "semantically overloaded": they collapse too many different types of information into the same parameters. Looking at Figure 2, G6 and G3 arguably have the most natural parse trees, in the sense that they invoke one dedicated production per base pair (as opposed to two, as in G4's stutter-step for each base pair, or the overloaded basepair/bifurcation production in G5).
Our results here probably underestimate the performance that could be wrung from any of these SCFGs if they were more systematically and carefully parameterized. Our parameterization here was based just on counts from a large rRNA database. SSU and LSU ribosomal RNAs, though large, are not representative of all RNA structures; the ideal training set would be a large number of evolutionarily unrelated RNA secondary structures. Additional performance can probably be achieved from these SCFGs by training on larger, more diverse datasets. One might also explore more sophisticated parameterization strategies. We have done some exploratory work using conditional maximum likelihood training [55] to find parameters that directly optimize the accuracy of structure predictions in the training rRNA data, rather than using maximum likelihood parameters, but did not obtain a significant performance increase. Another potentially promising strategy would be to "crosstrain" the parameters, incorporating the thermodynamic parameters in some way as prior information to smooth some of the more poorly determined probabilistic parameters, analogous to how probabilistic data is increasingly being used to augment the energy rules [7].
Comparison to Mathews et al. benchmark
The sensitivity of mfold and RNAstructure was reported to be 73% on a different benchmark dataset, which is quite a bit higher than the 56% as obtained here [7]. This concerned us, so before drawing any conclusions from our results, we did additional work to verify our benchmarking of the thermodynamic methods, using mfold as a representative method.
Dave Mathews kindly provided us the benchmark set used in [7]. Because some of the sources of his structure data were privileged, Mathews could not grant us permission to freely distribute this benchmark set (which is why we report performance results on a new benchmark set we are comfortable distributing). We measure mfold at 67% sensitivity on Mathews' dataset instead of the 73% sensitivity reported in Mathews et al. [7]. Our procedure differs from theirs in two steps. First, we count only exactly correct base pair predictions, whereas Mathews et al. count a base pair as correct even if there is a slight (1 nt) slip in the prediction. Second, we report the sensitivity over all base pairs (total correct base pairs / total base pairs predicted in the whole benchmark), whereas they calculate an average of this value over each sequence. We found that each of these differences contributes about a 3% difference in the measured sensitivities, with our procedures systematically producing more conservative measures than Mathews et al.
The difference between the 56% we report and the 67% obtained by applying our method to Mathews' dataset lies in the choice of secondary structures. Most importantly, prediction accuracy varies significantly from RNA family to RNA family. We have excluded tRNAs from our benchmark set because they are small and reasonably predicted by both methods (the G6S grammar achieves roughly 80% sensitivity on the Mathews' tRNAs, and Mathews reports 83% for energy minimization [7].) Instead, we included tmRNAs, which tend to be difficult to predict (mfold and G6S achieve around 45% sensitivity) because of the presence of pseudoknots which account for 21% of base pairs in the family. These differences account for some of the difference, since Mathews et al. included tRNAs but did not use tmRNAs.
We also looked carefully at the overlap between the 16 RNase P's in the Mathews benchmark and the 225 RNase P's in ours: 9 are unique to the Mathews set; 218 are unique to ours; and 7 occur in both sets. mfold predicts the 9 structures unique to the Mathews set with 65% sensitivity and the 218 structures unique to our dataset at 56% sensitivity, so there may be some bias towards more difficult-to-predict structures in our set. In addition, there are differences between the secondary structures in our dataset and in Mathews' set even for exactly the same sequences. We compared the structures for the 7 RNase Ps included in both sets and found that these matched in only about 90% of their base pairs. mfold predicted those structures annotated in the Mathews benchmark at 60% sensitivity, whereas it predicted those in ours (e.g. structures as annotated in Jim Brown's current RNase P database) at 56% sensitivity. The secondary structures of many bacterial and archaeal RNase P's were revised in 2001 in light of additional comparative sequence analysis [56]. This suggests (not unexpectedly) that there is a slight bias towards mfold in benchmarks. RNA secondary structures are derived from a combination of manual comparative analysis and computational prediction, so there is some logical circularity when benchmarking (that is, we are benchmarking mfold on structures that sometimes may have been produced in part by mfold.) This bias disappears gradually as comparative evidence accumulates and structures are refined. We therefore feel we can adequately account for the absolute differences in prediction accuracy as measured by the two benchmarks.
The most relevant question is whether different prediction methods are ranked the same on either data set. This appears to be the case. For example, on the Mathews benchmark set measured by our procedures, the G3 grammar has a sensitivity of 41%, G6 has a sensitivity of 55%, and mfold is at 67%, which is essentially the same relative difference as shown in Table 3. We therefore believe that the relative performances reported in Table 3 are reasonably fair and largely independent of our choice of test structures. The benchmark set we used in Table 3 is available from our web site.
