

Abstract
We describe the renal biopsy findings in a 14-year-old girl with Neimann-Pick disease. The renal biopsy showed chronic changes involving all components of the parenchyma, including focal global glomerulosclerosis, tubular atrophy, interstitial fibrosis and vascular sclerosis. On light microscopy, significant findings included foamy podocytes, vacuolated tubular epithelial cells and collections of foam cells in the interstitium. Electron microscopy was confirmatory which showed myelin-like inclusions in podocytes, endothelial cells, tubular epithelial cells and small nerves. The findings are similar to Fabry's disease, except that small nerve involvement appears to be unique to Neimann Pick disease.
Keywords: myelin inclusions, Neimann-Pick disease, zebra bodies
Introduction
Neimann-Pick disease (NPD) is a devastating inherited condition resulting from the enzymatic deficiency of sphingomyelinase, which leads to lysosomal accumulation of sphingomyelin [1]. Two variants of this condition are recognized, types A and B. In the most common type, type A, there is progressive and marked accumulation of sphingomyelin in the spleen, liver, gastrointestinal tract, lymph nodes, bone marrow, tonsils, lungs and heart. In type A, there is also massive accumulation within the central nervous system often leading to death within 3 years of life [1].
Renal involvement of NPD has not been previously described. Many similarities exist between the renal involvement in this condition and, another glycogen storage disease, Fabry's disease. We present a case in whom renal insufficiency was thought secondary to NPD and provide a histological description.
Case report
The patient was a 14-year-old female with a history of NPD type A/B diagnosed at 6 months of age at which time the sphingomyelinase level was 0.30 U/g (<1% normal). Manifestations of her condition became apparent at 2 months of age when hepatosplenomegaly was noted. Sequestration syndrome from splenomegaly developed at 15 months of age, which led to splenectomy. End-stage liver disease with severe portal hypertension and massive ascites eventually developed by 4 years of age, and she received a cadaveric orthotopic liver transplant. At 10 years of age, she had progressive respiratory distress and fatigue. Aortic insufficiency, moderate mitral valve insufficiency and poor systolic function were discovered at age 10, and she subsequently underwent a Ross procedure and mitral valve annuloplasty. In addition to cardiac and liver manifestations of NPD, she had neurological deficits. She had global developmental delay with a developmental level of ∼3 years of age.
At 14 years of age, decreasing renal function was noted. Her creatinine gradually increased from 0.5 mg/dL after cardiac surgery at age 10 up to 1.07 mg/dL (table 1). Although her creatinine was elevated, her blood pressure was well controlled, she did not have any lower extremity oedema or weight gain, and no haematuria was detected. Her medications included cyclosporin, penicillin VK, digoxin, lisinopril, furosemide and fluoxetine. There was no reported history of nonsteroidal anti-inflammatory use. Due to worsening serum creatinine, and the concern that the worsening renal function could be secondary to cyclosporin toxicity, a renal biopsy was performed.
Renal biopsy results
The specimen submitted for light microscopy contained both renal cortex and medulla. There were six glomeruli present, of which four were globally sclerosed. Glomeruli with segmental sclerosis were not present. The Bowman's space was distended, and the podocytes appeared foamy with prominent vacuolization (Figure 1A). The mesangium showed a mild increase in the matrix like material. Proliferative or inflammatory features were not present. In addition, double contours, basement membrane spikes or pinholes were not present along the capillary walls. In tubules and interstitium, some of the tubules had a vacuolated and foamy cytoplasm. Many tubules also contained hyaline casts. Collections of foam cells along with few chronic interstitial infiltrates were also noted in the interstitium (Figure 1B). Extensive tubular atrophy and interstitial fibrosis were present. Arteries and arterioles showed mild-to-moderate sclerosis of the intima. Immunofluorescence studies were negative for immune deposits, although two glomeruli (out of seven) showed segmental coarse accumulation of C3, suggesting foal and segmental glomerulosclerosis. The ultrastructural examination of the glomeruli showed large visceral epithelial cells (podocytes) containing many myelin-like inclusions, with a lamellated appearance (zebra bodies) (Figure 2A). The myelin-like inclusions were surrounded by a single layer of the membrane indicating their presence in lysosomes. The glomerular basement membranes were thickened in many loops, and in some loops, the basement membranes showed multiple layers of the basement membrane-like material. Few cellular elements were noted entrapped within the thickened glomerular basement membranes. There was also extensive effacement of the foot processes of these cells. The endothelial cells showed focal loss of fenestrations, they did not contain myelin-like inclusions. The mesangium showed a mild increase in the matrix material. The examination of tubular epithelial cells also showed numerous myelin-like inclusions (Figure 2B). Endothelial cells of the peritubular capillaries were also noted to contain the myelin-like inclusions (Figure 2C). Finally, these were also noted in many small nerves (Figure 2D).
