

Abstract
Human trisomy 21, which results in Down syndrome (DS), is one of the most complicated congenital genetic anomalies compatible with life, yet little is known about the molecular basis of DS. It is generally accepted that chromosome 21 (Chr21) transcripts are overexpressed by about 50% in cells with an extra copy of this chromosome. However, this assumption is difficult to test in humans due to limited access to tissues, and direct support for this idea is available for only a few Chr21 genes or in a limited number of tissues. The Ts65Dn mouse is widely used as a model for studies of DS because it is at dosage imbalance for the orthologs of about half of the 284 Chr21 genes. Ts65Dn mice have several features that directly parallel developmental anomalies of DS. Here we compared the expression of 136 mouse orthologs of Chr21 genes in nine tissues of the trisomic and euploid mice. Nearly all of the 77 genes which are at dosage imbalance in Ts65Dn showed increased transcript levels in the tested tissues, providing direct support for a simple model of increased transcription proportional to the gene copy number. However, several genes escaped this rule, suggesting that they may be controlled by additional tissue-specific regulatory mechanisms revealed in the trisomic situation.
Trisomy 21 results in Down syndrome (DS), and is the most frequent aneuploidy that survives gestation. At 1 in 700 live births, it represents a major health problem affecting tens of thousands of individuals each year. Many aspects of development are affected by the inheritance of three instead of two copies of chromosome 21 (Chr21; Epstein 1995). However, the mechanisms by which dosage imbalance disrupts development are not well understood (Kahlem and Yaspo 2000). It is widely assumed that transcript levels for the ∼284 Chr21 genes (http://chr21.molgen.mpg.de/chr21_catalog9.html) are increased in DS proportionally to the gene dosage, that is, ∼50% greater than normal, but this hypothesis has never been demonstrated systematically. Although it is reasonable to postulate that transcription rates would be 50% higher with a third copy of Chr21 genes present, it is less clear that the levels of stable transcripts in a cell should be proportional to gene copy number. Many factors determine messenger RNA half-life, which is highly variable for different genes. Initial studies attempting to analyze the Chr21 transcriptome in DS have been reported recently. A global up-regulation of Chr21 genes was found in RNAs from the cerebral cortex and from astrocytic cell lines derived from DS fetuses (Mao et al. 2003). Transcriptome analysis of pooled amniocytes collected from fetuses carrying constitutive trisomy 21 showed only a small change (×1.1-fold) in individual expression levels of 79 Chr21 genes (FitzPatrick et al. 2002). However, this conclusion was based on a single array hybridization, weakening quantitative evaluation of this data set. That study highlights a central difficulty in evaluating the DS transcriptome; that is, to access sufficient amounts of the desired tissues.
Mouse models of human genetic conditions provide access to all tissues at all stages of development. The Ts65Dn mouse is an established model of trisomy 21 manifesting a number of phenotypes that parallel those seen in DS (Reeves et al. 2001). Serial analysis of gene expression (SAGE) of adult brain of Ts65Dn mice has been reported, which however gave little information on the level of expression of the Chr21 orthologous genes (Chrast et al. 2000). Only 15 triplicated genes were detected by this technique, in part because the level of expression of most known triplicated genes was too low to achieve statistical significance. Investigating temporal and spatial gene expression patterns should contribute to the understanding of their function and of their involvement in phenotypes. We previously cloned 158 orthologs to Chr21 genes (referred to herein as “mmu21” genes) and established their expression map in euploid mice by a combination of in situ hybridization and EST mining (http://chr21.molgen.mpg.de/hsa21/; Gitton et al. 2002). Here, we analyzed the transcriptome of nine tissues from adult Ts65Dn mice and euploid littermates by hybridization on cDNA arrays based on the mmu21 gene collection, confirming many results by quantitative real-time PCR (qPCR). We found that most of the mmu21 genes were expressed in the brain, in contrast to skeletal muscle, where relatively few were expressed. The majority of triplicated genes showed elevated transcript levels consistent with gene dosage (i.e., ∼50% increased), suggesting that there is little regulation of gene expression at the level of transcription. A few genes at dosage imbalance showed down-regulation, compensation, or strong overexpression, suggesting that additional regulatory mechanisms influence the transcription of these genes.
RESULTS
As a first step in understanding the molecular basis of DS pathogenesis, we performed a transcriptome analysis of a wide range of tissues from Ts65Dn mice. These mice have segmental trisomy of mouse chromosome 16, with ∼128 genes at dosage imbalance (Reeves et al. 1995). The distal boundary of the T(16;17)65Dn chromosome is the most distal gene on mouse Chr16, now known to be Znf295 (Davisson et al. 1990; Akeson et al. 2001). The proximal breakpoint was previously defined as falling between Ncam2 and Gabpa (Akeson et al. 2001), which are separated by about 3.4 Mb on MMU16. To refine this localization, we isolated 12 BAC clones from the RPCI-23 library that were distributed across the proximal breakpoint region. These were used to narrow the breakpoint region to about 100 kb by FISH analysis of metaphase spreads prepared from Ts65Dn-derived ES cells. The most distal BAC that was present in two copies and absent from the T65Dn chromosome was clone 134F19 (Fig. 1). The next clone giving a strong positive signal on the T65Dn marker chromosome was 359P19. The breakpoint is thus proximal to Mrpl39, as predicted by the elevated expression of three genes proximal to Gabpa in trisomic mice (see below).
Figure 1.

