

Abstract
A transcriptional regulatory network encompasses sets of genes (regulons) whose expression states are directly altered in response to an activating signal, mediated by trans-acting regulatory proteins and cis-acting regulatory sequences. Enumeration of these network components is an essential step toward the creation of a framework for systems-based analysis of biological processes. Profile-based methods for the detection of cis-regulatory elements are often applied to predict regulon members, but they suffer from poor specificity. In this report we describe Regulogger, a novel computational method that uses comparative genomics to eliminate spurious members of predicted gene regulons. Regulogger produces regulogs, sets of coregulated genes for which the regulatory sequence has been conserved across multiple organisms. The quantitative method assigns a confidence score to each predicted regulog member on the basis of the degree of conservation of protein sequence and regulatory mechanisms. When applied to a reference collection of regulons from Escherichia coli, Regulogger increased the specificity of predictions up to 25-fold over methods that use cis-element detection in isolation. The enhanced specificity was observed across a wide range of biologically meaningful parameter combinations, indicating a robust and broad utility for the method. The power of computational pattern discovery methods coupled with Regulogger to unravel transcriptional networks was demonstrated in an analysis of the genome of Staphylococcus aureus. A total of 125 regulogs were found in this organism, including both well-defined functional groups and a subset with unknown functions.
Micro-organisms respond rapidly to changing conditions by activating programs of gene expression. The discovery of the regulatory networks involved in these adaptations is one of the grand challenges in modern molecular biology. As such, high-throughput laboratory approaches have been widely used to profile patterns of gene expression and detect potential target sites for sequence-specific DNA-binding transcription factors (Cao et al. 2002; Conway and Schoolnik 2003). In parallel, computational methods are increasingly applied to identify potential regulatory networks (Shen-Orr et al. 2002; Mwangi and Siggia 2003). Successful bioinformatics methods for elucidating the networks have generally used a two-step process. In the first stage, classes of cis-regulatory elements (cis-REs) in an organism are identified—most commonly by pattern-discovery methods. Subsequently, sets of genes that potentially constitute regulons are defined as those genes that contain instances of a cis-RE pattern within their regulatory regions.
Identification of classes of transcription-factor binding sites and other cis-REs involved in gene expression is central to computational studies of gene regulation. Given a set of coregulated genes, statistical methods can be used to extract cis-REs on the basis of their overrepresentation in the regulatory regions (Blanchette and Tompa 2002; Aerts et al. 2003; Zheng et al. 2003). These initial sets of coregulated genes may be obtained by compiling gene-specific experimental studies, or as output from genome-scale screens. As a direct result of the increasing pool of genome sequences, such sets are increasing constituted by orthologous genes under the assumption that orthologs across species of moderate evolutionary distance remain subject to the control of the same cis-RE. The latter method, called phylogenetic footprinting, has proven effective in studies of transcriptional regulation in Escherichia coli and related γ-proteo-bacteria (McGuire et al. 2000; Laikova et al. 2001; McCue et al. 2001; Panina et al. 2001, 2003; Rajewsky et al. 2002; Panina et al. 2003), bacteria from the Bacillus/Clostridium cluster (Rodionov et al. 2001; Terai et al. 2001) and Archea (Gelfand et al. 2000a).
Putative regulons are defined as sets of genes containing cis-REs in their regulatory regions. Motif models obtained in the pattern discovery phase are used to scan a genome to detect all genes containing the putative cis-REs. The algorithms used to define these sets are recognized to generate numerous false predictions, even with specific models for cis-REs (Gelfand et al. 2000b) and optimized parameter settings (Robison et al. 1998). On the basis of the principles that motivated phylogenetic foot-printing, several methods have been developed to enhance the specificity of regulon predictions. Tan et al. (2001) assigned higher confidence values to predicted Crp and FNR regulon members of E. coli that had orthologs in predicted Crp and FNR regulons in Haemophilus influenzae (Tan et al. 2001). Manson McGuire and Church (2000) predicted regulons on the basis of conserved operons and used the presence of a shared regulatory site as additional evidence for their regulon prediction. Rodionov et al. (2002a) used the idea of conserved regulons to direct site searches with known motifs to members of conserved regulons. These qualitative studies indicated the potential power of conservation analysis to improve regulon predictions.
In this study, we describe Regulogger, an algorithm that uses comparative genome analysis to increase the accuracy of regulon predictions. Regulogger builds on the assumption that predicted regulon members are more reliable when orthologous genes contain similar cis-REs. To quantify the degree of conservation of the regulatory signal, Regulogger calculates for each predicted regulon member a relative conservation score (RCS) on the basis of the fraction and number of orthologs that share the same cis-RE. Regulon members that do not have a conserved cis-RE are considered false-positive predictions, whereas members that have orthologs with the same cis-RE are considered true positives. Application of Regulogger to a predicted regulon thus produces a set of genes whose sequence and regulatory signal is conserved across multiple genomes. Such a set is defined as a regulog (Fig. 1). A quantitative assessment of Regulogger, using 48 transcription factors from E. coli, revealed a greater than fivefold increase in specificity of regulon predictions without significant sensitivity decrease. To demonstrate the utility of Regulogger in combination with phylogenetic footprinting to reveal regulatory networks, we applied the method to the human pathogen Staphylococcus aureus. The results quantitatively demonstrate that regulon conservation analysis, as implemented in Regulogger, is a powerful method to study and discover regulatory networks.
Figure 1.

Outline of the Regulogger method. First a putative regulon in the target genome (Genome A) is predicted by searching the entire genome for genes with a particular cis-RE in their upstream region. This predicted regulon in genome A is shown at the top. Regulogger identifies regulons in other genomes (B, C, D, and E) that are regulated by the same cis-RE. On the basis of the fraction of orthologs in other genomes (indicated in this figure by the same letter) that are regulated by the same cis-RE, a relative conservation score (RCS) is calculated. The RCS is shown above the genes in the final regulog. The height of the box for each gene correlates to the RCS for that gene, and thus indicates the confidence of the predictions. Predicted regulon members that have an RCS of 0 are regarded as false-positive predictions and are not present in the final regulog.
RESULTS
Phylogenetic Footprinting
Selection of Input Data and Settings for Phylogenetic Footprinting
The first step in the analysis of transcriptional regulatory networks is the definition of a set of sequences involved in transcriptional regulation (Fig. 2). To this end, we used phylogenetic footprinting—the detection of conserved patterns in upstream sequences of orthologous genes in related genomes. The selection of genomes for phylogenetic footprinting, shown in Figure 3, was based on two criteria. First, the genomes should contain sufficient orthologs with the proteins of the biological target, S. aureus, to allow for the identification of most cis-REs. Second, the diversity in the upstream sequences of orthologous genes should be sufficiently high to ensure that conserved patterns are not based on a subset of highly similar genome sequences. The latter criteria is based on the observation that, for highly similar sequences, conservation patterns are more likely to be produced by chance rather than by retained biological function (McCue et al. 2002; Rajewsky et al. 2002). Most of the orthologs to S. aureus genes were found in Bacillus subtilis, Bacillus halodurans, and Listeria monocytogenes (Table 1). A total of 1818 S. aureus proteins (71% of all annotated proteins) had at least one ortholog across the set of studied organisms. The average identity of the alignments of the orthologous regulatory regions was only 49%, which allows for the identification of functional sequences as conserved spots in a randomly mutated background (McCue et al. 2002).
