

Case report
A 65-year-old male pre-dialysis Turkish patient with diabetic nephropathy received a Cimino fistula of the left lower arm, which occluded before use within the following weeks after the operation. A polytetrafluorethylene (PTFE) loop was created at the left forearm. The first sessions of dialysis were successful; however, 2 weeks after starting dialysis using unfractionated heparin, the loop occluded. Platelet count dropped at this time, but not significantly. As thrombectomy of the loop was unsuccessful, a new PTFE loop at the left upper arm was created. After 2 months without any complications, the PTFE loop occluded again. A loop thrombectomy with revision of the protheto-venous anastomosis was performed initially and after 5 days, as the loop failed again. During both revisions, intraoperative angiography showed no stenosis of the central venous system draining of the left upper limb, and blood flow was well >600 mL/min. As there was no personal or family history of former thrombotic events, no examination for thrombophilic disorders was conducted at this time. Since the loop failed again after 8 days, a brachiocephalic fistula at the right arm was built. As recurrent clotting complications of the Shaldon catheters were recognized during dialysis, a tunnelled-cuffed catheter on the left side was applied to provide dialysis access until complete maturation of the fistula. In the further course, the tunnelled cuffed catheter had to be changed two times due to clotting complications and remained open only after blocking with urokinase. Platelet count did not drop significantly during each event (Figure 1).
Fig. 1.
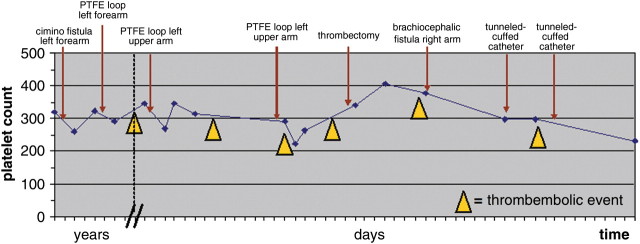
Platelet count and thromboembolic events of vascular access over the course of time.
Laboratory testing for thrombophilia risk factors was performed (Table 1). Abnormal findings included heterozygosity for the prothrombin G20210A mutation, an increased level of von Willebrand factor (vWF) (ristocetin cofactor activity: 289%; vWF antigen: 348%) and elevated plasma factor VIII coagulant activity (273%). A test for lupus anticoagulants was only weakly positive, and cardiolipin antibodies could not be detected. As there was no significant drop in platelet count and the diagnosed thrombophilias were sufficient to explain the recurrent thromboembolic events, no investigation for heparin induced thrombocytopenia antibodies (HIT antibodies) was conducted, and an anticoagulation with phenprocoumon was started. Overlapping, a full anticoagulation with certoparin adapted to body weight was given, until a stable international normalized ratio (INR) within the range of 2–3 was achieved. Dialysis was conducted with unfractionated heparin. Despite sufficient anticoagulation, recurrent clotting complications, especially recurrent thrombotic occlusion of the dialysis machine lines and dialyser, occurred during dialysis. Hence, we conducted a heparin-induced platelet aggregation assay (HIPA), which detected a heparin-dependent activation of donor platelets. Further dialysis was performed with danaparoid (initial dose of 3750 units). Further dosing had to be adapted according to the anti-Xa level. No more thrombotic events occurred from that point, and the fistula was successfully used after 8-week maturation period.
Table 1.
Established risk factors for thrombophilia and the constellation in our case [8,15]
| Pathology | Relative risk of thromboembolic events | Constellation in our case |
|---|---|---|
| Factor V Leiden mutation | 2.2-7 | - |
| Prothrombin gene mutation | 2.8 | + |
| Antithrombin III deficiency | 8.1 | - |
| Protein C deficiency | 7.3 | - |
| Protein S deficiency | 8.5 | - |
| Increased von Willebrand factor activity | * | + |
| Elevation of factor VIII:C | 4.8 | + |
| Lupus anticoagulants | * | (+) |
*no data available.
Discussion and review of literature
Vascular access thrombosis in patients undergoing maintenance haemodialysis is an important factor for morbidity and mortality. The major predisposing factor for thrombosis is anatomic stenosis, most commonly at the venous anastomoses or draining vein, being responsible for >80% of thromboses [1]. Vascular access stenosis is initiated by endothelial cell injury, which leads to endothelial and fibromuscular hyperplasia due to the upregulation of adhesion molecules on the endothelial cell surface and subsequent leucocyte adherence. Adhesion is followed by a release of chemotactic and mitogenic factors stimulating vascular smooth muscle cells to proliferate and migrate [2]. Other causes of thromboses are, among others, arterial stenoses and non–anatomic problems such as excessive post-dialysis fistula compression, hypotension and hypovolaemia, which account for ~15% of the cases [1].