Boundaries of the proximal T(16;17)65Dn breakpoint were determined by FISH. Left: Schematic illustrating the synteny between HSA21 and MMU16, and the translocation chromosome MMU1716 with the genes at the boundaries of the triplicated segment. Chromosomal position of the two BACs used for the FISH analysis is indicated. Right: Metaphase spreads from T(16;17)65Dn mouse; BAC 134F19 (top) is in red and BAC 359P19 (bottom) is in green. Arrows point to the MMU1716 chromosome.
To compare the expression profiles of the Chr21 orthologs in euploid and trisomic mice, we designed a mouse cDNA expression array interrogating 136 mmu21 genes. Seventy-seven of these are present in three copies in Ts65Dn, from Mrpl39 to Znf295 (Fig. 1). RNA pools from four adult male Ts65Dn mice and four male euploid littermates were prepared from nine tissues (lung, skeletal muscle, midbrain, cerebellum, cortex, liver, testis, heart, and kidney) dissected from 3–4-mo-old mice. Directly labeled first-strand cDNA probes from each tissue were hybridized to the arrays in at least two independent hybridizations. Filters were scored semi-automatically, and data were analyzed with dedicated software (Steinfath et al. 2001). A total of 446 nontriplicated genes (384 unigenes and 62 mmu21 genes) served as controls (see Methods and Suppl. protocols). Data are displayed in Supplemental Table 1.
Expression of mmu21 Genes in Nine Tissues from Control Mice
We detected 112 out of 136 mmu21 genes (82%) in at least one of the nine tissues tested in control mice. Fifteen of the 136 genes (11%) were detected in all nine tissues, consistent with a role as housekeeping genes (Suppl. Table 1). Skeletal muscle showed the least molecular complexity, where we detected expression for only 15% of the mmu21 genes (Fig. 2), reflecting its relatively simple cyto-architecture. Cerebral cortex showed the highest molecular complexity, indicating that Chr21 genes play an important role in the maintenance of this tissue. Genes from all functional categories are expressed in the cortex and in the midbrain, whereas ion channels and intracellular signaling molecules are rarely expressed in the liver and skeletal muscle (Fig. 2). Among the 24 genes showing no significant signal on arrays (Suppl. Table 1), most correspond to relatively rare or spatially restricted transcripts, but three, Znf294, Gart, and Lss, were shown previously to have widespread expression (Gitton et al. 2002). We analyzed eight of these 24 genes by qPCR: Adamts5, Znf294, C21orf63, Gart, Dyrk1a, Mx1, Tff3, and Tmprss3. qPCR is more sensitive than array hybridization, and all eight genes were detected in at least one tissue (Suppl. Table 1). We confirmed that many of these were expressed at low levels, in contrast to genes detected on arrays (Suppl. Fig. 1).
Figure 2.

Functional distribution of the 136 mmu21 genes in nine tissues of euploid mice. The 136 Chr21 orthologs (mmu21) were distributed into 10 categories of biological processes (left bar; Hill et al. 2002). Bars show the functional distribution of the genes expressed in each tissue, as percent of the total number of mmu21 genes. The second left bar (all brain) represents the accumulated data from the three brain regions.
Molecular Signatures of the mmu21 Genes in Control and Trisomic Mice
We used hierarchical clustering of the array data to display the expression patterns of mmu21 genes in nine tissues of the control and trisomic mice. The relative expression level of a given gene in each tissue was estimated using the average expression value obtained for this gene across all control tissues as a reference (see Methods). The mmu21 genes exhibited characteristic expression profiles in the nine tissues tested here, which could be distinguished and clustered by their molecular signatures. These patterns organized the tissues into several distinct groups (Fig. 3). For instance, the three brain regions (cortex, cerebellum, and midbrain) clustered together, a feature that was also observed after genome-wide gene profiling (Miki et al. 2001). This might reflect, in part, similarities in their composition (e.g., neurons and astrocytes) and their common embryonic origins. Heart and skeletal muscle also shared significant similarities in patterns of gene expression. We obtained a similar order in the clustering of the tissues by analyzing the expression data of the Unigene control genes (data not shown).
Figure 3.

Hierarchical clustering showing the expression levels of mmu21 genes across nine tissues of the control mice and Ts65Dn mice. For each clone we calculated the logarithm (base 2) of the ratio between the normalized intensity in the specific tissue and the average of intensities of this clone across the nine control tissues. Spot intensities below the average of intensities across all tissues give log-ratio values ranging from –12.6 to 0, and are represented by a color gradient spanning from light to dark green. Conversely, spot intensities above the average of intensities across all tissues give log-ratio values ranging from 0 to +12.6, and are represented by a color gradient spanning from dark to light red. Thirty-one clones (24 genes) that did not show significant expression values across all control tissues were excluded from the clustering; 130 clones (112 genes; rows) and nine tissues of control and TS65Dn mice respectively (columns) were clustered using the average-linkage hierarchical clustering method with Pearson correlation as similarity measure (J-Express V 2.1; www.molmine.com). Additionally, clones with the most similar expression profiles to Dscam (with respect to the Pearson correlation) are displayed: 10% closest (13 clones, left column), 15% closest (20 clones, middle column), and 20% closest (26 clones, right column). Note that in hierarchical clustering procedures, clones with similar expression profiles can be split to different parts of the dendrogram (e.g., Olig2) and vice-versa (e.g., Abcg1). Genes referenced in the text are highlighted as follows: *, Sh3bgr; Black bars: A: Group of genes highly expressed in the kidney and the liver (from Ifnar 2 to Pttg1ip, P-value = 1.05 10–4) of controls and trisomics. B: Group of genes with predominant expression in the testis of controls and trisomics. C: Cluster of genes highly expressed in the kidney and the liver (from Col18a1 to Ftcd, P-value = 5.84 10–3) of controls and trisomics. An interactive version of Figure 3 is given in Supplemental Figure 3.
