

Case report
A 71-year-old woman presented at the outpatient clinic for the first time in 1973 with diffuse bone pain mainly in both wrists, shoulders and the spine. Clinical examination showed limb deformities (genu varum of the left leg, genu valgum of the right leg) and short stature (142 cm). At that time, she had already experienced multiple spontaneous fractures of the right and left femur. There was no family history of bone disorders (mother and father as well as seven brothers and sisters were healthy).
Laboratory tests at that moment showed: sodium 141 mmol/L (normal range 133–145), potassium 3.9 mmol/L (3.5–5.1), chloride 104 mmol/L (95–108), bicarbonate 23 mmol/L (23–29), calcium 9.8 mg/dL (8.4–10.2), phosphate 1.7 mg/dL (2.4–4.5), serum creatinine 0.72 mg/dL and alkaline phosphatase 120 U/L (53–141). Hyperparathyroidism was excluded. Urine analysis showed a daily phosphate excretion of 597 mg (300–1300), a daily calcium excretion of 39 mg (100–250) and a daily creatinine excretion of 852 mg (660–2200), corresponding to a fractional renal excretion of phosphate of 67%. At that time, the diagnosis of a congenital disorder with hypophosphataemia due to renal phosphate wasting, resulting in osteomalacia was considered. Treatment with oral phosphate and vitamin D replacement was initiated, resulting in a normalization of her serum calcium and phosphate. The following years, a few episodes of symptomatic hypo- and hypercalcaemia (with nausea and vomiting) could not be prevented. Figure 1 shows the evolution over the years of the serum calcium and phosphate levels. Figure 2 shows the evolution of the daily urinary excretion of calcium and phosphate.
Fig. 1.

Evolution of serum calcium and phosphate levels over the years.
Fig. 2.

Evolution of daily urinary excretion of calcium and phosphate.
In 1988, she developed hyperparathyroidism with a serum PTH (parathyroid hormone) level of 305 pg/mL (PTH-COOH-terminal assay, normal range 10–80). Ultrasonography showed a left retrothyroidal, hypoechogenic nodule with a diameter of 10 mm, which was proven to be a parathyroid adenoma at subsequent surgery. Her postoperative serum PTH level was 91.9 pg/mL (PTH-intact assay, normal range 10–65). In 1994, the serum intact PTH level rose to 714 pg/mL, and an asymptomatic hypercalcaemia of 11.4 mg/dL was noted. Ultrasonography showed a large node (diameter 18 mm) in the dorsal, inferior side of the right thyroid lobe and a smaller node (diameter 6 mm) in the dorsal, inferior side of the left thyroid lobe. A total parathyroidectomy was performed, with subcutaneous reimplantation of one small parathyroid gland piece, superficial to the left musculus sternocleidomastoideus. Pathological examination of all resected parathyroid tissue showed diffuse multinodular hyperplasia. Postoperatively, the serum PTH level dropped to 5 pg/mL. In the year 2000, the PTH serum level rose again to 337 pg/mL, while an asymptomatic hypercalcaemic episode (serum calcium level 12.5 mg/dL) was noted, leading to surgical removal of the remaining parathyroid tissue.
In the 90s, she developed chronic kidney disease stage IV (serum creatinine level between 1.5 and 2.5 mg/dL, corresponding to a creatinine clearance of 20–30 ml/min) (Figure 3), associated with a minimal proteinuria (0.3 g/24 h). Repeated renal ultrasonography during follow-up showed bilateral small kidneys (7–8 cm in length), with a thin hyperechoic cortex. In June 2007, a CT scan of the abdomen showed bilateral diffuse, small medullary calcifications in both kidneys (Figure 4). Moreover, an 8-cm tumoural mass was noted in the inferior pole of the left kidney. In July 2007, a retroperitoneoscopic radical left nephrectomy was performed. Postoperatively, chronic intermittent haemodialysis had to be initiated. Pathological examination of the left kidney showed a renal clear cell carcinoma, without invasion of the surrounding tissues, compatible with a Fuhrman histological grade 3 lesion and an anatomic pT2N0M0 tumour (2002 TNM staging system). Pathological examination of the surrounding kidney tissue showed important interstitial fibrosis and tubular atrophy. There was an interstitial lymphocytic infiltrate. Moreover, several interstitial calcifications were present (Figure 5). Since the resection of the renal clear cell carcinoma was curative, no adjuvant therapy was given. Up until now, 8 months postoperatively, no tumour recurrence has occurred.
Fig. 3.

Evolution of serum creatinine levels. Development of chronic renal failure.
Fig. 4.

