

Background
A renal biopsy in a case of systemic lupus erythematosus (SLE) nephritis presenting with both nephrotic syndrome (NS) and acute renal failure (ARF) demonstrated atypical fibrillary deposits in the glomerular mesangium and subendothelium. The size and morphology of fibrils resembled those of fibronectin (FN) glomerulopathy [1,2], as also confirmed by immunoperoxidase staining. As no familial history of FN disorders was noted, this case may represent the first report of a new kidney disease entity in the context of SLE.
Case report
A 62-year-old man without previous known signs and symptoms of kidney disease presented with the abrupt onset of nephrotic syndrome (NS). On admission, massive limb and scrotal oedema, bilateral pleural effusion and mild ascites were noted, without other relevant physical signs. BP was 115/70 mmHg. Renal function was normal, with serum creatinine (sCr) of 1.05 mg/dL (92.82 μmol/L) and BUN of 29 mg/dL (10.35 mmol/L). Urinalysis showed >100 RBC/hpf and abundant hyaline and granular casts. Proteinuria was 20–33 g/day, with typical hypoalbuminaemia. Serum C3 and C4 were normal at 0.98 and 0.2 g/L, respectively. No anaemia or other haematological abnormalities were present. Antinuclear Abs were positive at a titre of 1:320 and homogeneous pattern, while anti-ds-DNA Abs were positive at 1:320 (212,00 WHO U/ml). Upon aggressive i.v. diuretic treatment (up to 250 mg furosemide/day) sCr and BUN increased over 8 days to 6.6 and 62 mg/dL (583.4 μmol/L and 22.13 mmol/L), respectively. The patient was then transferred to our Nephrology ward, where renal vein thrombosis was ruled out by Doppler ultrasound, and accurate fluid management stabilized renal function. A renal biopsy was immediately obtained.
Renal histology
The light microscopy specimen contained 12 glomeruli, mostly enlarged, swollen and with mild focal mesangial proliferation. Abundant PAS+, fucsine + amorphous matrix was deposited in pericapillary areas (Figure 1). Direct immunofluorescence on four glomeruli showed a ‘full house’ pattern, with intense, finely granular subendothelial, intramembranous and mesangial IgG, C3, κ and λ light chains. Immunoperoxidase was briskly positive for the total FN. Congo red staining was negative.
Fig. 1.
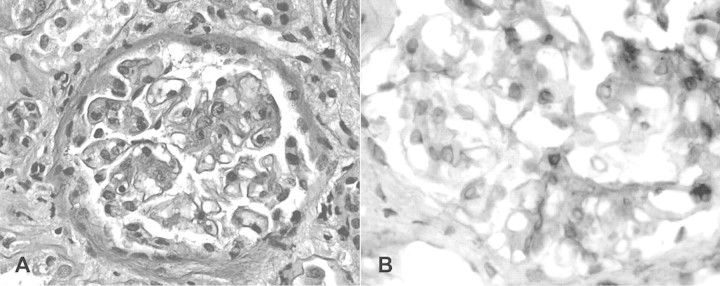
Renal biopsy showing mild mesangial and focal GBM thickening (panel A, PAS stain), partly resulting from the deposition of fibronectin (panel B, immunoperoxidase). Light microscopy, original magnification 200×.
Transmission electron microscopy (EM) on six glomeruli showed extensive foot process effacement, associated with irregular width of the GBM. Intramembranous, subendothelial and mesangial electron-dense deposits had a typical fibrillary pattern; capillary loops were often collapsed by fibrils. The average width of the fibrils measured by a Soft Imaging System software (Münster, Germany) was 13.7 ± 1.8 nm (Figure 2).
Fig. 2.