A number of genes were expressed predominantly, though not exclusively, in single tissues. For example, Sh3bgr was strongly expressed in heart and skeletal muscle as described (Egeo et al. 2000). We observed a group of three genes showing predominant expression in testis, namely Tsga2, whose gene product is involved in spermatogenesis (Taketo et al. 1997), C21orf56, and Fam3b, which may also play a role in sperm function. Indeed, C21orf56 and Tsga2 match ESTs that are derived nearly exclusively from testis (Gitton et al. 2002). Kidney and liver share two significant clusters of highly expressed genes (Fig. 3), containing several transcripts known to exert a key function in either of these tissues. For instance, Cbs catalyses the condensation of homocysteine and serine to cystathionine, mainly in liver cells (Kraus et al. 1978), and Ftcd is an enzyme linking histidine catabolism to folate metabolism that is mainly expressed in the liver as estimated by EST mining (Gitton et al. 2002). Col18a1 was reported to be highly expressed in endothelial/epithelial cells of the liver (Lietard et al. 2000), kidney, and choroid plexus (Muragaki et al. 1995). Pttg1ip is a signal transduction semaphorine-like molecule predominantly expressed in the kidney. Midbrain and cortex displayed very similar profiles of highly expressed genes, and many of these were rather weakly expressed in the cerebellum. Among genes predominantly active in the brain, Dscam is a cell surface protein acting as an axon guidance receptor (Schmucker et al. 2000). Pearson's correlation coefficient identified cohorts of genes that had expression profiles most similar to Dscam. We calculated the 10%, 15%, and 20% most similar cohorts (see Methods). The “20%” cohort includes C21orf5, C21orf66, Pcnt2, Son, Hunk, Smt3h1, Dscr3, Pfkl, Tmem1, Olig2, App, Tiam1, Synj1, Clic6, Abcg1, Hrmt1l1, Adarb1, Pcp4, S100b, Dscam, Ttc3, Kcnj6, and Kiaa0184 (Fig. 3). As a group, most of these genes are well conserved throughout evolution, are expressed during prenatal development, and demonstrate patterned expression in the brain, suggesting their relevance for the development of the central nervous system (Gitton et al. 2002). Several of these (App, Synj1, Tiam1, Dscam, S100b, Kcnj6, Olig2, Adarb1, and Pcp4) are known to play key roles in brain development and function (Reeves et al. 1994; Hess 1996; Melcher et al. 1996; Leeuwen et al. 1997; Yamakawa et al. 1998; Saito et al. 2001; Lu et al. 2002).
Figure 3 shows that tissues from trisomic mice always clustered with the corresponding euploid tissue, even for the closely matched pairs, demonstrating that transcriptome alterations in Ts65Dn are smaller than the transcriptome differences between tissues. Thus, trisomy appears to predominantly affect levels of expression but not tissue specificity of mmu21 genes.
Gene Dosage Effects in the Ts65Dn Mice
The prevalent hypothesis of gene dosage effect on transcription is that genes represented in three copies will be overexpressed by a factor of 1.5 (Epstein 1986). We observed a general trend consistent with this idea for most of the triplicated MMU16 genes, whereas most of the control genes show a Ts versus Eu ratio close to 1.0 in all tissues (Fig. 4). Assuming that the trend line for diploid genes is close to one, the level of overexpression was corrected by the value of slope obtained for UniGene controls. We found that the global overexpression of the trisomic genes was close to 1.5-fold in cortex (×1.63), heart (×1.73), testis (×1.51), and liver (×1.6), whereas somewhat lower values were seen in cerebellum (×1.37), midbrain (×1.30), lung (×1.27), and kidney (×1.23). Overexpression was collectively less pronounced in skeletal muscle (×1.16) but was still higher in trisomic than in euploid mice.
Figure 4.

Linear regression plots comparing trisomic and control samples. For each plot corresponding to a given tissue, the linear regression for the triplicated genes is in green, and that of the UniGene sample is in blue. Each clone was plotted using the average of its normalized spot intensities obtained in the replicated hybridizations, with the value in control tissue on the x-axis and with the value in Ts65Dn tissue on the y-axis. Bars show the interval [μ–σ,μ+σ] where μ and σ are the mean and standard deviation, respectively, for each data point across the replicated experiments. We excluded from the graphics outlier spots with intensities ±4 std. dev. above the mean intensity (e.g., Wrb). (S) is the slope for the regression line, (I) the intercept, and (C) the correlation. Scales range from 0–100.000, except for lung and skeletal muscle, which range from 0–50.000.
Array data identified several trisomic genes that escape the 1.5-fold overexpression rule, and a few disomic genes that were dysregulated in trisomic tissues (discussed below). To validate the array data, we analyzed 39 mmu21 genes (29 triplicated and 10 duplicated) by qPCR in several tissues (Suppl. Table 1). We observed good concordance with qPCR for 78% of the genes detected on arrays; that is, the expression ratios in trisomic versus controls were found to be within the same range by the two methods (Fig. 5; Suppl. Fig. 2, Suppl. Table 1). The array analysis was sufficiently precise that elevated expression of Atp5a, Jam2, and Mrpl39 in the trisomic mice led to a refined definition of the translocation breakpoint on the T65Dn chromosome (Fig. 1). Discordant data were most often found for moderately or weakly expressed genes, for example in skeletal muscle.