CT scan of the abdomen. CT scan of the abdomen shows diffuse, small medullary calcifications in both kidneys (shown is the left kidney on a sagittal section).
Fig. 5.
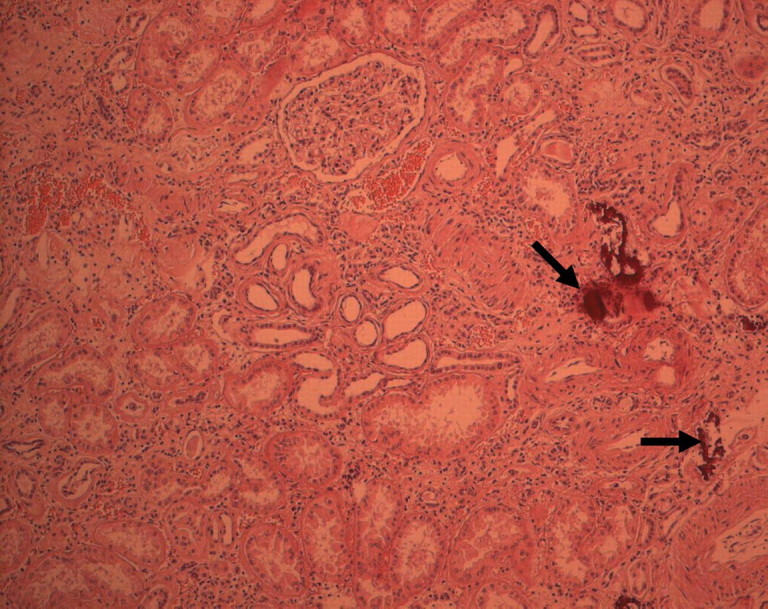
Histological examination of the left kidney. The kidney tissue surrounding the renal clear cell carcinoma shows important interstitial fibrosis and tubular atrophy. There is an interstitial lymphocytic infiltrate. Several interstitial calcifications are present (arrows).
The patient has one daughter, one grandson and one granddaughter. None of them have bone abnormalities.
Questions
What syndrome may explain the hypophosphataemia and osteomalacia? What is the reason for the development of hyperparathyroidism and chronic kidney disease?
Answers to the quiz of the preceding case
Our patient has X-linked hypophosphataemic rickets (XLH), due to a heterozygous mutation c.1805G>A (p.Trp602X) in exon 18 of the PHEX gene (phosphate-regulating endopeptidase homolog, X-linked) identified by sequence analysis. She developed hyperparathyroidism due to the treatment with oral phosphate. The chronic kidney disease was due to nephrocalcinosis, as shown on a CT scan (Figure 4) and by histological examination (Figure 5).
Discussion
This case clearly shows the evolution of XLH, a dominantly inherited disorder with a prevalence of approximately one case per 20 000 individuals. It is characterized by growth retardation, rachitic and osteomalacic bone disease, hypophosphataemia and renal defects in phosphate reabsorption and vitamin D metabolism.
XLH is an X-linked dominant disorder with clinical manifestations in heterozygous female and hemizygous male patients. It is caused by renal phosphate wasting due to an abnormal proximal renal tubular function, which results in an increased renal clearance of inorganic phosphorus and hypophosphataemia. The gene responsible for XLH was identified on chromosome Xp22.1 and was named PHEX [1]. The gene encodes a cell-surface-bound protein-cleaving enzyme that is predominantly expressed in bone and teeth and that degrades and inactivates hormone-like substances. These hormone-like substances are also called phosphatonins and include fibroblast growth factor 23 (FGF23), matrix extracellular phosphoglycoprotein (MEPE) and frizzled-related protein 4 (FRP4). Mutations in the PHEX gene result in failure to inactivate the phosphatonins, leading to abnormally high circulating concentrations of these factors. This in turn results in decreased expression of the sodium phosphate cotransporter that is responsible for phosphate reabsorption in the proximal tubule [2,3]. A wide variety of gene defects in XLH—including nonsense, splice site and missense mutations and gene deletions—involving almost the entire coding region have been described. In our patient, the entire PHEX coding sequence (22 exons and exon flanking boundaries) was evaluated by direct sequencing after PCR amplification of the genomic target sequences. Sequence analysis revealed the presence of a pathogenic nonsense mutation c.1805G>A (p.Trp602X) in exon 18. To our knowledge, this is the first time this specific mutation has been described [4,5].
In addition, the disease is characterized by normal 25-hydroxyvitamin D and low or inappropriately normal serum 1,25-dihydroxyvitamin D serum level, due to aberrant regulation of renal 25-hydroxy-D-1α-hydroxylase activity [2]. Other clinical features of XLH are enthesopathy (calcification of tendons, ligaments and joint capsules), late dentition, dental abscesses and early tooth decay (secondary to poor mineralization of the interglobular dentine), premature cranial synostosis, hypertension and left ventricular hypertrophy.
To prevent growth retardation and correct the rickets and osteomalacia, XLH patients are currently treated with a combination of oral phosphate and vitamin D. However, careful monitoring of the patients is necessary (at least annual monitoring of serum phosphorus, calcium, alkaline phosphatase, creatinine and PTH), since important treatment complications like hyperparathyroidism and nephrocalcinosis may occur during this therapy, as demonstrated in our patient.
