

Abstract
As genomes evolve, they undergo large-scale evolutionary processes that present a challenge to sequence comparison not posed by short sequences. Recombination causes frequent genome rearrangements, horizontal transfer introduces new sequences into bacterial chromosomes, and deletions remove segments of the genome. Consequently, each genome is a mosaic of unique lineage-specific segments, regions shared with a subset of other genomes and segments conserved among all the genomes under consideration. Furthermore, the linear order of these segments may be shuffled among genomes. We present methods for identification and alignment of conserved genomic DNA in the presence of rearrangements and horizontal transfer. Our methods have been implemented in a software package called Mauve. Mauve has been applied to align nine enterobacterial genomes and to determine global rearrangement structure in three mammalian genomes. We have evaluated the quality of Mauve alignments and drawn comparison to other methods through extensive simulations of genome evolution.
The recent determination of numerous bacterial and eukaryotic genome sequences poses new challenges for comparative sequence analysis. In addition to identifying local changes in the sequences of individual genes, the availability of genome sequences provides a basis for comparison of the structure and organization of genomes as a whole. Genomes are known to undergo several types of large-scale evolutionary events. Gene duplication can result in the existence of paralogous genes, whereas gene loss may remove a copy and obscure the assumption of orthology. Reordering of genetic elements occurs by mechanisms such as repeated inversion or translocation. Horizontal transfer introduces new genetic elements into bacterial genomes (Hacker and Carniel 2001). Furthermore, the rates and patterns of each event depend on the particular set of genomes being compared. For example, observations of gene duplication and repetitive sequences are much more common among higher eukaryotes than bacteria, whereas genome rearrangements can be readily observed between both closely related and divergent organisms of all types (Tillier and Collins 2000; Eichler and Sankoff 2003). Genome comparison systems must account for all of these evolutionary phenomena to provide a complete picture of genetic differences among organisms.
Early sequence comparison methods were designed to identify nucleotide substitutions and small insertions and deletions by computing an alignment of pairs of short sequences. Such early techniques as Needleman-Wunsch global alignment and Smith-Waterman local alignment use methods whose computation time scales as O(n2), where n is the length of input sequences. Numerous multiple sequence alignment and comparison methods are based on dynamic programming algorithms similar to Smith-Waterman and Needleman-Wunsch (Thompson et al. 1994; Morgenstern et al. 1996; Morgenstern 1999; Notredame et al. 2000; Lee et al. 2002). Such pairwise and multiple sequence alignment methods suffer the limitation that application to long (typically n > 10 kb) sequences is prohibitively timeconsuming (Ureta-Vidal et al. 2003).
The availability of genome sequences demands methods for aligning long genomic DNA sequences. Several heuristic approaches to align long sequences have been developed under the assumption that highly similar subsequences can be found quickly and are likely to be part of the correct global alignment. These local alignments are used to anchor a global alignment, reducing the number of possible global alignments considered during a subsequent O(n2) dynamic programming step. Some spurious local alignments are typically found because of random sequence similarity, particularly when using a sensitive local alignment method. A method for selecting alignment anchors must be used to filter out spurious matching regions. Alignment tools such as MUMmer, GLASS, AVID, and WABA align pairs of long sequences, implementing various methods to discover local alignments (Delcher et al. 1999; Batzoglou et al. 2000; Kent and Zahler 2000; Morgenstern 2000; Bray et al. 2003). Similar multiple sequence alignment methods for long sequences have been developed and implemented in software packages such as MAVID, MLAGAN, and MGA (Hohl et al. 2002; Bray and Pachter 2003; Brudno et al. 2003a). All of these pairwise and multiple sequence aligners assume the input sequences are free from significant rearrangements of sequence elements, selecting a single collinear set of alignment anchors.
Recently, methods have been developed to perform pairwise genome comparison in the presence of rearrangements. Shuffle-LAGAN, a variant of the LAGAN alignment system, was the first genome comparison method described that explicitly deals with genome rearrangements during the alignment process (Brudno et al. 2003b). Like other genome alignment methods, Shuffle-LAGAN uses an anchored alignment approach. Rather than selecting a single collinear set of anchors, Shuffle-LAGAN selects anchors collinear in the first sequence with rearrangements permitted in the other sequence. Although Shuffle-LAGAN's alignment approach works for pairwise comparison, an extension of the method to multiple genome sequences has not yet been suggested.