Figure 5.

Comparison of trisomic/euploid ratios obtained by real-time PCR and arrays for the three brain regions. Histograms showing the correlation between array and qPCR results for cerebellum, cortex, and midbrain. Blue bars represent mean ratios of three independent array hybridizations, and red bars represent mean ratios of two independent qPCR experiments. Error bars were calculated by averaging standard deviations of the independent experiments. Genes represented by 1–3 clones on the arrays were independently compared to the qPCR result. (*) beside a gene name indicates an array ratio (blue bar) with a significant P-value (<0.05). Correlation histograms for all tissues are available in Supplemental Figure 2.
Higher-Order Transcript Regulation in Trisomy
Trisomic genes that escaped the “1.5-fold rule” were either compensated (transcript levels not different from euploids), underexpressed, or elevated with a ratio substantially greater than 1.5, in one or more trisomic tissues. Nineteen of those genes were tested by qPCR. Nine genes were down-regulated or compensated in a single tissue while following the 1.5-fold trend in other tissues (Suppl. Table 1), suggesting some form of feedback on transcription or other mechanisms controlling transcript levels. Usp16, a ubiquitin processing protease, and Atp5o, a subunit of ATP synthase, were down-regulated in testis (×0.52 and ×0.75, respectively). Dscr5, a gene of unknown function, was down-regulated in skeletal muscle (×0.58). Sh3bgr followed the expected trend in cortex (×1.5), but was down-regulated in midbrain (×0.55). Other genes showed expression ratios close to one. C21orf7, a putative TGF-β-like activated kinase, and Tmprss2, a transmembrane serine protease, were compensated in kidney (×0.97 and ×1.02). Dscr2, Sod1, and C21orf7 were compensated in testis (×0.91, ×1.08, and ×1.15, respectively). Finally, Mrps6, a mitochondrial ribosomal protein, was compensated in skeletal muscle (×1.07).
Thirteen genes at dosage imbalance displayed a trisomic/euploid ratio >2 (Suppl. Table 1). Examples are Bace2 in testis (×3.43) and in cortex (×2.15), Dscr5 in liver (×2.75), App in skeletal muscle (×3.99), Mx2 in heart (×2.26), Il10rb in testis (×2.73), Son in lung (×2.21), and Cbr1 in kidney (×2.13). We also found increased overexpression of Kcne2 in midbrain (×3.39); Mrps6 (×2.47), Sh3bgr (×2.61); and C21orf63 (×3.93) in kidney; Fam3b in lung (×2.93); and Mx1 in skeletal muscle (×3.7) and heart (×2.7).
Although the analysis of genes mapping elsewhere in the genome was not the scope of the present study, we found several control genes showing a ratio of Ts versus Eu greater or smaller than 1, possibly reflecting potential cascade regulations or secondary effects due to the partial trisomy 16. For example, we confirmed by qPCR that two disomic mmu21 genes were dysregulated: C21orf56 in testis (×0.16) and Tff3 in liver (×17.8). Among the 384 UniGene control genes, array data indicated several dysregulated genes, such as mt-Rnr2 (16S ribosomal RNA) up-regulated in testis, Male enhanced antigen-1 down-regulated in testis, Hba-a1 (hemoglobin α chain) down-regulated in cerebellum and midbrain, and ras-related protein Ral-B, down-regulated in cortex (Suppl. Table 2).
DISCUSSION
The postulate that genes present in triplicate in the genome would be overexpressed by 50% compared with disomic genes has never been formally demonstrated in a systematic fashion, in a panel of tissues from a mammal with trisomy. As a step towards understanding the molecular consequences of trisomy, we analyzed the levels of expression of 136 mmu21 transcripts in nine tissues of Ts65Dn trisomic mice and euploid littermates. We found that 82% of the tested orthologs of human Chr21 were expressed in at least one tissue. Previous expression maps of Chr21 orthologs in mice demonstrated that 94%–98% of all mmu21 genes are expressed in adult mice, using more sensitive but nonquantitative approaches (Gitton et al. 2002; Reymond et al. 2002). Hierarchical clustering of quantitative array data in this study clearly demonstrated that the mmu21 genes are differentially expressed in the nine tested tissues, while confirming previous conclusions that housekeeping genes represent a relatively small fraction of this gene set.
Our observations suggest that global changes in Chr21 transcript levels in DS are somewhat more complicated than believed previously. As predicted, most genes at dosage imbalance in Ts65Dn mice were up-regulated by about 50%. That is, their steady-state transcript levels were directly proportional to the number of copies of the corresponding structural genes across multiple tissues, consistent with previous observations reported for several individual Chr21 genes. However, a significant fraction of genes were differentially regulated in a few tissues, suggesting additional mechanisms affecting gene expression in specific cell types. The assumption that 50% more template will result in 50% more steady-state RNA has been central to DS research for many years. Nonetheless, it is a somewhat surprising observation on several levels. First, this result indicates that there is little or no feedback to regulate steady-state mRNA levels in response to gene copy number by controlling either transcription or any of the other steps determining mRNA quantity in the cell (e.g., splicing, RNA processing, or degradation). Rather, template availability appears to determine transcript levels for a given gene, even though the actual level of transcript varies among tissues. Second, transcription for many genes is tightly regulated with regard to tissue type and developmental stage. For a number of gene products (e.g., transcription factors, cell surface receptors and their ligands, signal transduction molecules, rate-limiting enzymes in metabolic pathways), the amount of protein produced is critical to cellular and higher functions. It is also curious that RNA levels are not particularly subject to feedback or another type of regulation from an evolutionary context, given the prevalence of aneuploidy and the significant perturbations that result. Trisomy 21 is the most frequent live-born aneuploidy, and even here, only a fraction of DS conceptuses survive to term. Prenatal mortality is nearly universal for the more common human trisomies of Chr13 and 18 and for any other full chromosome trisomies.