Figure 2.

Schematic representation of the strategy to identify regulogs in S. aureus. From the genomic sequence, protein-coding regions are identified. For all proteins, orthologs in other genomes are defined. These ortholog sets are used for phylogenetic footprinting, in which Gibbs sampling is run on upstream regions of sets of orthologous genes to obtain putative regulatory motifs (e.g., binding sites). Low-scoring patterns are filtered and patterns with similar sequences are clustered. For each pattern, the putative regulon in S. aureus is defined. These predicted regulons are filtered with the Regulogger method described in Figure 1. This produces a set of regulogs, conserved regulons, in S. aureus.
Figure 3.

Phylogenetic relationship of the organism used in this study, based on the 16sRNA sequence. The genomes of B. subtilis, B. halodurans, and L. monocytogenes were used for application Regulogger to the regulons in S. aureus. The genomes of E. coli, Y. pestis, V. cholerae, H. influenzae, and P. aeruginosa were used for validation of Regulogger.
Table 1.
Characteristics of Species That Were Used for Phylogenetic Footprinting With S. aureus
|
Average identity between aligned upstream regions of orthologous genesa,b
|
Number of orthologs with S. aureus (total no. of ORFs in genome)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Target genome | ||||||||
| Query genome | Spyg | Lmon | Saur | Llac | Spne | Bhal | Bsub | |
| Spyg | 100 | 48 | 50 | 50 | 52 | 47 | 47 | 1180 (1697) |
| Lmon | 49 | 100 | 51 | 50 | 50 | 48 | 49 | 1493 (2846) |
| Saur | 50 | 51 | 100 | 51 | 50 | 48 | 49 | — (2594) |
| Llac | 50 | 50 | 51 | 100 | 51 | 47 | 48 | 1302 (2267) |
| Spne | 50 | 48 | 49 | 49 | 100 | 46 | 46 | 1204 (2094) |
| Bhal | 47 | 48 | 48 | 48 | 48 | 100 | 49 | 1523 (4066) |
| Bsub | 48 | 49 | 49 | 48 | 48 | 49 | 100 | 1593 (4112) |
Because the sets of orthologous upstream sequences were based on the COG classification of the genes (see Methods), the composition of the sets between two genomes depends on which of the two genomes is used as the query genome and which is used as the target genome. This asymmetrical definition of orthologous upstream sequences leads to an asymmetrical calculated average identity between genomes.
The standard deviation from the mean was, in all cases, around 10%.
Phylogenetic Footprinting Applied to the BSUB Study Set
The application of a Gibbs sampling algorithm to detect conserved patterns has been described by McCue et al. (2001). In that study, orthologous gene sequences from the gram-negative γ-proteobacteria, having an average G+C of around 50%, were used. To validate whether the algorithm with our particular settings would perform well on the gram-positive set used in this study, having a low G+C content of typically around 35%, we validated the phylogenetic footprinting procedure using a reference set of transcription factors from B. subtilis, the BSUB set. This reference set was obtained from data in the DBTBS database (Ishii et al. 2001) as described in the Methods section. For each transcription factor of the BSUB set, the genes in B. subtilis known to be subject to its control were collected and orthologous regulatory regions extracted. Gibbs sampling was applied to identify putative cis-REs. The resulting pattern was compared with the BSUB-binding profile, yielding a P-value for similarity. The P-value for the alignment score was obtained by aligning the pattern for the transcription factor with 200 patterns obtained with randomly chosen ortholog sets. Of the detected patterns, 24% matched to the reference profile with P < 0.05. The patterns that score with a higher P-value are not necessarily spurious, as they may correspond to alternative cis-REs, which are bound by different trans-acting factors. We anticipate, on the basis of these results, that analysis of the S. aureus genes with orthologs will produce patterns for most functional cis-REs.
Phylogenetic Footprinting Applied to the Genome of S. aureus
To identify a set of putative cis-REs in S. aureus, we analyzed overrepresented patterns in the promoters of 1818 sets of orthologous proteins using the genomes shown in Table 1. This genome-wide analysis yielded 1430 putative cis-REs. To partially distinguish functionally relevant patterns from those present by chance, we compared the observed average maximum a posteriori (MAP) values (see Methods) with the distribution of average MAP-values for 500 sets of random sequences with the same length distribution. These random sequences were created such that they had, on average, an identity of 49% in an alignment, which is equal to the average identity of the sequences in the real data set (Table 1). The resulting data (Fig. 4) demonstrate that patterns detected in orthologous regulatory regions have, on average, a higher significance than patterns that are obtained from random sequences. From the distribution of average MAP-values obtained with the sets of random sequences, we selected a minimum average MAP-value threshold of 1.5. Application of this threshold to the set of 1430 patterns yielded 318 significant patterns. These were regarded as putative cis-REs and used for further analysis. At this threshold, 97% of the patterns obtained from the random sequences are discarded.
Figure 4.

Phylogenetic footprinting on the genome of S. aureus. (Gray) The distribution of the average MAP-values that were obtained by performing Gibbs sampling on orthologous regulatory regions; (Black) the distribution of scores that were obtained using randomized upstream cis-REs with the same AT content, length, and average identity as the real orthologous regulatory regions.
As a first-step to investigate the properties of the putative cis-REs, the 318 derived matrices from S. aureus were clustered on the basis of their similarity. The analysis produced 125 clusters with distinct regulatory motifs. In a comparison of the derived motifs with profiles from the BSUB reference set, 43 motifs matched one or more of the known transcription-factor binding profiles. The remaining 82 patterns may thus contain potentially novel regulatory motifs from S. aureus.
Identification of Putative Regulons
To define sets of potentially coregulated genes, all operons of the subject gram-positive genomes (Table 1) were searched for putative regulatory sites in their upstream region by use of the 318 binding profiles generated with the phylogenetic footprinting procedure. The best match was determined for each profile in each promoter, and the corresponding P-value was calculated on the basis of randomized data. The frequency of scores was determined for a range of P-value cut-off values for the matrix score. Using a conservative threshold (P < 0.05), the average regulon size was equal to 4.5% of the number of ORFS in the genome. For B. subtilis and E. coli, this percentage would correspond to a regulon size of 150 genes. Analysis of the known B. subtilis regulons of the BSUB reference set showed that regulons consist, on average, of 0.25% of all ORFS, which translates to an average of 10 ORFS per regulon. This is consistent with the analysis of McCue et al. (2002), who compiled a set of 453 experimentally verified genes regulated by a total of 48 transcription factors of E. coli. In our ECO reference set, which partly overlaps the set used by McCue et al. (2002), the average regulon size is 16.7 ORFS. The number of genes in the predicted regulons is almost an order of magnitude larger than laboratory-based reference sets, indicating that a large fraction of the predictions consist of false positives.
Identification of Regulogs
Validation of Regulogger
To discriminate true regulon members from spurious predictions, we developed Regulogger, a method that filters out false positives on the basis of the conservation of regulons across multiple genomes. Application of Regulogger to predicted regulons produces sets of genes for which the regulatory signal is conserved across genomes—the regulogs (Fig. 1).