Although patients on dialysis form a high-risk group due to their increased exposure to heparin, there are a few studies assessing the incidence and the impact of HIT in dialysis patients. Existing data concerning prevalence are conflicting, and it is unclear whether the presence of antibodies is associated with an increased risk for vascular access thrombosis [3,4]. Nevertheless, positive antibody testing has a significant impact on morbidity and mortality regardless of thromboembolic events [5].
There are two types of heparin-associated thrombocytopaenia. Type 1 is characterized by a fall in platelet count of <30% starting within the first 2 days of treatment with heparin, which returns to normal even under a continued administration of heparin. It is not associated with an increased risk for thromboembolism. The pathomechanism seems to be non-immune and due to a direct effect on platelet activation by inhibition of the adenylatecyclase [6].
Type 2 is the more serious immune-mediated form and is characterized by a fall in platelet count of >50% with an onset usually after the fifth day of treatment and the building of platelet-rich white clots. Atypical forms without a significant drop in platelet count have been reported, especially in patients undergoing chronic haemodialysis [7]. The incidence of emergence of HIT II after exposure to heparin ranges from <1 and up to 6.5% [8]. Longer duration of therapy, use of unfractionated heparin, surgery (especially in the case of orthopaedic surgery) and female sex are strongly associated with higher risk [9]. Pathophysiologically, the emergence of HIT antibodies is provoked by a complex of heparin and platelet factor 4 (PF4), which activates platelets through an interaction with the Fc-gamma RIIa (CD32) receptor. The activated platelets aggregate to white clots, leading to thrombopaenia and thrombosis [10]. The complex also activates endothelial cells resulting in an increased release of chemokines and adhesion molecules [11].
There are only limited data about the role of thrombophilia for thrombosis of vascular accesses in patients with end-stage renal disease. Despite the lack of prospective studies, retrospective data suggest an impact of thrombophilias for vascular access thromboses that might be more important than previously thought [12].
It is distinguished between inherited and acquired thrombophilias. Inherited thrombophilias include antithrombin, protein C and protein S deficiencies, the prothrombin gene variant, and factor V Leiden mutation, which may be defined as a poor response to the anticoagulant action of activated protein C. The latter both have the highest incidences accounting for >50% of cases, but are associated with mild thrombophilia, whereas protein C and protein S deficiencies are associated with a moderate and antithrombin deficiency with a high risk for thromboembolic events. Other conditions, which are associated with an increased thromboembolic risk, are a plasminogen and factor XII deficiency and a dysfibrinogenaemia [13].
The presented patient with end-stage renal disease experienced recurrent thrombotic events of vascular access. Thrombophilia screening revealed the prothrombin gene variant G20212A, an increased level of von Willebrand factor and elevated plasma factor VIII coagulant activity, so that anticoagulation with phenprocoumon was administered, under which the patient continued to suffer from thromboembolic events during dialysis. After detecting HIT antibodies and using danaparoid instead of unfractionated heparin for dialysis, no further thromboembolic events were recognized.
As the patient still developed thromboembolic complications even under sufficient oral anticoagulation, the presence of anti-PF4 antibodies with a positive HIPA test might have been the decisive factor for the hypercoagulable state of the patient, even in the absence of a significant drop in platelet count. The anticoagulation with phenprocoumon could be an attributing factor, predisposing to microthrombosis via protein C depletion during acute HIT.
In the clinical situation, an examination for HIT antibodies is rarely conducted and usually only in the case of a fall of platelet count. Additional factors such as surgical trauma, which are associated with an increase in platelet count, might lead to a pseudo-stable platelet count despite of clinically significant HIT. Recurrent filter clotting even in the absence of a drop of the platelet count is often associated with the presence of anti-PF4/heparin antibodies with a significant impact on haemodialysis quality [8].
The screening of asymptomatic patients for inherited thrombophilias is only recommended in individual high-risk situations, as there is only a limited benefit that could justify lifelong prophylactic anticoagulation [14].
The incidence and impact of HIT antibodies in dialysis patients have not been elucidated completely, and there are no recommendations for antibody screening. After the first recurrence of a thromboembolic event without any hint for an anatomic reason or recurrent filter clotting, it might be useful to screen not only for inherited thrombophilias but also for anti-PF4 antibodies even in the case of stable platelet count to avoid vascular access-related complications and further interventions, which is costly and an important cause of morbidity and hospitalization. An atypical HIT in patients on haemodialysis should therefore be suspected in the case of unexplained clotting complications or thrombosis and should be verified by a functional HIT antibody testing (HIPA). A positive HIT antibody ELISA without clinical manifestation is a common situation in haemodialysis patients and must be interpreted only in the clinical context. Many patients have HIT antibodies without a clinically apparent HIT. The ELISA can also be negative in the case of a positive HIPA. In this case, antibodies against a different epitope are suspected. As the HIPA in our case is positive and HIT clinically suspected, an ELISA gives no further information, neither positive nor negative.