Fig. 1.
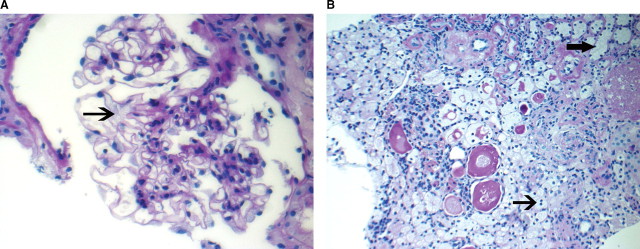
Light microscopy: Sections showing (A) glomerulus with foamy podocyte (arrow), (B) foam cells in the interstitium (thin arrow) and vacuolated foamy tubular epithelial cells (thick arrow) (×40).
Fig. 2.
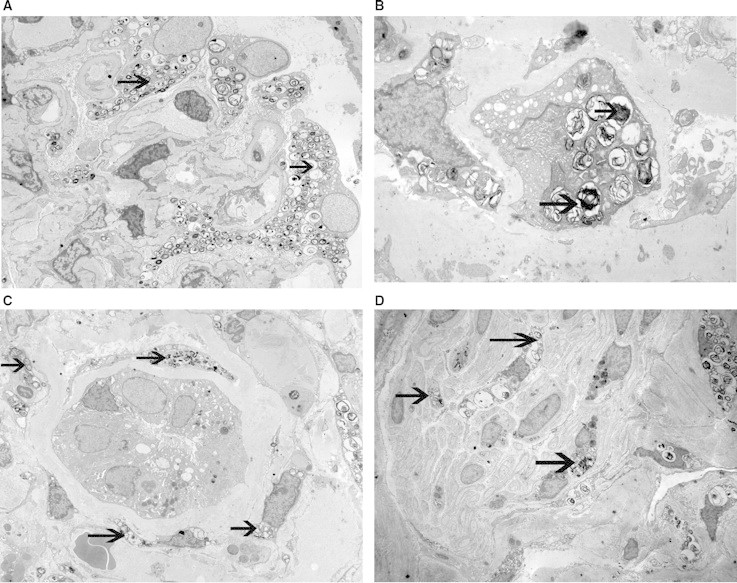
Electron microscopy showing myelin-like inclusions in (A) podocytes, (B) tubular epithelial cells, (C) endothelial cells of peritubular capillaries and (D) small nerves (A: ×2500, B: ×2500, C: ×2500, D: ×1850).
Table 1.
Laboratory values
| Serum creatinine | 1.07 mg/dL |
| Creatinine clearance (Cockcroft-Gault) | 41.6 mL/min |
| BUN | 47 mg/dL |
| Sodium | 134 mEq/L |
| Potassium | 5.2 mEq/L |
| Chloride | 105 mEq/L |
| CO2 | 22 mEq/L |
| Phosphorus | 3.9 mg/dL |
| Cyclosporin level | 197 ng/mL |
Diagnosis: Niemann-Pick disease, with focal global glomerulosclerosis, extensive tubular atrophy and interstitial fibrosis, and moderate arterial and arteriolar sclerosis.
Discussion
NPD types A and B are two related disorders more common in Ashkenazi Jews in which there is an inherited deficiency of spingomyelinase [1]. This enzymatic deficiency results in lysosomal accumulation of sphingomyelin. The original clinical description of Neimann-Pick disease was made in 1914 after an 18-month young girl with massive splenomegaly died after rapid neurologic deterioration [2]. Neimann described the first recognized clinical case, and Pick described the presence of visceral foam cells characteristic in this condition. Later, abnormal sphingomyelin accumulation in patients with this disease was discovered.
In the 1960s, NPD was divided into four subtypes, A, B, C and D, but eventually a distinct difference was made between type A, B and C. Additionally, it was found that there was no biochemical or pathological difference between types C and D other than the geographic location of patients affected; therefore, type D is no longer used for classification [2]. Although previously grouped together with NPD types A and B, NPD type C is unrelated and has a different pathogenic mechanism. In NPD-C, the primary defect involves intracellular cholesterol esterification and transport resulting in the accumulation of unesterified cholesterol [1,2].