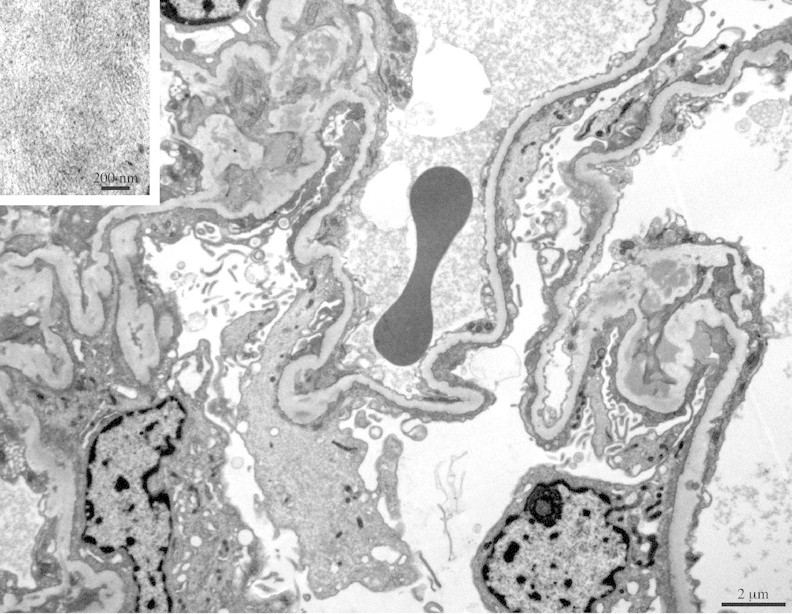
Transmission electron microscopy shows focal enlargement of GBM, mostly due to subendothelial electron-dense deposits of immunoglobulins. Note larger deposits of poorly formed, short fibrils suggesting a weaving pattern (inset). Uranyl acetate/lead citrate.
The histopathological findings were thus consistent with an FN-type fibrillary deposition in the context of lupus nephritis class II with active lesions.
Follow-up
Intravenous pulse methylprednisolone was administered, 1 g/day for 3 consecutive days, followed by daily oral prednisone, 1 mg/kg BW and cyclophosphamide, 2 mg/kg BW/day p.o. After 1 week, the patient was discharged with sCr and BUN of 4.2 and 43 mg/dL (371.2 μmol/L and 15.3 mmol/L), respectively. Other medications were furosemide, 25 mg tid; hydrochlorothiazide, 25 mg/day and enalapril maleate, 20 mg/day. One month later, renal function further improved to a calculated (Cockroft-Gault) GFR of 44 ml/min, with sCr of 1.9 mg/dL (167.9 μmol/L). Proteinuria was still in the nephrotic range, averaging 22 g/day. Because of marked leukopenia (total WBC 1250, neutrophils 665/mm3) cyclophosphamide was discontinued at Day 38 and replaced with cyclosporine 3 mg/kg BW. Within 15 days, WBC count was back to 10,650/mm3, sCr and BUN were 1.0 and 24 mg/dL, respectively, with a substantial reduction of proteinuria to 1.59 g/day. Prednisone is being gradually tapered to a present dose of 25 mg/day.
Discussion
Glomerular disorders caused by deposition of non-amyloid fibrils are heterogeneous [1–3]. The structural lesions are not characteristic, so that mesangial expansion and/or proliferation, membranous or membranoproliferative patterns can be observed. Fibrils usually appear as a mildly PAS-positive material surrounding glomerular capillaries and often bulging into the mesangium. On light microscopy it is often difficult to distinguish such deposits from those of light chain or diabetic glomerulopathies. Only EM may identify the deposits, based on the ultrastructure and width of the fibrils [4,5].
Fibrillary glomerulonephritis is the most common form, accounting for nearly 85% of the cases; Congo red or thioflavine T-negative fibrils have an average width of 15–20 nm, as opposed to amyloid fibrils of 8–10 nm, with a similar random distribution [3–5]. Immunotactoid glomerulopathy is characterized by orderly deposition of microtubules with larger diameter, i.e. 30–60 nm, often associated with a circulating monoclonal immunoglobulin or paraprotein [5,6]. There may be a relationship between fibrillary glomerulopathies and hepatitis C infection with cryoglobulins.
A unique form of fibrillary glomerulopathy is associated with deposition of FN, usually as a hereditary disease [1–3]. FN is a dimeric glycoprotein with two nearly identical 250 kDa subunits. It is a component of several normal matrices, involved in the control of cellular proliferation and cell-to-cell interactions, including platelet adhesion and thrombogenesis [7,8]. Increased FN expression in diabetes or glomerulonephritis usually reflects local mesangial or epithelial cell production of the insoluble, cellular form. FN glomerulopathy results instead from massive deposition of the soluble, plasma isoform [8]. For unknown reasons, in certain individuals circulating FN tends to accumulate along the glomerular capillaries, often as a PAS-positive homogeneous mesangial material [1–3,8]. EM typically reveals large to massive electron-dense finely granular or fibrillar subendothelial deposits. Randomly arranged fibrils are ∼12 nm in width and 125 nm in length [1–3,8]. The clinical picture is one of a steroid-resistant NS, slowly progressing to end-stage renal failure. FN nephropathy tends to cluster in certain families, suggesting a mutation of the FN gene or a regulatory protein. One such ligand could be uteroglobin, as experimental uteroglobin-deficient mice have enhanced glomerular FN deposition. Other candidate genes belong to the regulator of complement activation cluster [9].