MultiPipMaker, based on BLASTZ, is a tool that can align multiple genomes to a single reference genome in the presence of rearrangements (Schwartz et al. 2003a). MultiPipMaker uses BLASTZ (Schwartz et al. 2003b) on each pair of reference and nonreference genomes to calculate pairwise local alignments. These local alignments are used to construct a rough global alignment that is iteratively refined. Because MultiPipMaker does not provide a mechanism for global alignment of regions not included in the initial local alignments, more divergent homologous regions between local alignments may remain unaligned. As such, MultiPipMaker can best be described as a multiple local aligner for genome sequences, rather than a global aligner. Furthermore, neither Shuffle-LAGAN nor MultiPipMaker provides a means to precisely identify the breakpoints of multiple genome rearrangements.
During the past several years, researchers from around the world published the finished genome sequences of several enterobacteria, nine of which we presently consider (Table 1). Previous studies have shown that these nine enterobacterial genomes have undergone significant horizontal transfer and numerous genome rearrangements since their divergence. However, a lack of effective tools has constrained comparison of the rates and patterns of large-scale evolutionary processes in these bacteria to pairwise and three-way studies.
Table 1.
The Published Genome Sequences of These Nine Enterobacteria Are a Target for the Alignment System Presented Here
| Species | Genome size | Reference |
|---|---|---|
| E. coli K12 MG1655 | 4,639,221 | Blattner et al. 1997 |
| E. coli O157:H7 EDL933 | 5,524,971 | Perna et al. 2001 |
| E. coli O157:H7 VT-2 Sakai | 5,498,450 | Hayashi et al. 2001 |
| E. coli CFT073 | 5,231,428 | Welch et al. 2002 |
| S. flexneri 2A 2457T | 4,599,354 | Wei et al. 2003 |
| S. flexneri 2A | 4,607,203 | Jin et al. 2002 |
| S. enterica Typhimurium LT2 | 4,857,432 | McClelland et al. 2001 |
| S. enterica Typhi CT18 | 4,809,037 | Parkhill et al. 2001 |
| S. enterica Typhi Ty2 | 4,791,961 | Deng et al. 2003 |
Numerous large-scale evolutionary events such as horizontal transfer and rearrangement are scattered throughout their genomes.
We describe a genome comparison method that identifies conserved genomic regions, rearrangements and inversions in conserved regions, and the exact sequence breakpoints of such rearrangements across multiple genomes. Furthermore, our comparison method performs traditional multiple alignment of conserved regions to identify nucleotide substitutions and small insertions and deletions (indels). We implemented our methods in a genome alignment package called Mauve. Mauve represents the first alignment system that integrates analysis of large-scale evolutionary events with traditional multiple sequence alignment. By integrating these previously separate analysis steps, Mauve provides additional ease-of-use and sensitivity over other systems when comparing genomes with significant rearrangements.
Like other genome alignment methods, Mauve uses anchoring as a heuristic to speed alignment. Unlike other multiple genome alignment systems, Mauve's anchor selection method relaxes the assumption that the genomes under study are collinear. Instead, Mauve identifies and aligns regions of local collinearity called locally collinear blocks (LCBs). Each locally collinear block is a homologous region of sequence shared by two or more of the genomes under study, and does not contain any rearrangements of homologous sequence. The algorithms described in this paper are limited to identifying LCBs that contain sequence elements conserved among all the genomes being aligned; in the general case, however, an LCB may be composed solely of sequence regions shared by a subset of the genomes. Remaining unaligned regions conserved among a subset of the genomes can be extracted and aligned using other methods.
The locally collinear blocks identified by Mauve's anchor selection algorithm are required to meet a user-specified minimum weight criteria as described in the Methods section. The weight of an LCB provides a measure of confidence that it is a true genome rearrangement rather than a spurious match. By selecting a high minimum weight during alignment, the user can identify genome rearrangements that are very likely to exist, whereas by selecting a lower minimum weight, the user can trade some specificity for sensitivity to smaller genome rearrangements.
