

Case
A 50-year-old man was seen for chronic renal failure of unknown origin. He had been diagnosed in 1981 and monitored continuously while his renal functions had deteriorated slowly; his current glomerular filtration rate was 17 ml/min. On this occasion, he was seen by a new doctor who observed that he was hard of hearing. The day was unusually bright and sunny; hence lateral cervical scars, 3 cm in length, were noted bilaterally (Figure 1). The patient recalled that these were from surgery in childhood. On even closer examination, bilateral pre-auricular pits were also found, and the patient could express some whitish material from one of these (Figure 2). Further history taking revealed that his father and grandmother had similar pits, and that his father had died with renal failure of unknown cause. The patient had four children: three of whom had pre-auricular pits and branchial sinuses but no known renal problems. One of his daughters had died in a car accident, and her kidneys had been ‘unsuitable’ for transplantation although details were unavailable (Figure 3).
Fig. 1.
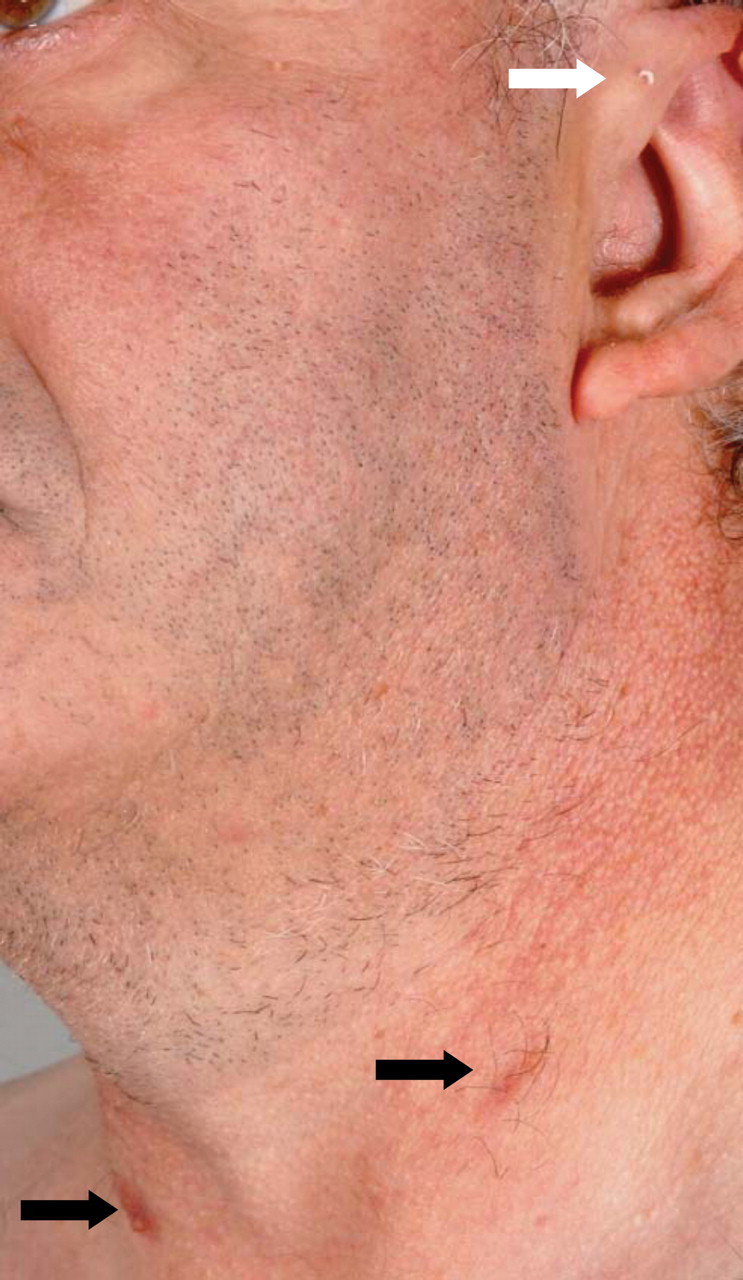
Cervical scars (black arrows) and pre-auricular pit (white arrow).