To assess the impact of Regulogger on predictive accuracy, we analyzed a reference set of 48 transcription factors of E. coli, the ECO set, for which binding sites and corresponding regulon members are known (see Methods). With each transcription factor from the ECO set, the genome sequences of E. coli and four additional γ-proteobacteria (Vibrio cholerae, Pseudomonas aeruginosa, Haemophilus influenzae, and Yersinia pestis) were analyzed to identify putative regulon and regulog members.
Because the main object of site searching combined with Regulogger is to detect true positive regulon members that can be targeted with experiments, we evaluated the accuracy of the methods with respect to the Positive Predictive Value (PPV) statistic (given by the ratio of the number of true positives vs. the number of predictions) and the sensitivity (Sn), given by the ratio of the number of true positives to the number of known positives. A more common definition of accuracy is given by the ratio of correct predictions (the number of true positives plus true negatives) to the total number of predictions. This definition of accuracy is uninformative, as the number of true negatives is orders of magnitude higher than the number of true positives, yielding accuracies close to 1, even with high false-positive rates and low sensitivities.
The efficiency of Regulogger (EfREG) was assessed by comparing the predictions on the basis of the site search and Regulogger against the reference regulons of E. coli. The EfREG was then calculated as follows:
 |
An EfREG exceeding 1 indicates that the regulog prediction is more accurate than regulon prediction. For 33 of 48 (69%) transcription factors, an EfREG exceeding 1 was obtained. The average EfREG for the entire ECO set was 4.2 (Table 2). The potential power of Regulogger is demonstrated by the results obtained for the pdh regulog. Application of Regulogger removed 98 of 101 false positives, while retaining the true positives, leading to a 25-fold increased PPV with the same sensitivity. The average regulon size decreased by an order of magnitude from 174 to 20 after application of Regulogger, which is close to the average regulon size of 16.7 in the ECO set. The average PPV of the predictions increased 5.3-fold (from 3.8% to 20%), with only a modest 1.7-fold decrease in sensitivity (from 53% to 32%). No correlation between the specificity (information content) or length of the matrices and the Regulogger efficiency was found (data not shown), indicating that Regulogger is efficient for a variety of matrices. When validated on the BSUB reference set, Regulogger gave comparable results with an average EfREG of 4.5 and EfREG ≥ 1 for 47% of the factors. The results of the latter analysis are available on our Web site at http://regulogs.cgb.ki.se/REGULOGS.
Table 2.
Predicted Regulons and Regulogs for 48 Transcription Factors of E. coli, the ECO set
|
TF
|
Known members
|
Predicted members
|
Sensitivity
|
PPV
|
EfREG
|
|||
|---|---|---|---|---|---|---|---|---|
| REGULON | REGULOG | REGULON | REGULOG | REGULON | REGULOG | |||
| pdhR | 4 | 102 | 4 | 0.25 | 0.25 | 0.01 | 0.25 | 25.5 |
| ilvY | 2 | 182 | 12 | 1 | 1 | 0.01 | 0.17 | 15.17 |
| oxyR | 5 | 137 | 11 | 0.6 | 0.6 | 0.02 | 0.27 | 12.45 |
| torR | 4 | 77 | 7 | 1 | 1 | 0.05 | 0.57 | 11 |
| metR | 4 | 218 | 21 | 0.75 | 0.75 | 0.01 | 0.14 | 10.38 |
| tyrR | 11 | 134 | 8 | 0.73 | 0.55 | 0.06 | 0.75 | 9.42 |
| nagC | 10 | 231 | 25 | 0.4 | 0.4 | 0.02 | 0.16 | 9.24 |
| glpR | 8 | 191 | 16 | 1 | 0.88 | 0.04 | 0.44 | 9.14 |
| iclR | 4 | 249 | 28 | 0.5 | 0.5 | 0.01 | 0.07 | 8.89 |
| malT | 10 | 156 | 8 | 0.6 | 0.4 | 0.04 | 0.5 | 8.67 |
| Irp | 29 | 252 | 17 | 0.14 | 0.1 | 0.02 | 0.18 | 8.34 |
| galR | 5 | 123 | 18 | 1 | 1 | 0.04 | 0.28 | 6.83 |
| modE | 3 | 81 | 12 | 1 | 1 | 0.04 | 0.25 | 6.75 |
| argR | 10 | 182 | 20 | 0.7 | 0.6 | 0.04 | 0.3 | 6.69 |
| cpxR | 12 | 307 | 50 | 0.17 | 0.17 | 0.01 | 0.04 | 6.14 |
| trpR | 12 | 72 | 3 | 0.33 | 0.17 | 0.06 | 0.67 | 6 |
| phoB | 23 | 199 | 22 | 0.22 | 0.17 | 0.03 | 0.18 | 5.79 |
| rpoE | 27 | 144 | 14 | 0.26 | 0.19 | 0.05 | 0.36 | 5.25 |
| fis | 22 | 233 | 20 | 0.14 | 0.09 | 0.01 | 0.1 | 5.18 |
| metJ | 5 | 93 | 12 | 1 | 0.8 | 0.05 | 0.33 | 4.96 |
| fur | 20 | 265 | 47 | 0.8 | 0.7 | 0.06 | 0.3 | 4.32 |
| lexA | 16 | 163 | 26 | 0.75 | 0.56 | 0.07 | 0.35 | 3.53 |
| arcA | 56 | 224 | 13 | 0.38 | 0.16 | 0.09 | 0.69 | 3.16 |
| fadR | 6 | 91 | 20 | 0.67 | 0.5 | 0.04 | 0.15 | 2.56 |
| gcvA | 4 | 110 | 6 | 0.75 | 0.25 | 0.03 | 0.17 | 2.04 |
| ompR | 11 | 152 | 9 | 0.27 | 0.09 | 0.02 | 0.11 | 1.88 |
| flhC | 33 | 95 | 13 | 0.24 | 0.12 | 0.08 | 0.31 | 1.83 |
| fruR | 13 | 193 | 19 | 0.77 | 0.31 | 0.05 | 0.21 | 1.63 |
| narL | 48 | 209 | 39 | 0.5 | 0.27 | 0.11 | 0.33 | 1.57 |
| purR | 27 | 203 | 48 | 0.74 | 0.44 | 0.1 | 0.25 | 1.52 |
| dnaA | 3 | 223 | 38 | 0.67 | 0.33 | 0.01 | 0.03 | 1.47 |
| fnr | 72 | 229 | 39 | 0.26 | 0.12 | 0.08 | 0.23 | 1.32 |
| rpoN | 26 | 265 | 38 | 0.27 | 0.12 | 0.03 | 0.08 | 1.28 |
| soxS | 7 | 206 | 23 | 0.43 | 0.14 | 0.01 | 0.04 | 1 |
| ada | 4 | 195 | 15 | 0 | 0 | 0 | 0 | 1 |
| marR | 8 | 185 | 24 | 0.38 | 0.12 | 0.02 | 0.04 | 0.86 |
| crp | 156 | 542 | 106 | 0.45 | 0.16 | 0.13 | 0.24 | 0.65 |
| araC | 9 | 132 | 13 | 0.56 | 0.11 | 0.04 | 0.08 | 0.41 |
| hns | 12 | 166 | 15 | 0.17 | 0 | 0.01 | 0 | 0 |
| cynR | 4 | 137 | 12 | 0.25 | 0 | 0.01 | 0 | 0 |
| cytR | 10 | 154 | 4 | 0.4 | 0 | 0.03 | 0 | 0 |
| hipB | 2 | 140 | 10 | 1 | 0 | 0.01 | 0 | 0 |
| cysB | 17 | 114 | 6 | 0.12 | 0 | 0.02 | 0 | 0 |
| lacl | 3 | 180 | 10 | 0.33 | 0 | 0.01 | 0 | 0 |
| cspA | 2 | 107 | 7 | 0.5 | 0 | 0.01 | 0 | 0 |
| fhlA | 14 | 169 | 6 | 1 | 0 | 0.08 | 0 | 0 |
| melR | 3 | 55 | 3 | 0.67 | 0 | 0.04 | 0 | 0 |
| deoR | 6 | 118 | 11 | 0.33 | 0 | 0.02 | 0 | 0 |
The predicted regulons were obtained by applying a site search to the genome of E. coli with a threshold score of P < 0.05. The regulogs were obtained by applying Regulogger to the obtained regulons. The genomes that were used for filtering were Yersinia pestis, Pseudomonas aeruginosa, Haemophilus influenzae, and Vibrio cholerae. The efficiency of Regulogger (EfREG) was calculated by comparing the specificity and sensitivity of the regulon and regulog predictions as described in the text.