Prior to Mauve, other methods have been developed to identify homologous regions of genome sequence in the presence of large-scale rearrangements, a problem also known as strip generation. Such methods typically use some metric to cluster matches between two or more genomes then evaluate which “clusters” represent homologous regions of interest rather than spurious matches. GRIMM-Synteny is one such method that clusters matches within some given gap distance and then removes clusters that span less than a given length of the chromosome (Pevzner and Tesler 2003a). FISH, another software package, implements a similar clustering method but uses a statistical framework to determine which clusters of matches are significant (Calabrese et al. 2003). Unlike Mauve, GRIMM and FISH do not identify strictly collinear clusters of matches necessary for genome alignment, nor do they perform recursive homology detection. However, with extensions similar in nature to the Mauve algorithm, GRIMM and FISH could become suitable methods for alignment anchor selection.
In addition to the Mauve alignment algorithm, a simple viewing system has been developed to display the rearrangement structure of several genome sequences. The viewer uses the first sequence to assign a reference orientation to LCBs in the remaining sequences. Thus, regions that are in the reverse-complement orientation relative to the first sequence appear inverted in the viewer. Because the boundaries of rearrangement have been determined, the viewer is able to draw a single line that logically connects the entire homologous collinear blocks from each genome. Previous visualization systems drew one line per local alignment, often yielding a confusing picture of complex rearrangement structures.
Finally, to make an informed decision when choosing between alignment tools, it is important to have not only an understanding of the algorithms used but also the empirical performance of the alignment system. Toward this end, we empirically characterized our alignment system and compared its performance with other well-known genome alignment systems. Manually validating a benchmark alignment on the genome scale is too labor-intensive. Instead, we developed a simple genome evolution simulation system that incorporates large- and small-scale evolutionary events. Because the evolutionary history is known, the simulator can generate the “correct” alignment in addition to the evolved sequences. We measured the ability of Mauve and other genome aligners to reproduce the “correct” alignment for the evolved sequences.
The Mauve alignment system and visualization environment are available for download from http://gel.ahabs.wisc.edu/mauve.
METHODS
The set of target genomes for our alignment system led us to consider several factors when designing an alignment algorithm. The alignment system must quickly align long genome sequences. Although parallel dynamic programming methods have been used with some success (Martins et al. 2001), anchored alignment approaches require only modest computational resources while having a tolerable impact on alignment quality (Ureta-Vidal et al. 2003).
The target genomes are known to have significant repetitive regions such as ribosomal RNA operons and prophages. When searching for anchors across multiple genomes, problems arise if a particular repetitive motif occurs numerous times in each sequence because it becomes unclear which combination of regions to align. For a repetitive element existing r times in each of G genomes, there will be rG possible alignment anchors, of which at most r represent truly orthologous anchors. As more genomes are aligned, the number of possible anchors grows exponentially while the number of anchors that can be included in an alignment of orthologous sequences remains constant. Mauve avoids this problem by using Multiple Maximal Unique Matches (multi-MUMs) of some minimum length k as alignment anchors. multi-MUMs are exactly matching subsequences shared by two or more genomes that occur only once in those genomes and that are bounded on either side by mismatched nucleotides. Because using multi-MUMs reduces anchoring sensitivity in conserved repetitive regions and regions that have undergone numerous nucleotide substitutions or indels, Mauve uses a recursive anchoring strategy that progressively reduces k, searching for smaller anchors in the remaining unmatched regions.
The enterobacterial genomes are known to have undergone significant genome rearrangements as described in their genome papers. Algorithms used by other global multiple alignment systems anchor their alignments by selecting the highest-scoring collinear chain of local alignments (Hohl et al. 2002; Bray and Pachter 2003). Such methods preclude identification of the rearrangements known to exist in our data set and many others. To successfully align our target genomes, the anchor selection method should identify consistent (collinear) subsets of local alignments to use as anchors while filtering out unlikely local alignments. Ideally, an algorithm would identify a maximum-weight set of anchors such that each collinear subset of anchors meets some minimum-weight criteria. Mauve uses a greedy breakpoint elimination algorithm to generate an approximate solution to the maximum-weight noncollinear anchoring problem.