Fig. 2.
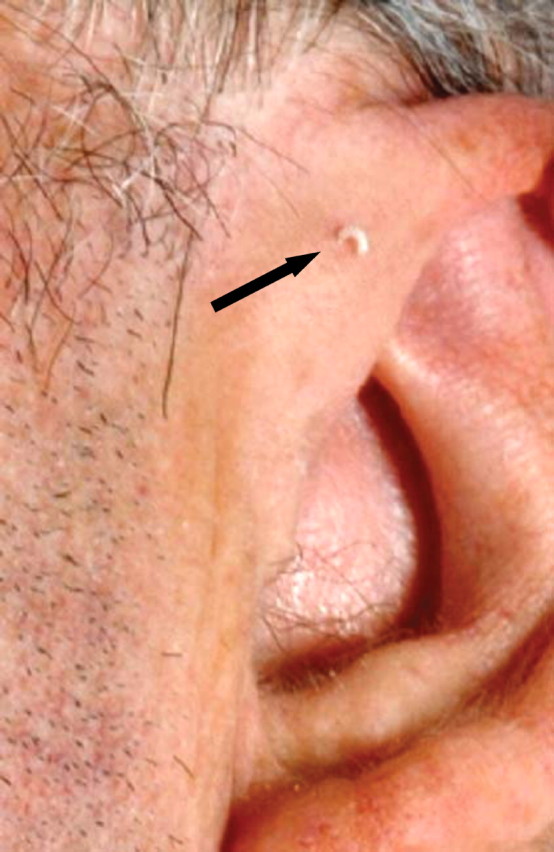
Pre-auricular pit with whitish discharge.
Fig. 3.
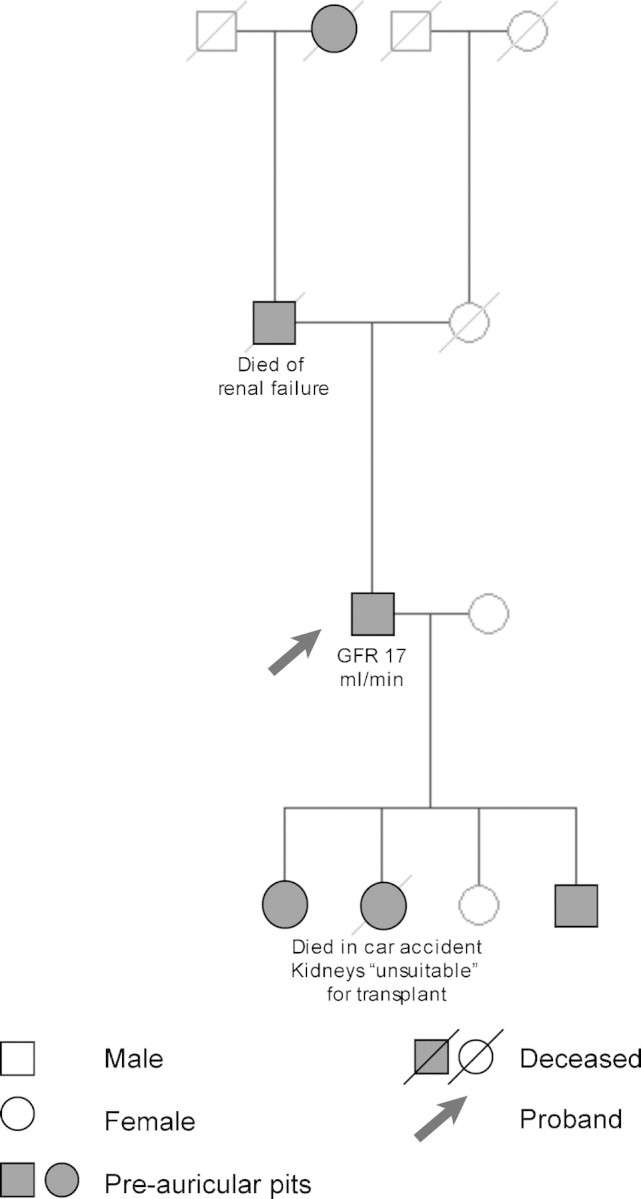
Pedigree of the patient (drawn with the Pedidraw™ online tool, http://pedidraw.gcnet.org.cn/, accessed 16 December 2008).