Minimal Dependence of Regulogger on Parameters
Three parameters potentially influence regulog predictions, that is, the site-score threshold, the conservation threshold, and the selection of genomes. The performance of Regulogger for the ECO set was assessed by plotting Receiver Operator Characteristics (ROC) curves, using different cut-off scores for the site-score threshold and conservation score. In an ROC curve, the sensitivity is plotted against the false-positive rate, and shows the tradeoff between sensitivity and specificity for a given method. Accurate methods are indicated by curves in the top left part of the ROC space, whereas curves close to the diagonal indicate less accurate methods.
The highest accuracy of Regulogger was obtained when a site-score threshold between P < 0.02 and P < 0.05 was used for constructing the regulons, with a maximum for P < 0.04. The accuracy for Regulogger decreased when regulons were constructed with the most stringent site-score threshold of P < 0.002 or with site scores of P > 0.1. However, even at these threshold values, the curves are well above the diagonal.
For site-score thresholds around P < 0.04, the false-positive rate is between 7% and 9% when the lowest threshold for the RCS, 0.25, is used. This value of the RCS corresponds to one of four orthologs having a binding site in common with the query gene. When only fully conserved binding sites are considered, that is, an RCS of 1, the false-positive rate decreases to ∼1.5%, with a decrease in sensitivity to 18%.
The EfREG for the range of potential setting of the site score threshold (between P < 0.01 and P < 0.05), robustly remained between 3 and 4.5 (Fig. 5). The maximum EfREG of 4.5 was obtained at a site-score threshold of P < 0.04. At this threshold, an EfREG ≥ 1.0 was obtained for 73% of the transcription factors.
Figure 5.

(A) Efficiency of Regulogger at different site-score thresholds used to predict regulons. Regulogger efficiency (EfREG) for the individual transcription factors was calculated on the basis of the sensitivity and specificity of the regulon and regulog predictions as described in the text. (B) ROC curve showing the sensitivity vs. false-positive rate of Regulogger. The ROC curves were calculated with different settings of the site-score threshold as indicated in the legend. The numbers in the figure indicate the various cut-off values for the RCS. The leftmost point in each curve corresponds to the most stringent cut off (RCS = 1); the rightmost point of each curve corresponds to an RCS of 0.25.
The above results were obtained by considering all genes that were reciprocal best hits to each other as orthologs. The influence of a more stringent or a more relaxed definition of orthologs on the EfREG was tested by applying Regulogger using two alternative ortholog definitions. A relaxed definition classified all proteins with a BLASTP expectation score below a defined threshold as orthologs. As a stringent definition, orthologs had to be reciprocal best hits with a BLASTP expectation score below a specified threshold. With both definitions, regulogs for a series of BLASTP expectation score thresholds were computed and compared with the regulog predictions on the basis of reciprocal best hits alone. Using the relaxed definition for regulogs, the average EfREG for the ECO set ranged from 3.15 (BLASTP expectation score threshold of 1e–5) to 4.2 (threshold of 1e–40). The latter efficiency score matches the value obtained when the regulog calculation was based on reciprocal best hits. Using a more stringent definition for regulog members, by imposing a BLASTP expectation score threshold in addition to the reciprocal best hit requirement, the same results were shown as the regulog definitions based on reciprocal best hits alone.
The choice of species can influence the performance of Regulogger in two ways. If the number of identifiable orthologs is small, the accuracy of Regulogger can decline. Furthermore, the evolutionary distance to E. coli of the respective genomes may impact Regulogger efficiency. Table 3 shows the average EfREG for the ECO set with all combinations of the E. coli genome with one or more subject genomes (Table 3). The percentage of transcription factors for which an EfREG of >1 was observed increased with an increasing number of genomes. It is noteworthy that Regulogger still produces improved predictions for 57% of the transcription factors when only two genomes (E. coli and Y. pestis) are analyzed. The average EfREG was maximal (4.92) when the genomes of Y. pestis and V. cholerae were used and dropped when the genomes of the more distantly related H. influenzae and P. aeruginosae (Fig. 3) were added to the genome set.
Table 3.
Regulogger Efficiency With Different Genome Sets
| Genomes used for Regulogger | Average EfREG | Percentage of factors with EfREG ≥ 1 |
|---|---|---|
| Ecol, Ypes, Paer, Hinf, Vcho | 4.18 | 0.73 |
| Ecol, Ypes, Vcho, Hinf | 4.44 | 0.69 |
| Ecol, Ypes, Vcho, Psae | 4.22 | 0.71 |
| Ecol, Vcho, Hinf, Psae | 4.14 | 0.58 |
| Ecol, Ypes, Hinf, Psae | 3.68 | 0.71 |
| Ecol, Ypes, Vich | 4.92 | 0.65 |
| Ecol, Vich, Hinf | 4.49 | 0.54 |
| Ecol, Ypes, Hinf | 3.73 | 0.62 |
| Ecol, Ypes, Psae | 3.37 | 0.69 |
| Ecol, Vcho, Psae | 3.61 | 0.54 |
| Ecol, Hinf, Psae | 3.09 | 0.44 |
| Ecol, Ypes | 3.77 | 0.58 |
| Ecol, Vcho | 3.72 | 0.42 |
| Ecol, Hinf | 2.60 | 0.33 |
| Ecol, Psae | 1.95 | 0.31 |
The regulons of E. coli to which Regulogger was applied were obtained by performing a site search with the 48 matrices from the ECO reference set using a threshold score of P < 0.05. The average Regulogger efficiency (EfREG) was calculated as described in the text.
The above results indicate that Regulogger is a robust filtering method. Although the efficiency may be fine-tuned, Regulogger is not critically dependent on parameters and provides good results for a range of genomes, site scores, and conservation thresholds.