To our knowledge, ours is the first report of glomerular FN deposition in the context of a fairly atypical form of lupus nephritis. Our patient presented with severe NS rapidly evolving into ARF, in the absence of other signs of autoimmune disease. Renal histology was hardly consistent with abrupt impairment of glomerular filtration, in the absence of endocapillary thrombosis, leukocyte infiltration, necrosis or extracapillary proliferation. Aggressive diuretic treatment may at least partly explain ARF on a pre-renal basis. Additionally, glomerular and tubulointerstitial swelling due to marked hyponychia may have contributed to a functional phenomenon. The distinguishing feature of amorphous fibrils of 13.7 ± 1.8 nm average width led to the recognition of a component of deposition disease, involving non-immunoglobulin material which stained positive for the total FN but not Congo red. Interestingly, the association of immunoglobulins and FN has been described in the cryoprecipitate from a patient with fibrillary glomerulonephritis [10]. In our case, an association with cryoglobulins is unlikely, as the patient was HCV-negative and had no detectable circulating cryoglobulins.
In conclusion, the description of a novel association between FN fibrillary glomerulopathy and SLE brings about the issue of a careful search for autoimmune disorders in patients presenting with glomerular deposition of paraproteins. Similarly, it would seem appropriate to screen all renal biopsies from SLE patients for immune and non-immune deposits by the routine use of EM, as recommended by many pathologists.
Acknowledgments
The work was supported by funding from the Ministry of University and Research of Italy (MIUR)—quota 60% Ricerche di Facoltà (to P.M.).
Conflict of interest statement. None declared.
References
- 1.Strom EH, Banfi G, Krapf R, et al. Glomerulopathy associated with predominant fibronectin deposits: a newly recognized hereditary disease. Kidney Int. 1995;48:163–170. doi: 10.1038/ki.1995.280. [DOI] [PubMed] [Google Scholar]
- 2.Gemperle O, Neuweiler J, Reutter FW, et al. Familial glomerulopathy with giant fibrillar (fibronectin-positive) deposits: 15-year follow-up in a large kindred. Am J Kidney Dis. 1996;28:668–675. doi: 10.1016/s0272-6386(96)90247-4. [DOI] [PubMed] [Google Scholar]
- 3.Brady H. Fibrillary glomerulopathy. Kidney Int. 1998;53:1421–1429. doi: 10.1046/j.1523-1755.1998.t01-1-00094.x. [DOI] [PubMed] [Google Scholar]
- 4.Bridoux F, Hugue V, Coldefy O, et al. Fibrillary glomerulonephritis and immunotactoid (microtubular) glomerulopathy are associated with distinct immunologic features. Kidney Int. 2002;62:1764–1775. doi: 10.1046/j.1523-1755.2002.00628.x. [DOI] [PubMed] [Google Scholar]
- 5.Rosenstock JL, Markowitz GS, Valeri AM, et al. Fibrillary and immunotactoid glomerulonephritis: distinct entities with different clinical and pathological features. Kidney Int. 2003;63:1450–1461. doi: 10.1046/j.1523-1755.2003.00853.x. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz MM, Korbet SM, Lewis EJ. Immunotactoid glomerulopathy. J Am Soc Nephrol. 2002;13:1390–1397. doi: 10.1097/01.asn.0000013397.06964.19. [DOI] [PubMed] [Google Scholar]
- 7.Pankow R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 8.Assmann KJ, Koene RA, Wetzels JF. Familial glomerulonephritis characterized by massive deposits of fibronectin. Am J Kidney Dis. 1995;25:781–791. doi: 10.1016/0272-6386(95)90555-3. [DOI] [PubMed] [Google Scholar]
- 9.Vollmer M, Krapf R, Hildebrandt F. Exclusion of the uteroglobin gene as a candidate for fibronectin glomerulopathy (GFND) Nephrol Dial Transplant. 1998;13:2417–2418. doi: 10.1093/ndt/13.9.2417. [DOI] [PubMed] [Google Scholar]
- 10.Rostagno A, Vidal R, Kumar A, et al. Fibrillary glomerulonephritis related to serum fibrillar immunoglobulin–fibronectin complexes. Am J Kidney Dis. 1996;28:676–684. doi: 10.1016/s0272-6386(96)90248-6. [DOI] [PubMed] [Google Scholar]