It will be interesting to see whether organisms that are less sensitive to trisomy exert more control over transcript levels. In Drosophila melanogaster, individuals trisomic for an entire chromosome arm can survive to late stages of pupal development (Devlin et al. 1982). The transcriptional activity of many loci appears to be negatively regulated or compensated in both larvae and adult flies with autosomal trisomy (Birchler et al. 1990). A reduction in the level of gene products per gene template by one-third from the diploid quantity has been observed for several enzymes, including Sod. Concentration-dependent repressors of these loci often reside in the duplicated chromosome arms, as described for the Adh gene regulator involving a closely linked cis-acting element (Devlin et al. 1988). However, it is thought that this reflects a consequence of control mechanisms that normally operate in diploids, rather than a mechanism evolved to compensate for the genetic imbalance in trisomy (Devlin et al. 1988).
Dosage compensation has been rarely described for mammalian autosomal aneuploidies. It was recently shown that a dosage compensation mechanism occurs for the Igf2r imprinted gene in segmental mouse trisomy 17 (Vacik and Forejt 2003). Clinical reports of both paternal and maternal uniparental disomy for Chr21 indicate that there are no imprinted loci among the Chr21 genes.
The results of our quantitative analysis suggest that regulatory mechanisms that can act positively or negatively occur in some tissues. In a simple model, a repressor at dosage imbalance would have a corresponding effect on the level of expression of the gene it regulates. The results might be markedly different when the gene and regulator are in cis (both at dosage imbalance), compared to genes in trans that are present in the normal two copies when the regulator is present in three copies. The nature of such modifiers is at present unknown, although antisense transcripts and microRNAs are good candidates (Lagos-Quintana et al. 2002). Further analysis of gene expression profiles in trisomic samples will be helpful in revealing such mechanisms.
It is difficult to predict from the expression profiles which genes will show dosage sensitivity in mammalian development. As yet we have little idea of the normal range of variability in gene expression that is tolerated (or required) by the organism. Identifying dosage-dependent factors will require understanding of the molecular and cellular functions of these genes and of the biochemical mechanisms that are involved. We know far less about the phenotypic consequences of a subtle up-regulation (e.g., 1.5-fold) of a given gene than for its depletion. Morphogens such as bicoid function in a dose-dependant fashion for which an increase of 50% of the gene product has significant impact on the phenotype, albeit less drastic than the one caused by a reduced amount of the gene product in Drosophila larvae (Driever and Nusslein-Volhard 1988). We found that most triplicated genes coding for DNA binding proteins, including transcription factors, chromatin proteins, and RNA binding proteins, are overexpressed by a factor of about 1.5-fold. These factors may exert a trans-acting effect by altering the stoichiometry of regulatory complexes setting the level of gene expression on target genes located elsewhere in the genome. For example, Tff3 and C21orf56, located on MMU17 and 10, respectively (and thus not at dosage imbalance), are strongly dysregulated in Ts65Dn mice, suggesting that modifiers may be encoded on the triplicated region of MMU16. Identifying their promoter targets and analyzing the expression profiles of the whole transcriptome would be essential to reveal expected cascade effects and finally to understand whether these genes cause a dose-dependent phenotype.
The systematic analysis of mmu21 gene expression profiles in a panel of tissues contributes to an understanding of how cells and organisms respond to structural gene dosage imbalance. Considering the phenotypes of the Ts65Dn mice, further analysis of the genes expressed in brain and testis will be a first priority. For instance, Ts65Dn males are sterile by arrest of spermatogenesis. There is most probably a change in cell type composition of the sterile testis, which could cause secondary change in gene expression profiles. Although gene expression profiles of whole tissues can only reflect the mean composition of several hundreds of cell types, it is relevant to investigate the effects of trisomy in a large panel of tissues and cell types.
Our analysis shows a global up-regulation of the genes present in three copies, but also uncovered rare variations in transcript levels among trisomic genes that may indicate discrete regulatory mechanisms that become apparent in trisomy. It will be interesting to investigate whether the down-regulation or compensation effects are observed at different stages of mouse development. The consequences of differentially expressed genes and regulators at dosage imbalance is an important factor in understanding phenotypic outcomes of trisomy, which may result from compensation and down-regulation as well as overexpression of Chr21 loci.
METHODS
FISH Analysis
Two BACs from the RPCI-23 library, 134F19 and 359P19, were obtained from BACPAC Resources. One microgram of BAC DNA was directly labeled with either Spectrum Green or Spectrum Orange in a nick translation reaction (Vysis) and pre-annealed with a 10-fold excess of mouse Cot-1 DNA (Gibco). Metaphase spreads were prepared from a mouse ES cell line containing the T65Dn chromosome (Davisson and Akeson 1987) and hybridized as described (Moore et al. 1999). Following hybridization, the slides were washed with 2× SSC at 72°C for 5 min, followed with a 1-min rinse in 2× SSC at room temperature. Chromosomes were counterstained with DAPI II (Vysis) and viewed with a Zeiss Axioskop using a SenSys CCD camera (Photometrics) and Quips Smart Capture imaging software (Applied Imaging).