Regulogger Applied to Regulons of S. aureus
Phylogenetic footprinting identified 318 cis-REs in the genome of S. aureus. Corresponding regulons were predicted using a site-score cut off of P < 0.03. To construct regulogs, Regulogger was applied to the genomes of S. aureus, B. subtilis, B. halodurans, and L. monocytogenes. For each regulog, the RCS was calculated. As some orthologous sets of genes produced similar cis-RE models, we merged regulogs derived with similar matrices (as scored by the described matrix comparison metric). This yielded a final set of 125 regulogs for S. aureus, and the matrix models for the associated classes of cis-REs. The 15 highest-scoring regulogs are reported (Table 4), and the full list containing 125 regulogs is available on our Web site. Within the predicted regulogs, members are ranked according to the RCS.
Table 4.
Regulogs Identified in S. aureus


The regulogs were constructed by performing Regulogger analysis on regulons in S. aureus using the genomes of B. subtilis, L. monocytogenes, and B. halodurans. The regulons in S. aureus were obtained by performing a site search with the matrices obtained by phylogenetic footprinting using a site score threshold P < 0.03. The regulogs are sorted on the basis of their scores, which represent average relative conservation score (RCS) of the regulog members. The regulogs with the highest scores are thus composed of members for which the regulatory signal is highly conserved across multiple genomes. The function of the regulogs was determined by assigning a functional category to each gene of S. aureus on the basis of the function of its ortholog in B. subtilis. The functional categories for the genes of B. subtilis were taken from the SubtilList webserver (http://genolist.pasteur.fr/SubtiList/). The functional category with the highest degree of over-representation is shown. For regulogs that were merged on the basis of the similarity of their corresponding matrices, the score of the highest scoring regulog is given.
Several of the predicted regulogs are consistent with characterized regulons. The highest scoring regulog consists of genes involved in nitrogen assimilation. In B. subtilis, TnrA activates the nrgAB operon and represses the glnRA operon in response toglutamine depletion by binding to a TGTNAN7TNACA consensus sequence (Wray Jr. et al. 2000). Other examples of regulogs that are controlled by known sequence-specific transcription factors include the fur regulog (nr. 9; Xiong et al. 2000), the fnr regulog (nr. 7; Nakano and Zuber 1998), the sos regulog (nr. 15; Hamoen et al. 2001), and the ctsr regulog (nr. 14; Kruger and Hecker 1998). Several known regulons subject to the regulation by stem-loop structures of RNA are found, in agreement with earlier studies (Terai et al. 2001). An example of this type of regulation is provided by the regulog containing the aminoacyl-tRNA synthetases (nr. 6). These synthetases belong to the family of T-box proteins. The transcription of these genes is regulated by a termination–antitermination system, in which distinct stem loops are formed dependent on the binding of charged or uncharged tRNA (Grundy et al. 1997). A similar mechanism, in which RNA secondary structures are formed in response to changing levels of thiamine, regulates the genes in the thi regulog, involved in thiamine synthesis (nr. 4 and nr. 5). A recent study showed that the thi-element, which is central to this mechanism, is highly conserved in both eubacteria and archaea (Rodionov et al. 2002b). A high-scoring regulog without an associated transcription factor, the nrd regulog (nr. 3) comprises genes that are involved in reduction of ribonucleotides. The regulation of these genes in E. coli is complicated and involves binding of Fis and DnaA, and the presence of an A/T-rich sequence upstream of a 45-bp inverted repeat (Jacobson and Fuchs 1998). The cis-RE of the regulog is an A/T-rich, nonpalindromic sequence, indicating that some of the regulatory mechanisms that are found in E. coli may also play a role in regulation in S. aureus. The remaining regulogs (e.g., nr. 2, nr. 3, nr. 8, nr. 10, nr. 11, and nr. 12) are novel—a corresponding transcription factor or experimentally verified regulatory sequence is not yet known.
Expanding Known Regulogs: The fur Regulog
The fur regulog was detected with a high RCS. The Fur regulatory protein regulates transcription of target operons that are involved in iron uptake via a 19-bp GATAATGATAATCATTATC consensus sequence, the Fur box. The fur regulon (Fig. 6) is well characterized in S. aureus (Xiong et al. 2000; Sebulsky and Heinrichs 2001), B. subtilis (Baichoo et al. 2002), and gram-negative bacteria (Panina et al. 2001). The predicted regulog includes known constituent genes, such as katA, sirA, sirB, and the fhuABG and ahp operons (Morrissey et al. 2000; Sebulsky et al. 2000; Horsburgh et al. 2001; Sebulsky and Heinrichs 2001). Additional regulog members, which have been previously linked to fur, but for which Fur regulation has not been experimentally verified, include proteins that play a role in iron uptake, such as rhizobactin siderophore biosynthesis proteins, thiodoxin reductase, and ferrichrome ABC transporters. A total of 20 regulog members are annotated as either unknown or conserved hypothetical proteins (Kuroda et al. 2001), eight of which have the maximum RCS of 1. These proteins are predicted with high confidence to be novel members of the fur regulog and are candidates for targeting with experimental methods. For one of these proteins, SA0997, a homolog in S. aureus RN6390 was shown to be iron regulated and able to bind human transferrin as a means of obtaining iron (Taylor and Heinrichs 2002).
Figure 6.

Schematic representation of the predicted fur regulog. The logo represents the pattern that was obtained by phylogenetic footprinting. The arrows that connect the pattern and the groups of genes under the control of the pattern indicate the relative conservation score of the gene, the thickest arrows belonging to the most-conserved genes, for which we thus have a high confidence that they belong to the regulog. Ovals indicate known members in S. aureus. Rectangles indicate genes that may be suspected to be in the Fur regulog, on the basis of their sequence similarity to proteins in other organisms that have been shown to be regulated by Fur. Hexagonal boxes represent new members of the regulog. This means that they are predicted by Regulogger to be regulated by Fur, but no experimental evidence exists.
DISCUSSION
Regulon conservation analysis is a potent tool for enhancing computational discovery of transcriptional networks. Building on methods for comparative genome analysis (Manson McGuire and Church 2000; Rodionov et al. 2001; Tan et al. 2001) we developed Regulogger, a robust quantitative method for the identification of conserved regulons. Regulogger offers improved performance, as it supports the discovery of regulons with various degrees of conservation and produces quantitatively ranked results. The later quantitative results facilitate targeting of experiments for cases of high confidence.
The impact of Regulogger and regulatory conservation analysis will ultimately be defined by the specificity of predictions for regulon members. Application of Regulogger to cis-RE models for 48 transcription factors of E. coli yielded a significant fivefold increase in the specificity of predicted regulon members. The actual specificity may be substantially higher than reported, as the annotated sets of genes constituting known regulons are incomplete.
The specificity will improve with better operon predictions. We defined operons with a simple heuristic—genes in the same orientation with an intergenic distance of <50 bp. This heuristic failed to place sirC with sirA and sirB in the fur regulog in S. aureus. The intergenic distance between sirC and the adjacent sirB gene is 113 bp. Operon predictions based on colocalization in multiple genome sequences have been described (Yada et al. 1999; Ermolaeva et al. 2001, Moreno-Hagelsieb and Collado-Vides 2002; Sabatti et al. 2002; Zheng et al. 2002; Bockhorst et al. 2003), which may further enhance the quality of the regulon analysis methods.