Conclusions
Our goal in this work was to explore how much SCFG design decisions impact RNA secondary structure prediction accuracy. Structural ambiguity is clearly a concern if structure prediction is the goal, as we showed with the reordering experiment. The results in Table 3 indicate that even among different unambiguous SCFG designs, design decisions matter quite a bit.
The prediction accuracy of relatively simple SCFGs is not too far from the accuracy of the energy minimization methods. SCFGs are easily trained from secondary structure databases, and different designs can be explored fairly easily. It might be useful to explore more sophisticated SCFG designs to see if an appropriately designed SCFG might even be able to outperform the existing energy minimization methods for single-sequence prediction. For instance, it would be interesting to know how an unambiguous SCFG mirror of the Zuker algorithm would perform, with probabilistic parameters for stacking, hairpin, bulge, and interior loop lengths, multifurcations, dangles, and coaxial stacking. Although one implementation of an SCFG mirror of the Zuker algorithm has been described [35], it used a structurally ambiguous grammar (it was not intended for secondary structure prediction per se; it was only used in summed Inside algorithm calculations where ambiguity doesn't matter, not in a CYK algorithm where ambiguity does matter).
Apparently without considering alternative designs, Knudsen and Hein had already described the simple and effective G6 grammar, and extended it to analysis of input multiple sequence alignments in the program Pfold [18,26]. Because our exploration has not been exhaustive, we can not say that G6 is the best possible simple grammar. However, after exploring various alternative SCFG designs, we confirm that the Knudsen/Hein grammar is an excellent, simple framework in which to develop some probabilistic RNA analysis methods.
The long term goal of this line of work is to extend a single sequence SCFG design into a pairwise SCFG to address the problem of structural alignment, combining structural and evolutionary information in the same model. Given that the pairwise algorithm will be  (MN4) in memory and (MN6) in time for grammars of M nonterminals and sequences of length N [27], we know that we will have computational complexity issues and therefore must seek the simplest reasonably performing grammars. While the G6 grammar is small and works well, the G7 and G8 grammars look easiest to extend to the pairwise SCFG case. We are currently implementing methods to extend them to pair-SCFGs for pairwise alignment and structure prediction.
(MN4) in memory and (MN6) in time for grammars of M nonterminals and sequences of length N [27], we know that we will have computational complexity issues and therefore must seek the simplest reasonably performing grammars. While the G6 grammar is small and works well, the G7 and G8 grammars look easiest to extend to the pairwise SCFG case. We are currently implementing methods to extend them to pair-SCFGs for pairwise alignment and structure prediction.
Authors' contributions
RDD wrote the code and carried out all the experiments. SRE suggested the idea of comparing grammar designs. RDD drafted the manuscript, and both authors collaborated closely in writing the final version.
Acknowledgments
Acknowledgments
RDD was supported by a Howard Hughes predoctoral fellowship and an Olin predoctoral fellowship. The work was also supported by funding from the Howard Hughes Medical Institute, Alvin Goldfarb, and NIH NHGRI HG01363. We thank Graeme Mitchison for discussions leading to the G4 grammar, Ivo Hofacker for discussions which led to the G5 grammar, Bjarne Knudsen for help with Pfold, David Mathews for help with RNAstructure and for providing the benchmark dataset from [7], and Elena Rivas for numerous discussions. Some of this work was conceived at an ESF and NIH funded workshop on computational RNA biology in Benasque, Spain, in summer 2003.
Contributor Information
Robin D Dowell, Email: robin@genetics.wustl.edu.
Sean R Eddy, Email: eddy@genetics.wustl.edu.