Total RNA Preparation and Purification
Total RNA was extracted with Trizol (Gibco) from tissues of Ts65Dn and euploid littermates males between 13 and 16 wks of age. Equal amounts of RNAs from four euploid or four trisomic animals were pooled to create the euploid or trisomic samples, respectively. RNA was treated with RNase-free Dnase I (Ambion), quantified by UV spectrophotometry, and its integrity was verified by gel electrophoresis. Identical sources of total RNA were used for the array hybridization and qPCR.
cDNA Arrays
cDNA clones for mmu21 genes (Gitton et al. 2002) were PCR-amplified and spotted in duplicate at high-density on nylon filters according to standard protocols (RZPD). Then, 384 nontriplicated UniGene mouse cDNA clones were spotted in duplicate and used for normalization purposes (accession nos. UniGene cDNAs are provided in Suppl. Table 2). Mapping information was available for 307 UniGene clones showing a homogeneous distribution across the 19 autosomes and the X chromosome (see Suppl. protocols). Filters were prehybridized at 65°C in 20 mL Church buffer (0.25 M sodium phosphate buffer pH 7.2, 1 mM EDTA, 1% bovine serum albumin, 7% SDS) supplemented with 200 μg salmon sperm DNA (Sigma) and 5 μg yeast tRNA (Sigma). First-strand cDNA probes were prepared from 10 μg DNA-free total RNA. RNA was annealed with 21 μM oligo p(dT)15 (Roche) for 10 min at 70°C and chilled on ice. Reverse transcription was carried out in 50 μL of a solution containing (50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 80 units RNAse inhibitor, 0.54 mM dATP, dTTP, dGTP, 0.32 μM dCTP, 0.26 μM alpha-33P-dCTP) with 400 units of Superscript II at 42°C for 90 min. The reaction was stopped by heating to 70°C for 15 min, and RNA was degraded in (300 mM NaOH, 0.33% SDS, 17.4 mM EDTA) at 68°C for 30 min followed by neutralization in (Tris-HCl pH 8.0 236 mM; HCl 135 mM). For blocking repetitive sequences, the radiolabeled probe was preannealed with 17 μg mouse-Cot1 DNA (Gibco) and 11 μg p(dA)40–60 (Amersham) in (0.1% SDS; 5× SSC). Probe was added to prehybridized filters, and hybridization was carried out in Church buffer at 65°C for 15 h. Two identical filters were hybridized simultaneously with each probe. Filters were washed to (0.1× SSC; 0.1% SDS) at 65°C for 20 min. Filters were exposed5dto BAS2325 screens (Fujifilm) and scanned with a BioImage Analyser BA S1800 (Fuji). Hybridizations were performed three times for all tissues except for cerebellum and heart (two times) and skeletal muscle (one time).
Data Analysis
Image analysis was carried out by a semi-automated procedure. A grid was placed manually (Visual grid, GPC Biotech) on each filter in order to determine the center of each spot. Afterwards, the pixel intensities were quantified within a predefined area around the center using a Gaussian spot shape (Steinfath et al. 2001). Data were normalized as described in the Supplementary protocols. In order to judge whether a gene is expressed in a given tissue, we incorporated a negative control signal distribution (Suppl. protocols). To evaluate differential expression of the genes, P-values of Wilcoxon's rank-sum test were used as a reference, because this distribution-free test does not require specific assumptions on the signal distributions (Herwig et al. 2001). Fold changes for trisomic versus controls were considered significant when the P-value was <0.05. Ratios were computed as mean intensities of Ts65Dn and control spots from replicated experiments and the statistical errors of these ratios were calculated to fall within the bounds
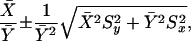 |
where X̄ and Ȳ were the averages across replicated experiments, and Sx and Sy were the respective standard errors of the means (SEM) calculated as
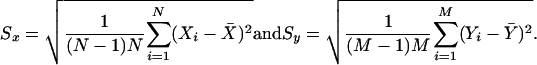 |
Reproducibility of signals across replicated experiments for each clone was measured with the coefficient of variation (CV), that is, the ratio of the mean (m), and the standard deviation (s) of these repeated signals. Clustering of gene expression profiles was done using the hierarchical method with average linkage as an update rule and Pearson correlation as a pairwise similarity measure (J-Express; www.molmine.com). In order to compute a P-value for a given group of data points, we used a straightforward resampling method (Suppl. protocols).
Real-Time Quantitative PCR
For each gene tested by qPCR, a primer pair was designed using the Primer Express software (PE Applied Biosystems). Sizes of amplicons ranged from 52 to 111 bp (Suppl. Table 3). Reverse transcription was carried out using standard conditions. In brief, 1 μg of DNase-treated total RNA sample was reverse-transcribed into first-strand cDNA using random hexamers and SuperscriptII reverse transcriptase (Invitrogen), and subsequently diluted with nuclease-free water (Promega) to 12.5 ng/μL equivalent total RNA.
Real-time PCR reactions were performed using the ABI Prism 7900HT Sequence Detection System (PE Applied Biosystems). Amplification plots and predicted threshold cycle (Ct) values (fractional cycle number at which the amount of amplified target reaches a fixed threshold) were obtained with the Sequence Detector Software (SDS version 2.0, PE Applied Biosystems). For each gene, the ratios Ts65Dn/controls were calculated using the comparative Ct method (User Bulletin #2, Applied Biosystems). The final ratios and standard deviations were calculated as the mean of the ratios and of the standard deviations of independent duplicated experiments, respectively.