The choice of species impacts the performance of Regulogger. Efficient filtering requires genomes to be sufficiently distant to the target genome and to one another to distinguish functional sequences as overrepresented in the promoters of orthologous genes. However, a balance must be made to ensure that a sufficient number of orthology relationships can be established. For example, regulon members in the hipB regulon in the ECO set had no defined orthologs in the genomes analyzed with Regulogger. Furthermore, the regulatory sites associated with a regulon must be retained between species. As an example of a limitation in TF-binding specificity, the LexA protein in E. coli recognizes a different target sequence than the LexA ortholog in gram-positive bacteria (Winterling et al. 1997). For specific transcription factors, for which knowledge about the evolution of the binding site and the corresponding regulon is available (Madan Babu and Teichmann 2003), the choice of genomes could be adjusted to achieve optimal efficiency with Regulogger.
Incorporating a measure of evolutionary distance between subject organisms may be advantageous. One of the strengths of Regulogger is that it quantitatively ranks regulog members according to an RCS. This ranking facilitates the discrimination of functionally relevant regulogs and regulog members for experimental study. In calculation of the RCS, evolutionary distance between organisms was not taken into account. A regulog that is conserved across a wide evolutionary distance may be more significant than regulogs conserved in two closely related species, which should motivate future algorithmic advances.
The human pathogen, S. aureus, was analyzed with Regulogger to accelerate regulatory network discovery. The analysis yielded 125 regulogs associated with distinct cis-RE, which is less than the 129 putative regulatory proteins in S. aureus (Kuroda et al. 2001). The list of putative cis-REs presented in Table 4 is not comprehensive. Furthermore, some of the regulogs have overlapping gene sets and are clearly regulated by the same regulatory mechanism. For example, two separate parts of the 38-bp long cis-RE for the genes belonging to the thi box family (Miranda-Rios et al. 2001) were separately identified (regulogs 4 and 5 in Table 4). Only 71% of the genes of S. aureus had one or more identified orthologs, and were thus amenable to phylogenetic footprinting. Analysis of the regulatory regions of the remainder of those genes may well yield additional motifs. Furthermore, the set of patterns may be enlarged by using different parameter settings in the Gibbs sampling procedure or by incorporating alternative pattern detection methods.
Comparative genome analysis has been demonstrated to be a powerful tool for deciphering regulatory networks. The regulon conservation algorithm, Regulogger, will accelerate the computational discovery of regulogs and assist laboratory characterization efforts through its quantitative ranking of regulog members.
METHODS
Genomes
The genomes of the following organisms were used in this study (abbreviations used are given in parenthesis): Bacillus subtilis (Bsub), Bacillus halodurans (Bhal), Streptococcus pneumoniae (Spne), Lactococcus lactis (Llac), Staphylococcus aureus (Saur), Lysteria monocytogenes (Lmon), Streptococcus pyogenes (Spyo), Escherichia coli (Ecol), Yersinia pestis (Ypes), Pseudomonas aeruginosa (Paer), Haemophilus influenzae (Hinf), and Vibrio cholerae (Vcho). The genomic sequences were downloaded from GenBank (http://www.ncbi.nlm.nih.gov).
Collection of Orthologous Regulatory Regions
A set of orthologous regulatory regions was identified by grouping regulatory regions for genes that code for orthologous proteins. As the definition of orthology relationships between genes remains an area of intense research and some controversy, we have elected to apply the term on the basis of the annotations in a widely used resource. For each query protein, a set of orthologous proteins was obtained using the COGs (clusters of orthologous groups) database, [http://www.ncbi.nlm.nih.gov/COG/ (Tatusov et al. 2001)]. In the cases when multiple proteins from the same genome were present in a COG, the protein with the highest BLASTP score with the query protein was defined as the ortholog.
To obtain the orthologous regulatory regions, the noncoding sequence of, at most, 250 bp upstream of the gene was collected. The sequence was truncated at the edge of upstream adjacent genes or, in the case of a head–head configuration, not allowed to enter the region 50 bp upstream of the adjacent gene. For genes within an operon, the upstream sequence of the first gene in the operon was taken as the regulatory region. Operons were defined as sets of genes transcribed in the same direction with an intergenic distance of <50 bp (Moreno-Hagelsieb and Collado-Vides 2002).
Pattern Detection
To detect conserved cis-REs in the dissimilar orthologous regulatory regions, we used the Gibbs motif sampler (Thompson et al. 2003). The Gibbs sampler was configured to detect zero, one, or two instances of the same pattern in either the forward or reverse strand of a sequence. The width was kept constant at 16 bp, and palindromicity of the sites was not required, in order to prevent a bias toward sites that are bound by dimeric proteins. The Gibbs sampler calculates for each pattern a maximum a posteriori (MAP) value, which is the probability of the pattern compared with a background model. Because the MAP-value is positively correlated with the number of sequences that contribute to the patterns (Sandelin et al. 2003), an average MAP-value, obtained by dividing the total MAP-value by the number of sequences that contributed to the pattern, was used to evaluate the significance of the patterns (McCue et al. 2002). To obtain the most significant pattern from a sequence set, the Gibbs sampling algorithm was run 10 times on each sequence set, and the pattern with the highest average MAP-value was retained as the putative cis-RE.
Construction of Reference Sets of Transcription Factors from Bacillus subtilis and Escherchia coli
We collected two sets of transcription factors, for which both the transcription-factor binding site and an experimentally verified regulon has been described. These sets were used to validate the phylogenetic footprinting and Regulogger methods. The BSUB set consisted of transcription factors of B. subtilis that were obtained from the DBTBS database (http://elmo.ims.u-tokyo.ac.jp/dbtbs/; Ishii et al. 2001). This data set is a compilation of binding sites for a total of 89 transcription factors and σ factors from B. subtilis. We selected transcription factors for which at least two nonoverlapping binding sites were known, and constructed PFMs for these transcription factors by performing Gibbs sampling on the known binding sequences. The Gibbs sampler was run with varying widths and with multiple cycles for each width. The pattern with the highest MAP-value was retained as the matrix for that specific factor. The final BSUB set consisted of 31 transcription factors. A second set consisted of transcription factors from E. coli and was constructed as follows. A set of PFMs for 68 transcription factors and σ factors was downloaded from the DPInteract database (http://arep.med.harvard.edu/dpinteract; Robison et al. 1998). A set of experimentally verified regulons was downloaded from http://www.weizmann.ac.il/mcb/UriAlon/; Shen-Orr et al. 2002). Combining both data sets yielded a final set of 48 transcription factors for E. coli, the ECO set. A full description of the transcription factors from the ECO and BSUB set can be found on our Web site.
Clustering of the Patterns Obtained by Phylogenetic Footprinting
Clustering of the set of patterns that were obtained by phylogenetic footprinting on the genome of S. aureus, the SAUREUS set, was done as follows. First, all 318 matrices of the SAUREUS set were aligned to each other and scored on the basis of their similarity by use of a Needleman-Wunsch algorithm that was modified to align matrices (Sandelin et al. 2003). The distribution of the scores approximates a normal distribution with a mean (m) of 0.54 and a standard deviation (sd) of 0.08. We then used an UPGMA algorithm to cluster the matrices together using a threshold for the score of m + 2.5 × sd. To determine whether clusters from the SAUREUS set were similar to patterns from the BSUB set, all matrices of each cluster were scored against the matrices from the BSUB set. A P-value was obtained on the basis of the distribution of scores obtained by comparing the BSUB matrix against a set of random matrices. This set of 16-bp wide matrices was generated by concatenating random columns from the matrices of the SAUREUS set. A cluster was regarded to be similar to a BSUB matrix when the average score between the BSUB matrix and the matrices of the cluster was below P < 0.01.