Traditionally, NPD-A has been categorized as the severe infantile form with extensive neurological involvement leading to early death, while NPD-B results in organomegaly without severe neurological involvement. In the most common type, NPD-A, the sphingomyelinase level is typically <1% normal, while the level of acid sphingomyelinase in NPD-B is ∼10% normal [3]. There are several reports of patients with an intermediate form with neurological manifestations, where patients survive longer than the first 2–3 years of life. Thus, it seems that the neurologic manifestations in NPD A/B or acid sphingomyelinase deficiency occur along a continuum [3]. Our patient appears to have not only a more intermediate form of NPD, as she has lived into her teenage years, but also has marked neurological manifestations.
Patients with NPD typically present with massive hepatosplenomegaly with or without neurological deficits. Pulmonary and cardiac involvement has also been reported [4]. Renal involvement in NPD has not been reported in the past. In fact, clinical disease has only been rarely described in lysosomal storage diseases other Fabry's disease.
Fabry's disease, a deficiency in α-galactoside, is the classic example of a lysosomal storage disease manifesting itself in the kidney [5,6]. A renal biopsy sometimes leads to the unexpected diagnosis of Fabry's disease. In Fabry's disease, light microscopy demonstrates exceptionally large and vacuolated glomerular cells [5]. The podocytes are affected the most, but the endothelial, mesangial and parietal epithelial cells lining Bowman's capsule demonstrate these features as well. The empty appearance of these cells in paraffin-embedded sections is artefactual, as the accumulated glycosides are removed during processing. In fresh or formalin-fixed frozen sections, the accumulated material appears birefringent, autofluorescent, sudanophilic and positive to oil red O and PAS. The characteristic light microscopy findings of Fabry's disease found in the glomerulus are also found in the renal tubules and the endothelial and skeletal muscle cells of the vasculature. Interstitial foam cells are also seen. Advanced Fabry's disease leads to glomerular mesangial sclerosis, tubular atrophy, interstitial fibrosis and sclerosis of the vasculature. Immunofluorescence is negative or nonspecific. Electron microscopy shows the characteristic ‘zebra bodies’, which are enlarged lysosomes filled with granular to lamellated membrane structures readily stained with osmic acid. These ‘zebra bodies’ are most often seen in podocytes, Bowman's epithelium, distal tubular epithelium and vascular myocytes, but a few inclusions may be present in any renal cell [5,6].
The light microscopy findings in our patient share many characteristics typical of those found in patients with Fabry's disease. Light microscopy showed large vacuolated podocytes in the glomerulus, vacuolated tubular epithelial cells and collection of interstitial foam cells. Like in Fabry's disease, electron microscopy demonstrated the characteristic myelin-like inclusions and ‘zebra bodies’ in podocytes, endothelial cells and tubular epithelial cells. An interesting finding in our case was the involvement of many small nerves that contained myelin-like inclusions, explaining the many neurological manifestations of the disease. Involvement of nerves has not been documented in Fabry's disease.
Our findings are important because to the best of our knowledge renal involvement in NPD has not been described. This case illustrates that NPD can extensively involve the kidney. Renal involvement of NPD should be considered in all patients with this condition, especially in those with renal insufficiency.
Conflict of interest statement. None declared.
References
- 1.Cotran R, Kumar V, Collins T. Robbins Pathologic Basis of Disease. 6th edn. Philadelphia, PA: W. B. Saunders Company; 1999. [Google Scholar]
- 2.Patterson MC. A riddle wrapped in a mystery: understanding Niemann-Pick disease, type C. Neurologist. 2003;9:301–310. doi: 10.1097/01.nrl.0000094627.78754.5b. [DOI] [PubMed] [Google Scholar]
- 3.Wasserstein MP, Aron A, Brodie SE, et al. Acid sphingomyelinase deficiency: prevalence and characterization of an intermediate phenotype of Niemann-Pick disease. J Pediatr. 2006;149:554–559. doi: 10.1016/j.jpeds.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 4.Ferretti G, Lantuejoul S, Brambilla E, et al. Case report: pulmonary involvement in Niemann-Pick Disease subtype B: CT findings. J Comput Assist Tomogr. 1999;20:990–992. doi: 10.1097/00004728-199611000-00023. [DOI] [PubMed] [Google Scholar]
- 5.Fischer EG, Moore MJ, Lager DJ. Fabry disease: a morphologic study of 11 cases. Mod Pathol. 2006;19:1295–1301. doi: 10.1038/modpathol.3800634. [DOI] [PubMed] [Google Scholar]
- 6.Fervenza FC, Torra R, Lager DJ. Fabry disease: an underrecognized cause of proteinuria. Kidney Int. 2008;73:1193–1199. doi: 10.1038/sj.ki.5002677. [DOI] [PubMed] [Google Scholar]