Administration of oral phosphate increases the plasma phosphate concentration, leading to a decrease in the plasma ionized calcium concentration (due to complexing of phosphate with calcium) and a decrease in the plasma calcitriol concentration (by removing the hypophosphataemic stimulus to its synthesis). Both, hypocalcaemia and the removal of the normal inhibitory effect of calcitriol on the PTH synthesis, result in (reversible) secondary hyperparathyroidism. Rarely, a progression to irreversible (‘tertiary’) hyperparathyroidism has been described, due to autonomous parathyroid hyperfunction, necessitating surgical intervention [6,7].
An excess of vitamin D can lead to hypercalcaemia and hypercalciuria. The combination of longstanding hypercalcaemia/hypercalciuria and secondary hyperparathyroidism can lead to the development of chronic hypercalcaemic nephropathy. In this disorder, calcification, degeneration and necrosis of the tubular cells results in cell sloughing and eventual tubular atrophy and interstitial fibrosis and calcification (nephrocalcinosis). These changes are most prominent in the renal medulla but can also be seen in the renal cortex. The interstitial calcium deposition can be detected by ultrasonography or by computed tomography in the earlier stages of the disease and by a plain film of the abdomen when severe renal parenchymal involvement is present. In 1991, a study of 24 children (age ranging from 1 to 16 years) with XLH showed on renal ultrasonography that 19 of them (79%) had signs of nephrocalcinosis [8].
Several adjuvant therapies, such as recombinant growth hormone [9,10], thiazide diuretics [11] and 24,25-dihydroxyvitamin D [12], are being studied to improve the efficacy or diminish side effects of the current therapy with phosphate and calcitriol. However, at this moment, no guidelines can be made yet, regarding these new therapies.
In our patient, we observed the development of tertiary hyperparathyroidism (necessitating parathyroidectomy) and nephrocalcinosis with chronic renal insufficiency, during the years of treatment with oral phosphate and vitamin D. Whether or not nephrocalcinosis promoted the subsequent development of renal cancer is a matter of speculation, since this association has not been previously described.
Conflict of interest statement. None declared.
References
- 1.HYP Consortium A gene (PEX) with homologies to endopeptidases mutated in patients with X-linked hypophosphatemic rickets. Nat Genet. 1995;11:130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 2.Drezner MK. PHEX gene and hypophosphatemia. Kidney Int. 2000;57:9–18. doi: 10.1046/j.1523-1755.2000.00807.x. [DOI] [PubMed] [Google Scholar]
- 3.Tenenhouse HS. X-linked hypophosphataemia: a homologous disorder in humans and mice. Nephrol Dial Transplant. 1999;14:333–341. doi: 10.1093/ndt/14.2.333. [DOI] [PubMed] [Google Scholar]
- 4.PHEXdb Search Engine Site [homepage on the internet] [Updated 9 October 2007; cited 18 January 2008]. Available at www.phexdb.mcgill.ca .
- 5.Lo FS, Kuo MT, Wang CJ, et al. Two novel PHEX mutations in Taiwanese patients with X-linked hypophosphatemic rickets. Nephron Physiol. 2006;103:157–163. doi: 10.1159/000092916. [DOI] [PubMed] [Google Scholar]
- 6.Mäkitie O, Kooh SW, Sochett E. Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets. Clin Endocrinol. 2003;58:163–168. doi: 10.1046/j.1365-2265.2003.01685.x. [DOI] [PubMed] [Google Scholar]
- 7.Wu CJ, Song YM, Shue WH. Tertiary hyperparathyroidism in X-linked hypophosphatemic rickets. Intern Med. 2000;39:468–471. doi: 10.2169/internalmedicine.39.468. [DOI] [PubMed] [Google Scholar]
- 8.Verge CF, Lam A, Simpson JM, et al. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med. 1991;325:1843–1848. doi: 10.1056/NEJM199112263252604. [DOI] [PubMed] [Google Scholar]
- 9.Huiming Y, Chaomin W. Recombinant growth hormone therapy for X-linked hypophosphatemia in children. Cochrane Database Syst Rev. 2005;25:CD004447. doi: 10.1002/14651858.CD004447.pub2. [DOI] [PubMed] [Google Scholar]
- 10.Baroncelli GI, Bertelloni S, Ceccareli C, et al. Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr. 2001;138:236–243. doi: 10.1067/mpd.2001.108955. [DOI] [PubMed] [Google Scholar]
- 11.Seikaly MG, Baum M. Thiazide diuretics arrest the progression of nephrocalcinosis in children with X-linked hypophosphatemia. Pediatrics. 2001;108:e6. doi: 10.1542/peds.108.1.e6. [DOI] [PubMed] [Google Scholar]
- 12.Carpenter TO, Keller M, Schwartz D, et al. 24,25 Dihydroxyvitamin D supplementation corrects hyperparathyroidism and improves skeletal abnormalities in X-linked hypophosphatemic rickets—a clinical research center study. J Clin Endocrinol Metab. 1996;81:2381–2388. doi: 10.1210/jcem.81.6.8964881. [DOI] [PubMed] [Google Scholar]