Identification of Regulons
To identify coregulated genes, the regulatory regions were searched for the presence of a high-scoring match to the (putative) cis-REs using a site-search method implemented in the TFBS modules (Lenhard and Wasserman 2002). In this method, a positional frequency matrix (PFM) representing a consensus sequence is converted to a positional weight matrix (PWM), which is used to score the sequence according to the scoring system of Berg and Von Hippel (Berg 1988). A P-value for this score was computed from the score distribution obtained with the PWM applied to 1000 randomized sequences with the same length and AT content as the original sequence. A regulon was then defined by the collection of genes containing significant motifs detected with the PWM using the indicated P-value threshold.
Construction of Regulogs
Regulogs were constructed by filtering the predicted regulons using Regulogger. This was done as follows: For each predicted regulon member, a RCS was calculated. The RCS is a measure of conservation of a gene and its corresponding regulatory site across multiple genomes and was calculated as follows.
If geneA is a regulon member predicted to be under the control of the cis-RE S, the RCS is given by:
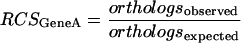 |
In this equation, orthologsobserved is the number of orthologs that are present in orthologous regulons in other genomes, that is, orthologs that are under the control of the same cis-RE. The term orthologsexpected is the total number of orthologs present in the genomes that are used with Regulogger.
For example, assume that the RCS is calculated for geneA in a particular genome, and that this gene has an ortholog in five genomes used in the analysis. If, in three of the five orthologous regulons, the ortholog to geneA is found, the RCS is 0.6. An RCS of 1 thus signifies a completely conserved presence of the regulatory signal upstream of orthologs across the genomes, whereas an RCS of 0 means a total separation of gene sequence and regulatory signal.
For the calculation of the RCS of an entire regulog, the average RCS of all predicted regulog members in the genomes used for the filtering was taken. To avoid circularity in calculation of the RCS for the entire regulog, genes that were used to construct the pattern for the regulating sequence of the regulog were not used for scoring.
Software and Data Resources
All sequence and matrix manipulations were performed with perl scripts using the Bioperl (Stajich et al. 2002) and TFBS modules (Lenhard and Wasserman 2002). The Gibbs sampler algorithm was obtained from the Lawrence group (Thompson et al. 2003; http://bayesweb.wadsworth.org/gibbs/gibbs.html). Alignment of orthologous regions was performed using ClustalX (Chenna et al. 2003). All described data are available from our Web site at http://regulogs.cgb.ki.se/REGULOGS.
Acknowledgments
We thank Albin Sandelin for advice on motif comparisons and Alex Hromockyj for early discussions about Regulogs. This work was financially supported by funds from the Karolinska-Pharmacia Genomics Collaboration and a Marie-Curie fellowship MCFI-2002-01638 to W.B.L.A.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2242604.
References
- Aerts, S., Thijs, G., Coessens, B., Staes, M., Moreau, Y., and De Moor, B. 2003. Toucan: Deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res. 31: 1753–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baichoo, N., Wang, T., Ye, R., and Helmann, J.D. 2002. Global analysis of the Bacillus subtilis Fur regulon and the iron starvation stimulon. Mol. Microbiol. 45: 1613–1629. [DOI] [PubMed] [Google Scholar]
- Berg, O.G. 1988. Selection of DNA binding sites by regulatory proteins: The LexA protein and the arginine repressor use different strategies for functional specificity. Nucleic Acids Res. 16: 5089–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette, M. and Tompa, M. 2002. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 12: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockhorst, J., Craven, M., Page, D., Shavlik, J., and Glasner, J. 2003. A Bayesian network approach to operon prediction. Bioinformatics 19: 1227–1235. [DOI] [PubMed] [Google Scholar]
- Cao, M., Kobel, P.A., Morshedi, M.M., Wu, M.F., Paddon, C., and Helmann, J.D. 2002. Defining the Bacillus subtilis sigma(W) regulon: A comparative analysis of promoter consensus search, run-off transcription/macroarray analysis (ROMA), and transcriptional profiling approaches. J. Mol. Biol. 316: 443–457. [DOI] [PubMed] [Google Scholar]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T.J., Higgins, D.G., and Thompson, J.D. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31: 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, T. and Schoolnik, G.K. 2003. Microarray expression profiling: Capturing a genome-wide portrait of the transcriptome. Mol. Microbiol. 47: 879–889. [DOI] [PubMed] [Google Scholar]
- Ermolaeva, M.D., White, O., and Salzberg, S.L. 2001. Prediction of operons in microbial genomes. Nucleic Acids Res. 29: 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand, M.S., Koonin, E.V., and Mironov, A.A. 2000a. Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res. 28: 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand, M.S., Novichkov, P.S., Novichkova, E.S., and Mironov, A.A. 2000b. Comparative analysis of regulatory patterns in bacterial genomes. Brief Bioinform. 1: 357–371. [DOI] [PubMed] [Google Scholar]
- Grundy, F.J., Haldeman, M.T., Hornblow, G.M., Ward, J.M., Chalker, A.F., and Henkin, T.M. 1997. The Staphylococcus aureus ileS gene, encoding isoleucyl-tRNA synthetase, is a member of the T-box family. J. Bacteriol. 179: 3767–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamoen, L.W., Haijema, B., Bijlsma, J.J., Venema, G., and Lovett, C.M. 2001. The Bacillus subtilis competence transcription factor, ComK, overrides LexA-imposed transcriptional inhibition without physically displacing LexA. J. Biol. Chem. 276: 42901–42907. [DOI] [PubMed] [Google Scholar]
- Horsburgh, M.J., Ingham, E., and Foster, S.J. 2001. In Staphylococcus aureus, fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol. 183: 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, T., Yoshida, K., Terai, G., Fujita, Y., and Nakai, K. 2001. DBTBS: A database of Bacillus subtilis promoters and transcription factors. Nucleic Acids Res. 29: 278–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, B.A. and Fuchs, J.A. 1998. Multiple cis-acting sites positively regulate Escherichia coli nrd expression. Mol. Microbiol. 28: 1315–1322. [DOI] [PubMed] [Google Scholar]
- Kruger, E. and Hecker, M. 1998. The first gene of the Bacillus subtilis clpC operon, ctsR, encodes a negative regulator of its own operon and other class III heat shock genes. J. Bacteriol. 180: 6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., Cui, L., Oguchi, A., Aoki, K., Nagai, Y., et al. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357: 1225–1240. [DOI] [PubMed] [Google Scholar]
- Laikova, O.N., Mironov, A.A., and Gelfand, M.S. 2001. Computational analysis of the transcriptional regulation of pentose utilization systems in the γ subdivision of Proteobacteria. FEMS Microbiol. Lett. 205: 315–322. [DOI] [PubMed] [Google Scholar]
- Lenhard, B. and Wasserman, W.W. 2002. TFBS: Computational framework for transcription factor binding site analysis. Bioinformatics 18: 1135–1136. [DOI] [PubMed] [Google Scholar]