Data were normalized using a geometric mean of three housekeeping genes: hypoxanthine guanine phosphoribosyl transferase (Hprt), phosphomannomutase 2 (Pmm2), and hydroxymethylbilane synthase (Hmbs). Validation experiments were carried out to assert the use of the comparative Ct method (see Suppl. protocols).
Acknowledgments
We thank G. Teltow for help with data management, and the German Resource Center for Genome Research (RZPD, Berlin) for clones and arrays. This work was supported in part by PHS awards HD24605 and HD38384 (to R.H.R.), and by grants from the EU (CT-2002-00816), the German Ministry of Education and Research (BMBF; NGFN National Genome Research Network), and the Max Planck Society to M.L.Y.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1951304.
Footnotes
[Supplemental material is available online at www.genome.org and at http://chr21.molgen.mpg.de. Array data are deposited in the Array Express database at EBI (http://www.ebi.ac.uk/arrayexpress), accession no. E-MEXP-102.]
References
- Akeson, E.C., Lambert, J.P., Narayanswami, S., Gardiner, K., Bechtel, L.J., and Davisson, M.T. 2001. Ts65Dn—Localization of the translocation breakpoint and trisomic gene content in a mouse model for Down syndrome. Cytogenet Cell Genet. 93: 270–276. [DOI] [PubMed] [Google Scholar]
- Birchler, J.A., Hiebert, J.C., and Paigen, K. 1990. Analysis of autosomal dosage compensation involving the alcohol dehydrogenase locus in Drosophila melanogaster. Genetics 124: 679–686. [PMC free article] [PubMed] [Google Scholar]
- Chrast, R., Scott, H.S., Papasavvas, M.P., Rossier, C., Antonarakis, E.S., Barras, C., Davisson, M.T., Schmidt, C., Estivill, X., Dierssen, M., et al. 2000. The mouse brain transcriptome by SAGE: Differences in gene expression between P30 brains of the partial trisomy 16 mouse model of Down syndrome (Ts65Dn) and normals. Genome Res. 10: 2006–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davisson, M.T. and Akeson, E.C. 1987. An improved method for preparing G-banded chromosomes from mouse peripheral blood. Cytogenet. Cell Genet. 45: 70–74. [DOI] [PubMed] [Google Scholar]
- Davisson, M.T., Schmidt, C., and Akeson, E.C. 1990. Segmental trisomy of murine chromosome 16: A new model system for studying Down syndrome. Prog. Clin. Biol. Res. 360: 263–280. [PubMed] [Google Scholar]
- Devlin, R.H., Holm, D.G., and Grigliatti, T.A. 1982. Autosomal dosage compensation Drosophila melanogaster strains trisomic for the left arm of chromosome 2. Proc. Natl. Acad. Sci. 79: 1200–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin, R.H., Holm, D.G., and Grigliatti, T.A. 1988. The influence of whole-arm trisomy on gene expression in Drosophila. Genetics 118: 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever, W. and Nusslein-Volhard, C. 1988. The bicoid protein determines position in the Drosophila embryo in a concentration-dependent manner. Cell 54: 95–104. [DOI] [PubMed] [Google Scholar]
- Egeo, A., Di Lisi, R., Sandri, C., Mazzocco, M., Lapide, M., Schiaffino, S., and Scartezzini, P. 2000. Developmental expression of the SH3BGR gene, mapping to the Down syndrome heart critical region. Mech. Dev. 90: 313–316. [DOI] [PubMed] [Google Scholar]
- Epstein, C.J. 1986. The consequences of chromosomal imbalance. Cambridge University Press, Cambridge, UK.
- Epstein, C.J. 1995. Down syndrome (trisomy 21). In The metabolic and molecular bases of inherited disease (ed. C.R. Scriver), pp. 749–794, McGraw-Hill, New York.
- FitzPatrick, D.R., Ramsay, J., McGill, N.I., Shade, M., Carothers, A.D., and Hastie, N.D. 2002. Transcriptome analysis of human autosomal trisomy. Hum. Mol. Genet. 11: 3249–3256. [DOI] [PubMed] [Google Scholar]
- Gitton, Y., Dahmane, N., Baik, S., Ruiz i Altaba, A., Neidhardt, L., Scholze, M., Herrmann, B.G., Kahlem, P., Benkahla, A., Schrinner, S., et al. 2002. A gene expression map of human chromosome 21 orthologues in the mouse. Nature 420: 586–590. [DOI] [PubMed] [Google Scholar]
- Herwig, R., Aanstad, P., Clark, M., and Lehrach, H. 2001. Statistical evaluation of differential expression on cDNA nylon arrays with replicated experiments. Nucleic Acids Res. 29: E117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, E.J. 1996. Identification of the weaver mouse mutation: The end of the beginning. Neuron 16: 1073–1076. [DOI] [PubMed] [Google Scholar]
- Hill, D.P., Blake, J.A., Richardson, J.E., and Ringwald, M. 2002. Extension and integration of the gene ontology (GO): Combining GO vocabularies with external vocabularies. Genome Res. 12: 1982–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlem, P. and Yaspo, M.L. 2000. Human chromosome 21 sequence: Impact for the molecular genetics of Down syndrome. Gene Funct. Dis. 1: 175–183. [Google Scholar]