References
- Eddy SR. Non-Coding RNA Genes and the Modern RNA World. Nat Rev Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- Zuker M. Calculating Nucleic Acid Secondary Structure. Curr Opin Struct Biol. 2000;10:303–310. doi: 10.1016/S0959-440X(00)00088-9. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Pieczenik G, Griggs JR, Kleitman DJ. Algorithms for Loop Matchings. SIAM J Appl Math. 1978;35:68–82. [Google Scholar]
- Waterman MS, Smith TF. RNA Secondary Structure: A Complete Mathematical Analysis. Math Biosci. 1978;42:257–266. doi: 10.1016/0025-5564(78)90099-8. [DOI] [Google Scholar]
- Zuker M, Stiegler P. Optimal Computer Folding of Large RNA Sequences Using Thermodynamics and Auxiliary Information. Nucl Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast Folding and Comparison of RNA Secondary Structures (The Vienna RNA Package) Monatsh Chem. 1994;125:167–188. [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Eddy SR, Durbin R. RNA Sequence Analysis Using Covariance Models. Nucl Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara Y, Brown M, Hughey R, Mian IS, Sjolander K, Underwood RC, Haussler D. Stochastic Context-Free Grammars for tRNA Modeling. Nucl Acids Res. 1994;22:5112–5120. doi: 10.1093/nar/22.23.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, Eddy SR, Krogh A, Mitchison GJ. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge UK: Cambridge University Press; 1998. [Google Scholar]
- Pace NR, Smith DK, Olsen GJ, James BD. Phylogenetic Comparative Analysis and the Secondary Structure of Ribonuclease P RNA – A Review. Gene. 1989;82:65–75. doi: 10.1016/0378-1119(89)90031-0. [DOI] [PubMed] [Google Scholar]
- Gutell RR, Power A, Hertz GZ, Putz EJ, Stormo GD. Identifying Constraints on the Higher-Order Structure of RNA: Continued Development and Application of Comparative Sequence Analysis Methods. Nucl Acids Res. 1992;20:5785–5795. doi: 10.1093/nar/20.21.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Larsen N, Woese CR. Lessons from an Evolving rRNA: 16S and 23S rRNA Structures from a Comparative Perspective. Microbiol Rev. 1994;58:10–26. doi: 10.1128/mr.58.1.10-26.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Lee JC, Cannone JJ. The Accuracy of Ribosomal RNA Comparative Structure Models. Curr Opin Struct Biol. 2002;12:301–310. doi: 10.1016/S0959-440X(02)00339-1. [DOI] [PubMed] [Google Scholar]
- Chiu DKY, Kolodziejczak T. Inferring Consensus Structure from Nucleic Acid Sequences. Comput Applic Biosci. 1991;7:347–352. doi: 10.1093/bioinformatics/7.3.347. [DOI] [PubMed] [Google Scholar]
- Muse SV. Evolutionary Analyses of DNA Sequences Subject to Constraints on Secondary Structure. Genetics. 1995;139:1429–1439. doi: 10.1093/genetics/139.3.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulko B, Haussler D. Using Multiple Alignments and Phylogenetic Trees to Detect RNA Secondary Structure. In Pac Symp Biocomput. 1996. pp. 350–367. [PubMed]
- Knudsen B, Hein J. RNA Secondary Structure Prediction Using Stochastic Context-Free Grammars and Evolutionary History. Bioinformatics. 1999;15:446–454. doi: 10.1093/bioinformatics/15.6.446. [DOI] [PubMed] [Google Scholar]
- Akmaev VR, Kelley ST, Stormo GD. Phylogenetically Enhanced Statistical Tools for RNA Structure Prediction. Bioinformatics. 2000;16:501–512. doi: 10.1093/bioinformatics/16.6.501. [DOI] [PubMed] [Google Scholar]
- Michel F, Westhof E. Modelling of the Three-Dimensional Architecture of Group I Catalytic Introns Based on Comparative Sequence Analysis. J Mol Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- McCaskill JS. The Equilibrium Partition Function and Base Pair Binding Probabilities for RNA Secondary Structure. Biopolymers. 1990;29:1105–19. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- Ding Y, Lawrence CE. A Statistical Sampling Algorithm for RNA Secondary Structure Prediction. Nucl Acids Res. 2003;31:7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabaska JE, Cary RB, Gabow HN, Stormo GD. An RNA Folding Method Capable of Identifying Pseudoknots and Base Triples. Bioinformatics. 1998;14:691–699. doi: 10.1093/bioinformatics/14.8.691. [DOI] [PubMed] [Google Scholar]
- Juan V, Wilson C. RNA Secondary Structure Prediction Based on Free Energy and Phylogenetic Analysis. J Mol Biol. 1999;289:935–947. doi: 10.1006/jmbi.1999.2801. [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Fekete M, Stadler PF. Secondary Structure Prediction for Aligned RNA Sequences. J Mol Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- Knudsen B, Hein J. Pfold: RNA Secondary Structure Prediction Using Stochastic Context-Free Grammars. Nucl Acids Res. 2003;31:3423–3428. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D. Simultaneous Solution of the RNA Folding, Alignment, and Protosequence Problems. SIAM J Appl Math. 1985;45:810–825. [Google Scholar]