Question
What is the diagnosis?
Answer
The diagnosis is branchio-oto-renal (BOR) syndrome [1]. The patient was referred for genetic counselling and hearing assessment. Investigation of his children was arranged.
Discussion
BOR syndrome [Online Mendelian Inheritance in Man (OMIM) 113 650, 610 896] is a developmental disorder mainly of the second branchial arch. It is characterized by sensori-neural, conductive or mixed hearing loss, structural defects of the inner and middle ear and branchial fistulas or cysts. Other manifestations are cleft palate, gustatory lacrimation and euthyroid goitre. Renal abnormalities vary in severity, may affect one or both kidneys and include renal hypoplasia or agenesis, pelvi-ureteric junction obstruction, vesicoureteric reflux and hydronephrosis.
BOR syndrome is autosomal dominant and genetically heterogeneous [2]. Disruption of genes that are crucial to the vertebrate development is the common theme: BOR1 syndrome (OMIM 113 650) is due to mutations in the EYA1 gene, the human homologue to the Drosophila eyes absent gene [3]. The SIX gene family, a human homologue to the Drosophila sine oculis, is affected in BOR2 syndrome (OMIM 610896) [4]. Interestingly, SIX and EYA1 interact during embryonic development [5]. This may provide a clue as to why BOR 1 and 2, although genetically different, cause a similar phenotype.
A clinical diagnosis of BOR syndrome can be established if an affected individual meets at least three major, or two major and two minor criteria, or one major criteria and an affected first-degree relative (Table 1) [1]. Our patient had three major criteria and affected relatives; hence a clinical diagnosis could be made with confidence. Unfortunately, the BOR syndrome is relatively unknown in adult nephrology. The neck of patients with vesicoureteral reflux should be examined for signs of BOR syndrome. This case and a sunny day (unusual in the North West of England) provided an interesting detour into the eyes absent and sine oculis genes in Drosophila. Previous doctors literally had no eye for the very subtle pre-auricular pits and the tell-tale cervical scars.
Table 1.
Diagnostic criteria for BOR syndrome [1]
| Major criteria | Minor criteria |
|---|---|
| Pre-auricular pits | External auditory canal anomalies |
| Deafness | Middle ear anomalies |
| Branchial sinuses/cysts/fistulas | Inner ear anomalies |
| Auricular deformity | Pre-auricular tags |
| Renal anomalies | Facial asymmetry or cleft palate |
Conflict of interest statement. None declared.
References
- 1.Chang EH, Menezes M, Meyer NC, et al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat. 2004;23:582–589. doi: 10.1002/humu.20048. [DOI] [PubMed] [Google Scholar]
- 2.Sanggaard KM, Rendtorff ND, Kjaer KW, et al. Branchio-oto-renal syndrome: detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses. Eur J Hum Genet. 2007;15:1121–1131. doi: 10.1038/sj.ejhg.5201900. [DOI] [PubMed] [Google Scholar]
- 3.Orten DJ, Fischer SM, Sorensen JL, et al. Branchio-oto-renal syndrome (BOR): novel mutations in the EYA1 gene, and a review of the mutational genetics of BOR. Hum Mutat. 2008;29:537–544. doi: 10.1002/humu.20691. [DOI] [PubMed] [Google Scholar]
- 4.Kochhar A, Orten DJ, Sorensen JL, et al. SIX1 mutation screening in 247 branchio-oto-renal syndrome families: a recurrent missense mutation associated with BOR. Hum Mutat. 2008;29:565. doi: 10.1002/humu.20714. [DOI] [PubMed] [Google Scholar]
- 5.Ruf RG, Xu PX, Silvius D, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA. 2004;101:8090–8095. doi: 10.1073/pnas.0308475101. [DOI] [PMC free article] [PubMed] [Google Scholar]