To assess the incidence and impact of heparin-induced anti-PF4 antibodies in patients undergoing maintenance haemodialysis, further prospective investigations with a large population are necessary. In this respect, longitudinal investigations and the change of hypercoagulable states among patients on long-term haemodialysis and the fate of the access seem to be of specific interest.
Teaching points
Consider hereditary thrombophilia in the case of a recurrent thrombosis in the absence of anatomic abnormalities of haemodialysis vascular access (Figure 2).
Heparin-induced thrombocytopaenia can be an underlying cause of recurrent thrombosis in the haemodialysis patient even in the absence of a drop in platelet count.
A functional test for HIT antibodies should be preferred to prove clinical relevance.
Heparin-induced thrombocytopaenia can be clinically suspected even in the case of a negative HIPA or ELISA.
Fig. 2.

Diagnostic approach to recurrent thrombosis of recurrent haemodialysis access.
Conflict of interest statement. None declaired
References
- 1.Schwab SJ, Harrington JT, Singh A, et al. Vascular access for haemodialysis. Kidney Int. 1999;55:2078–2090. doi: 10.1046/j.1523-1755.1999.00409.x. [DOI] [PubMed] [Google Scholar]
- 2.Stracke S, Konner K, Köstlin I, et al. Increased expression of TGF-beta1 and IGF-I in inflammatory stenotic lesions of haemodialysis fistulas. Kidney Int. 2002;61:1011–1019. doi: 10.1046/j.1523-1755.2002.00191.x. [DOI] [PubMed] [Google Scholar]
- 3.O'Shea SI, Sands JJ, Nudo SA, et al. Frequency of anti-heparin-platelet factor 4 antibodies in haemodialysis patients and correlation with recurrent vascular access thrombosis. Am J Hematol. 2002;69:72–73. doi: 10.1002/ajh.10032. [DOI] [PubMed] [Google Scholar]
- 4.Nakamoto H, Shimada Y, Kanno T, et al. Role of platelet factor 4-heparin complex antibody (HIT antibody) in the pathogenesis of thrombotic episodes in patients on haemodialysis. Hemodial Int. 2005;9:2–7. doi: 10.1111/j.1542-4758.2005.01163.x. [DOI] [PubMed] [Google Scholar]
- 5.Peña dela Vega L, Miller RS, Benda MM, et al. Association of heparin-dependent antibodies and adverse outcomes in haemodialysis patients: a population-based study. Mayo Clin Proc. 2005;80:995–1000. doi: 10.4065/80.8.995. [DOI] [PubMed] [Google Scholar]
- 6.Henríquez F, Rodríguez A, Mora C, et al. Heparin-induced thrombocytopenia in haemodialysis. Case report and review of the literature. Nefrologia. 2006;26:734–737. [PubMed] [Google Scholar]
- 7.Lasocki S, Piednoir P, Ajzenberg N, et al. Anti-PF4/heparin antibodies associated with repeated haemofiltration-filter clotting: a retrospective study. Crit Care. 2008;12:84. doi: 10.1186/cc6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood. 2005;106:2710–2715. doi: 10.1182/blood-2005-04-1546. [DOI] [PubMed] [Google Scholar]
- 9.Warkentin TE, Eikelboom JW. Who is (still) getting HIT? Chest. 2007;131:1620–1622. doi: 10.1378/chest.07-0425. [DOI] [PubMed] [Google Scholar]
- 10.Castelli R, Cassinerio E, Cappellini MD, et al. Heparin induced thrombocytopenia: pathogenetic, clinical, diagnostic and therapeutic aspects. Cardiovasc Hematol Disord Drug Targets. 2007;7:153–162. doi: 10.2174/187152907781745251. [DOI] [PubMed] [Google Scholar]
- 11.Blank M, Shoenfeld Y, Tavor S, et al. Anti-platelet factor 4/heparin antibodies from patients with heparin-induced thrombocytopenia provoke direct activation of microvascular endothelial cells. Int Immunol. 2002;14:121–129. doi: 10.1093/intimm/14.2.121. [DOI] [PubMed] [Google Scholar]
- 12.Knoll GA, Wells PS, Young D, et al. Thrombophilia and the risk for haemodialysis vascular access thrombosis. J Am Soc Nephrol. 2005;16:1108–1114. doi: 10.1681/ASN.2004110999. [DOI] [PubMed] [Google Scholar]
- 13.Tripodi A, Mannucci PM. Laboratory investigation of thrombophilia. Clin Chem. 2001;47:1597–1606. [PubMed] [Google Scholar]
- 14.Middeldorp S, Meinardi JR, Koopman MM, et al. A prospective study of asymptomatic carriers of the factor V Leiden mutation to determine the incidence of venous thromboembolism. Ann Intern Med. 2001;135:322–327. doi: 10.7326/0003-4819-135-5-200109040-00008. [DOI] [PubMed] [Google Scholar]
- 15.Martinelli I, Mannucci PM, De Stefano V, et al. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood. 1998;92:2353–2358. [PubMed] [Google Scholar]