To align the intervening regions of sequence between anchors, our method uses the progressive dynamic programming approach of CLUSTAL W (Thompson et al. 1994). In progressive alignment, a phylogenetic guide tree specifies the optimal progression of sequences to align when building the multiple alignment. Rather than recalculating a guide tree during each alignment of intervening regions, Mauve infers a single global phylogenetic tree. Not only does using a single average genome phylogeny save compute time, but recent results show it may yield a more robust phylogeny (Rokas et al. 2003).
The alignment algorithm can be summarized as follows:
Find local alignments (multi-MUMs).
Use the multi-MUMs to calculate a phylogenetic guide tree.
Select a subset of the multi-MUMs to use as anchors—these anchors are partitioned into collinear groups called LCBs.
Perform recursive anchoring to identify additional alignment anchors within and outside each LCB.
Perform a progressive alignment of each LCB using the guide tree.
The following sections give an overview of each step in the alignment process.
Finding Multi-MUMs
Mauve finds multi-MUMs using a simple seed-and-extend hashing method similar to that used by GRIL (Darling et al. 2004). In addition to finding matching regions that exist in all genomes, the algorithm identifies matches that exist in only a subset of the genomes being aligned. Although the seed-and-extend algorithm has time complexity O(G2n + Gn logGn), where G is again the number of genomes and n the average genome length, it is very fast in practice. Finding multi-MUMs typically consumes less than a minute per bacterial-size genome, and 3–4 h per mammalian genome on a standard workstation computer. Appendix A in the Supplemental material contains a detailed description of the matching algorithm.
Formally we define each multi-MUM as a tuple 〈L, S1,..., SG〉, where L is the length of the multi-MUM, and Sj is the left-end position of the multi-MUM in the j-th genome sequence. We denote the resulting set of multi-MUMs as M = {M1... MN}. The i-th multi-MUM in M is referred to as Mi. To refer to the length of Mi, we use the notation Mi·L, and similarly, we refer to the left end of Mi in the j-th genome sequence using the notation Mi·Sj. If multi-MUM Mi includes a region in the reverse complement orientation in sequence j, we define the sign of Mi·Sj to be negative. Finally, if multi-MUM Mi does not exist in sequence j, we define Mi·Sj to be 0—the leftmost position in any genome is 1 (or –1).
Calculating a Guide Tree
The method described to find multi-MUMs differs from that used by GRIL in that it can identify multi-MUMs in subsets of the genomes under study. Mauve exploits the information provided by subset multi-MUMs as a distance metric to construct a phylogenetic guide tree using Neighbor Joining (Saitou and Nei 1987).
Specifically, the ratio of base pairs shared between two genomes to their average genome length provides an estimate of sequence similarity. This similarity estimate is converted to a distance value for the Neighbor Joining distance matrix by subtracting it from one. Because multi-MUMs can overlap each other, calculating the similarity metric requires that overlaps among multi-MUMs are resolved such that each matching residue counts only once. To resolve an overlap, one match remains unchanged while the overlapping portion of the other match gets trimmed off and its remaining portion can still be counted. Mauve resolves overlaps in favor of the higher multiplicity match, where multiplicity(Mi) is defined as the number of genomes for which Mi·Sj ≠ 0. If the multiplicity of two overlapping matches is identical, the overlap is resolved in favor of the longer match.
Because the anchor selection method described below operates only on MUMs with multiplicity(Mi) = G, the guide tree is calculated prior to anchor selection so that it can take advantage of multi-MUMs with multiplicity(Mi) < G.
Selecting a Set of Anchors
In addition to local alignments that are part of truly homologous regions, the set of multi-MUMs M may contain spurious matches arising due to random sequence similarity. This step attempts to filter out such spurious matches while determining the boundaries of locally collinear blocks. An LCB can be considered a consistent subset of the multi-MUMs in M. Formally, an LCB is a sequence of multi-MUMs lcb ⊆ M, lcb = {M1, M2,..., M|lcb|} that satisfies a total ordering property such that Mi · Sj ≤ Mi + 1 · Sj holds for all i, 1 ≤ i ≤ |lcb|, and all j, 1 ≤ j ≤ G.7 For a given set of multi-MUMs, the minimum partitioning of M into collinear blocks can be found through breakpoint analysis (Blanchette et al. 1997). Because breakpoint analysis requires that matching regions exist in all genomes under study, multi-MUMs with multiplicity <G are removed from M before performing this step of the algorithm.