- Madan Babu, M. and Teichmann, S.A. 2003. Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res. 31: 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson McGuire, A. and Church, G.M. 2000. Predicting regulons and their cis-regulatory motifs by comparative genomics. Nucleic Acids Res. 28: 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue, L., Thompson, W., Carmack, C., Ryan, M.P., Liu, J.S., Derbyshire, V., and Lawrence, C.E. 2001. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 29: 774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue, L.A., Thompson, W., Carmack, C.S., and Lawrence, C.E. 2002. Factors influencing the identification of transcription factor binding sites by cross-species comparison. Genome Res. 12: 1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire, A.M., Hughes, J.D., and Church, G.M. 2000. Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res. 10: 744–757. [DOI] [PubMed] [Google Scholar]
- Miranda-Rios, J., Navarro, M., and Soberon, M. 2001. A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc. Natl. Acad. Sci. 98: 9736–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Hagelsieb, G. and Collado-Vides, J. 2002. A powerful non-homology method for the prediction of operons in prokaryotes. Bioinformatics 18: S329–S336. [DOI] [PubMed] [Google Scholar]
- Morrissey, J.A., Cockayne, A., Hill, P.J., and Williams, P. 2000. Molecular cloning and analysis of a putative siderophore ABC transporter from Staphylococcus aureus. Infect. Immun. 68: 6281–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwangi, M.M. and Siggia, E.D. 2003. Genome wide identification of regulatory motifs in Bacillus subtilis. BMC Bioinformatics 4: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, M.M. and Zuber, P. 1998. Anaerobic growth of a “strict aerobe” (Bacillus subtilis). Annu. Rev. Microbiol. 52: 165–190. [DOI] [PubMed] [Google Scholar]
- Panina, E.M., Mironov, A.A., and Gelfand, M.S. 2001. Comparative analysis of FUR regulons in γ-proteobacteria. Nucleic Acids Res. 29: 5195–5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panina, E.M., Vitreschak, A.G., Mironov, A.A., and Gelfand, M.S. 2003. Regulation of biosynthesis and transport of aromatic amino acids in low-GC Gram-positive bacteria. FEMS Microbiol. Lett. 222: 211–220. [DOI] [PubMed] [Google Scholar]
- Rajewsky, N., Socci, N.D., Zapotocky, M., and Siggia, E.D. 2002. The evolution of DNA regulatory regions for proteo-γ bacteria by interspecies comparisons. Genome Res. 12: 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison, K., McGuire, A.M., and Church, G.M. 1998. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J. Mol. Biol. 284: 241–254. [DOI] [PubMed] [Google Scholar]
- Rodionov, D.A., Mironov, A.A., and Gelfand, M.S. 2001. Transcriptional regulation of pentose utilization systems in the Bacillus/Clostridium group of bacteria. FEMS Microbiol. Lett. 205: 305–314. [DOI] [PubMed] [Google Scholar]
- Rodionov, D.A., Mironov, A.A., and Gelfand, M.S. 2002a. Conservation of the biotin regulon and the BirA regulatory signal in Eubacteria and Archaea. Genome Res. 12: 1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov, D.A., Vitreschak, A.G., Mironov, A.A., and Gelfand, M.S. 2002b. Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem. 277: 48949–48959. [DOI] [PubMed] [Google Scholar]
- Sabatti, C., Rohlin, L., Oh, M.K., and Liao, J.C. 2002. Co-expression pattern from DNA microarray experiments as a tool for operon prediction. Nucleic Acids Res. 30: 2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin, A., Hoglund, A., Lenhard, B., and Wasserman, W.W. 2003. Integrated analysis of yeast regulatory sequences for biologically linked clusters of genes. Funct. Integr. Genomics 3: 125–134. [DOI] [PubMed] [Google Scholar]
- Sebulsky, M.T. and Heinrichs, D.E. 2001. Identification and characterization of fhuD1 and fhuD2, two genes involved in iron-hydroxamate uptake in Staphylococcus aureus. J. Bacteriol. 183: 4994–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebulsky, M.T., Hohnstein, D., Hunter, M.D., and Heinrichs, D.E. 2000. Identification and characterization of a membrane permease involved in iron-hydroxamate transport in Staphylococcus aureus. J. Bacteriol. 182: 4394–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr, S.S., Milo, R., Mangan, S., and Alon, U. 2002. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 31: 64–68. [DOI] [PubMed] [Google Scholar]
- Stajich, J.E., Block, D., Boulez, K., Brenner, S.E., Chervitz, S.A., Dagdigian, C., Fuellen, G., Gilbert, J.G., Korf, I., Lapp, H., et al. 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12: 1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, K., Moreno-Hagelsieb, G., Collado-Vides, J., and Stormo, G.D. 2001. A comparative genomics approach to prediction of new members of regulons. Genome Res. 11: 566–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Natale, D.A., Garkavtsev, I.V., Tatusova, T.A., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D., and Koonin, E.V. 2001. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29: 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J.M. and Heinrichs, D.E. 2002. Transferrin binding in Staphylococcus aureus: Involvement of a cell wall-anchored protein. Mol. Microbiol. 43: 1603–1614. [DOI] [PubMed] [Google Scholar]
- Terai, G., Takagi, T., and Nakai, K. 2001. Prediction of co-regulated genes in Bacillus subtilis on the basis of upstream elements conserved across three closely related species. Genome Biol. 2: RESEARCH0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, W., Rouchka, E.C., and Lawrence, C.E. 2003. Gibbs Recursive Sampler: Finding transcription factor binding sites. Nucleic Acids Res. 31: 3580–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterling, K.W., Levine, A.S., Yasbin, R.E., and Woodgate, R. 1997. Characterization of DinR, the Bacillus subtilis SOS repressor. J. Bacteriol. 179: 1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray Jr., L.V., Zalieckas, J.M., and Fisher, S.H. 2000. Purification and in vitro activities of the Bacillus subtilis TnrA transcription factor. J. Mol. Biol. 300: 29–40. [DOI] [PubMed] [Google Scholar]
- Xiong, A., Singh, V.K., Cabrera, G., and Jayaswal, R.K. 2000. Molecular characterization of the ferric-uptake regulator, fur, from Staphylococcus aureus. Microbiology 146: 659–668. [DOI] [PubMed] [Google Scholar]
- Yada, T., Nakao, M., Totoki, Y., and Nakai, K. 1999. Modeling and predicting transcriptional units of Escherichia coli genes using hidden Markov models. Bioinformatics 15: 987–993. [DOI] [PubMed] [Google Scholar]
- Zheng, Y., Szustakowski, J.D., Fortnow, L., Roberts, R.J., and Kasif, S. 2002. Computational identification of operons in microbial genomes. Genome Res. 12: 1221–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J., Wu, J., and Sun, Z. 2003. An approach to identify over-represented cis-elements in related sequences. Nucleic Acids Res. 31: 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.ncbi.nlm.nih.gov; NCBI home page.
- http://www.ncbi.nlm.nih.gov/COG; COG.
- http://elmo.ims.u-tokyo.ac.jp/dbtbs; DBTBS.
- http://arep.med.harvard.edu/dpinteract; DPInteract.
- http://www.weizmann.ac.il/mcb/UriAlon; Alon Lab.
- http://bayesweb.wadsworth.org/gibbs/gibbs.html; Gibbs Sampler home page.
- http://regulogs.cgb.ki.se/REGULOGS; Regulogs additional information.
- http://genolist.pasteur.fr/SubtiList; Subtilist Web server.