- Gorodkin J, Heyer LJ, Stormo GD. Finding the Most Significant Common Sequence and Structure Motifs in a set of RNA Sequences. Nucl Acids Res. 1997;25:3724–3732. doi: 10.1093/nar/25.18.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lück R, Gräf S, Steger G. Construct: a Tool for Thermodynamic Controlled Prediction of Conserved Secondary Structure. Nucl Acids Res. 1999;27:4208–4217. doi: 10.1093/nar/27.21.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodkin J, Stricklin SL, Stormo GD. Discovering Common Stem-Loop Motifs in Unaligned RNA Sequences. Nucl Acids Res. 2001;29:2135–2144. doi: 10.1093/nar/29.10.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes I, Rubin GM. Pairwise RNA Structure Comparison with Stochastic Context-Free Grammars. In Pac Symp Biocomput. 2002. pp. 163–174. [DOI] [PubMed]
- Perriquet O, Touzet H, Dauchet M. Finding the Common Structure Shared by Two Homologous RNAs. Bioinformatics. 2003;19:108–116. doi: 10.1093/bioinformatics/19.1.108. [DOI] [PubMed] [Google Scholar]
- Mathews DH, Turner DH. Dynalign: an Algorithm for Finding the Secondary Structure Common to two RNA Sequences. J Mol Biol. 2002;317:191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- Ji Y, Xu X, Stormo GD. A Graph Theoretical Approach to Predict Common RNA Secondary Structure Motifs Including Pseudoknots in Unaligned Sequences. Bioinformatics. 2004. [DOI] [PubMed]
- Rivas E, Eddy SR. Secondary Structure Alone is Generally Not Statistically Significant for the Detection of Noncoding RNAs. Bioinformatics. 2000;6:583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- Rivas E, Eddy SR. Noncoding RNA Gene Detection Using Comparative Sequence Analysis. BMC Bioinformatics. 2001;2:8. doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaynes ET. Probability Theory: The Logic of Science. Cambridge University Press; 2003. [Google Scholar]
- MacKay DJC. Information Theory, Inference, and Learning Algorithms. Cambridge University Press; 2003. [Google Scholar]
- Sipser M. Introduction to Theory of Computation. Brooks Cole Pub Co; 1996. [Google Scholar]
- Hopcroft JE, Ullman JD. Introduction to Automata Theory, Languages, and Computation. Addison-Wesley; 1979. [Google Scholar]
- Harrison MA. Introduction to Formal Language Theory. Addison-Wesley; 1978. [Google Scholar]
- Ding Y, Lawrence CE. Statistical Prediction of Single-Stranded Regions in RNA Secondary Structure and Application to Predicting Effective Antisense Target Sites and Beyond. Nucl Acids Res. 2001;29:1034–1046. doi: 10.1093/nar/29.5.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegerich R. Explaining and Controlling Ambiguity in Dynamic Programming. In: Giancarlo R, Sankoff D, editor. In Proceedings of the 11th Annual Symposium on Combinatorial Pattern Matching, 1848. Montréal, Canada: Springer-Verlag, Berlin; 2000. pp. 46–59. [Google Scholar]
- Walter A, Turner D, Kim J, Lyttle M, Muller P, Mathews D, Zuker M. Coaxial Stacking of Helixes Enhances Binding of Oligoribonucleotides and Improves Predictions of RNA Folding. PNAS. 1994;91:9218–9222. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Walter AE, Turner DH. Thermodynamics of Coaxially Stacked Helixes With GA and CC Mismatches. Biochemistry. 1996;35:13753–13761. doi: 10.1021/bi960913z. [DOI] [PubMed] [Google Scholar]
- Rivas E, Eddy SR. A Dynamic Programming Algorithm for RNA Structure Prediction Including Pseudoknots. J Mol Biol. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- Wuyts J, Rijk PD, de Peer YV, Winkelmans T, Wachter RD. The European Large Subunit Ribosomal RNA Database. Nucl Acids Res. 2001;29:175–177. doi: 10.1093/nar/29.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts J, de Peer YV, Winkelmans T, Wachter RD. The European Database on Small Subunit Ribosomal RNA. Nucl Acids Res. 2002;30:183–185. doi: 10.1093/nar/30.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW. The Ribonuclease P Database. Nucl Acids Res. 1999;27:314. doi: 10.1093/nar/27.1.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblad MA, Gorodkin J, Knudsen B, Zwieb C, Samuelsson T. SRPDB: Signal Recognition Particle Database. Nucleic Acids Res. 2003;31:363–364. doi: 10.1093/nar/gkg107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwieb C, Gorodkin J, Knudsen B, Burks J, Wower J. tmRDB (tmRNA Database) Nucleic Acids Res. 2003;31:446–447. doi: 10.1093/nar/gkg019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA Family Database. Nucl Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Disney DH, Childs MD, Schroeder JL, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra MJ, Turner DH. Predicting Thermodynamic Properties of RNA. Meth Enzymol. 1995;25:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- Krogh A. Two Methods for Improving Performance of an HMM and Their Application for Gene Finding. Proc Int Conf on Intelligent Systems in Molecular Biology. 1997;5:179–186. [PubMed] [Google Scholar]
- Harris JK, Haas ES, Williams D, Frank DN, Brown JW. New Insight Into RNase P RNA Structure From Comparative Analysis of the Archaeal RNA. RNA. 2001;7:220–232. doi: 10.1017/S1355838201001777. [DOI] [PMC free article] [PubMed] [Google Scholar]