Given a minimum weight criteria MinimumWeight ≥ 0, Mauve uses a greedy breakpoint elimination algorithm to remove low-weight collinear blocks of M. As part of step 3 above, Mauve performs the following substeps repeatedly until all collinear blocks in M meet the minimum weight requirement:
Substep 1. Determine a partitioning of M into collinear blocks CB.
Substep 2. Calculate the weight, w(cbi) of each collinear block cbi ε CB.
Substep 3. Let z = mincbεCBw(cb).
Substep 4. Stop if z ≥ MinimumWeight.
Substep 5. Identify the collinear subsets MinCB ⊆ CB that satisfy w(cbi) = z.
Substep 6. For each cb ε MinCB, remove each multi-MUM M ε cb from M.
Substep 7. Go to substep 1.
Here w(cb) is defined as ΣMi ε cbMi·L. Substep 1 is identical to the method used by GRIL for partitioning M into collinear subsets and is described in Supplemental Appendix B.
To provide a fair measure of weight, each nucleotide in an LCB should count only once toward its weight. For this reason, breakpoint determination uses the set of nonoverlapping multi-MUMs that remain after guide tree calculation. By default, the MinimumWeight parameter is set to 3k, where k is the seed length used during the initial search for multi-MUMs. We chose 3k as a default minimum weight because it appears to filter the majority of spurious matches in data sets we have evaluated. Figure 1 illustrates the process of identifying collinear blocks of multi-MUMs and how removing a low-weight collinear region can eliminate a breakpoint. The resulting collinear sets of anchors delineate the LCBs that are used to guide the remainder of the alignment process.
Figure 1.

A pictorial representation of greedy breakpoint elimination in three genomes. (A) The algorithm begins with the initial set of matching regions (multi-MUMs) represented as connected blocks. Blocks below a genome's center line are inverted relative to the reference sequence. (B) The matches are partitioned into a minimum set of collinear blocks. Each sequence of identically colored blocks represents a collinear set of matching regions. One connecting line is drawn per collinear block. Block 3 (yellow) has a low weight relative to other collinear blocks. (C) As low-weight collinear blocks are removed, adjacent collinear blocks coalesce into a single block, potentially eliminating one or more breakpoints. Gray regions within collinear blocks are targeted by recursive anchoring.
Recursive Anchoring and Gapped Alignment
The initial anchoring step may not be sensitive enough to detect the full region of homology within and surrounding the LCBs. In particular, repetitive regions and regions with frequent nucleotide substitutions are likely to lack sufficient anchors for complete alignment. Using the existing anchors as a guide, two types of recursive anchoring are performed repeatedly. First, regions outside of LCBs are searched to extend the boundaries of existing LCBs and identify new LCBs. In Figure 1C, this corresponds to searching the white regions outside LCBs. Second, unanchored regions within LCBs are searched for additional alignment anchors. This corresponds to searching the gray regions within LCBs in Figure 1C.
When searching for additional anchors outside existing LCB boundaries, two factors contribute to Mauve finding additional anchors. First, Mauve uses a smaller value of the match seed size k. Second, because only the regions outside existing LCB boundaries are searched, regions not unique in the entire genome may be unique within regions outside LCBs. Not only can the range of existing LCBs be extended by searching regions outside LCB boundaries, but also new LCBs that meet the minimum weight requirement can be identified as well. To perform the search, the outside sequences in each genome are concatenated into a single sequence per genome. We refer to the set of concatenated sequences as S and the concatenated sequence from the j-th genome as Sj. Multi-MUMs of minimum length k are found, where
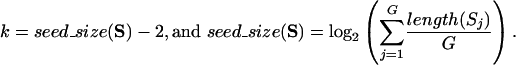 |
Because the left-end coordinates of each new multi-MUM are defined in terms of the concatenated sequence, they must be transposed back into the original coordinate system. Also, any matches spanning two concatenated subsequences must be split. The transposed multi-MUMs are added to M, and iterative removal of low-weight collinear subsets is performed as above. The process of searching regions outside LCBs is repeated until ΣcsεCSw(cs) remains the same during two successive iterations of the search.