- Kraus, J., Packman, S., Fowler, B., and Rosenberg, L.E. 1978. Purification and properties of cystathionine β-synthase from human liver. Evidence for identical subunits. J. Biol. Chem. 253: 6523–6528. [PubMed] [Google Scholar]
- Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. 2002. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12: 735–739. [DOI] [PubMed] [Google Scholar]
- Leeuwen, F.N., Kain, H.E., Kammen, R.A., Michiels, F., Kranenburg, O.W., and Collard, J.G. 1997. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J. Cell Biol. 139: 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietard, J., Theret, N., Rehn, M., Musso, O., Dargere, D., Pihlajaniemi, T., and Clement, B. 2000. The promoter of the long variant of collagen XVIII, the precursor of endostatin, contains liver-specific regulatory elements. Hepatology 32: 1377–1385. [DOI] [PubMed] [Google Scholar]
- Lu, Q.R., Sun, T., Zhu, Z., Ma, N., Garcia, M., Stiles, C.D., and Rowitch, D.H. 2002. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109: 75–86. [DOI] [PubMed] [Google Scholar]
- Mao, R., Zielke, C.L., Zielke, H.R., Pevsner, J. 2003. Global up-regulation of chromosome 21 gene expression in the developing Down syndrome brain. Genomics. 81: 457–467. [DOI] [PubMed] [Google Scholar]
- Melcher, T., Maas, S., Herb, A., Sprengel, R., Higuchi, M., and Seeburg, P.H. 1996. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J. Biol. Chem. 271: 31795–31798. [DOI] [PubMed] [Google Scholar]
- Miki, R., Kadota, K., Bono, H., Mizuno, Y., Tomaru, Y., Carninci, P., Itoh, M., Shibata, K., Kawai, J., Konno, H., et al. 2001. Delineating developmental and metabolic pathways in vivo by expression profiling using the RIKEN set of 18,816 full-length enriched mouse cDNA arrays. Proc. Natl. Acad. Sci. 98: 2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, C.S., Lee, J.S., Birren, B., Stetten, G., Baxter, L.L., and Reeves, R.H. 1999. Integration of cytogenetic with recombinational and physical maps of mouse chromosome 16. Genomics 59: 1–5. [DOI] [PubMed] [Google Scholar]
- Muragaki, Y., Timmons, S., Griffith, C.M., Oh, S.P., Fadel, B., Quertermous, T., and Olsen, B.R. 1995. Mouse Col18a1 is expressed in a tissue-specific manner as three alternative variants and is localized in basement membrane zones. Proc. Natl. Acad. Sci. 92: 8763–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, R.H., Yao, J., Crowley, M.R., Buck, S., Zhang, X., Yarowsky, P., Gearhart, J.D., and Hilt, D.C. 1994. Astrocytosis and axonal proliferation in the hippocampus of S100b transgenic mice. Proc. Natl. Acad. Sci. 91: 5359–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, R.H., Irving, N.G., Moran, T.H., Wohn, A., Kitt, C., Sisodia, S.S., Schmidt, C., Bronson, R.T., and Davisson, M.T. 1995. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 11: 177–184. [DOI] [PubMed] [Google Scholar]
- Reeves, R.H., Baxter, L.L., and Richtsmeier, J.T. 2001. Too much of a good thing: Mechanisms of gene action in Down syndrome. Trends Genet. 17: 83–88. [DOI] [PubMed] [Google Scholar]
- Reymond, A., Marigo, V., Yaylaoglu, M.B., Leoni, A., Ucla, C., Scamuffa, N., Caccioppoli, C., Dermitzakis, E.T., Lyle, R., Banfi, S., et al. 2002. Human chromosome 21 gene expression atlas in the mouse. Nature 420: 582–586. [DOI] [PubMed] [Google Scholar]
- Saito, T., Guan, F., Papolos, D.F., Lau, S., Klein, M., Fann, C.S., and Lachman, H.M. 2001. Mutation analysis of SYNJ1: A possible candidate gene for chromosome 21q22-linked bipolar disorder. Mol. Psychiatry 6: 387–395. [DOI] [PubMed] [Google Scholar]
- Schmucker, D., Clemens, J.C., Shu, H., Worby, C.A., Xiao, J., Muda, M., Dixon, J.E., and Zipursky, S.L. 2000. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 101: 671–684. [DOI] [PubMed] [Google Scholar]
- Steinfath, M., Wruck, W., Seidel, H., Lehrach, H., Radelof, U., and O'Brien, J. 2001. Automated image analysis for array hybridization experiments. Bioinformatics 17: 634–641. [DOI] [PubMed] [Google Scholar]
- Taketo, M.M., Araki, Y., Matsunaga, A., Yokoi, A., Tsuchida, J., Nishina, Y., Nozaki, M., Tanaka, H., Koga, M., Uchida, K., et al. 1997. Mapping of eight testis-specific genes to mouse chromosomes. Genomics 46: 138–142. [DOI] [PubMed] [Google Scholar]
- Vacik, T. and Forejt, J. 2003. Quantification of expression and methylation of the Igf2r imprinted gene in segmental trisomic mouse model. Genomics 82: 261–268. [DOI] [PubMed] [Google Scholar]
- Yamakawa, K., Huot, Y.K., Haendelt, M.A., Hubert, R., Chen, X.N., Lyons, G.E., and Korenberg, J.R. 1998. DSCAM: A novel member of the immunoglobulin superfamily maps in a Down syndrome region and is involved in the development of the nervous system. Hum. Mol. Genet. 7: 227–237. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://chr21.molgen.mpg.de; Human Chromosome 21 group home page at the MPIMG.
- http://www.ebi.ac.uk/arrayexpress; ArrayExpress database at EBI.