In addition to missing anchors outside the boundaries of LCBs, the initial anchoring pass may have lacked the sensitivity to find anchors in large regions within each LCB. Because progressive alignment requires relatively dense anchors (at least one anchor per 10 kb of sequence), Mauve performs recursive anchoring on the intervening regions between each pair of existing anchors. Not only does this step anchor more divergent regions of sequence, it also locates anchors in conserved repeats because many k-mers that are not unique in the whole genome are likely to be unique within the intervening regions between existing anchors. Unlike other genome aligners that perform a fixed number of recursive passes with a predetermined sequence of anchor sizes, Mauve calculates a minimum anchor size based on the length of the intervening sequence and stops recursive anchoring when either no additional anchors are found or when the intervening region is shorter than a fixed length, defaulting to 200 bp. During each recursive anchor search, a single collinear set of new anchors in the same orientation as the flanking anchors is selected to cover the region between flanking anchors. For each search, k is calculated as above: k = seed_size(S), where S is the set of intervening sequences, one per genome. By dynamically calculating the value of k, Mauve ensures that k is sized appropriately for the intervening region. Selecting a k too large prevents discovery of multi-MUMs in polymorphic regions, whereas selecting a k too small increases the likelihood that k-mers will not be unique in the intervening region.
Armed with a complete set of alignment anchors, Mauve performs a CLUSTAL W progressive alignment using the genome guide tree calculated previously. The progressive alignment algorithm is executed once for each pair of adjacent anchors in every LCB, calculating a global alignment over each LCB. Tandem repeats <10 kb in total length are aligned during this phase. Regions >10 kb without an anchor are ignored.
RESULTS
The Mauve genome alignment procedure results in a global alignment of each locally collinear block that has sequence elements conserved among all the genomes under study. Nucleotides in any given genome are aligned only once to other genomes, suggesting orthology among aligned residues. Mauve makes no attempt to align paralogous regions. The remaining unaligned regions may be lineage-specific sequence or rearranged or paralogous repetitive regions and can be identified as such during subsequent processing with other tools. Large (>10 kb) regions introduced to a subset of the genomes by horizontal transfer are not aligned by Mauve because they do not have alignment anchors conserved among all sequences. Both large and small regions existing in only a subset of the genomes and that also underwent local rearrangement remain unaligned.
Evaluating Alignment Quality
Without a “correct” alignment of the nine enterobacterial genomes, the calculated alignment generated by Mauve cannot be evaluated for accuracy. In fact, no manually curated multiple alignment benchmark data sets account for genome-scale evolutionary events such as inversion, rearrangement, and horizontal transfer. Despite the lack of a manually curated correct alignment, we can estimate the alignment accuracy by modeling evolution and aligning simulated data sets.
The inferential power yielded by evaluating alignment accuracy using simulated evolution is only as strong as the degree to which the simulation faithfully represents the actual evolutionary processes that governed the history of the genomes under study. Keeping that fact in mind, we constructed a simplistic model of genome evolution that we believe captures the major types, patterns, and frequencies of events in the history of the enterobacterial genomes. Given a rooted phylogenetic tree and an ancestral sequence, we would like to generate evolved sequences for each internal and leaf node of the tree, along with a multiple sequence alignment of regions conserved throughout the simulated evolution. To effectively represent genome evolution, the simulation must include nucleotide substitutions and indels in addition to genome-scale events such as horizontal transfer, inversion, and rearrangement.
Nucleotide substitutions are ostensibly the best understood and most ubiquitous evolutionary mechanism. We use the HKY model of nucleotide substitution implemented in the Monte Carlo simulation package called Seqgen (Rambaut and Grassly 1997). Small insertions and deletions (indels) are modeled as occurring with uniform frequency and distribution throughout the genomes, with a size sampled from a Poisson distribution with mean value 3 bp. When studying the differences between Escherichia coli O157:H7 EDL933 and K-12 MG1655, it became clear that a small number of horizontal transfers introducing large regions of sequence have occurred, whereas the majority of transfers introduced small sequence regions. Our model includes large horizontal transfer events uniformly distributed in length between 10 kb and 60 kb. The size of small horizontal transfer events is sampled from an exponential distribution with mean value 200 bp. Horizontal transfer is implemented by simultaneously evolving a set of “donor” genomes according to the same tree from which horizontally transferred sequence can be sampled.
Our model does not explicitly implement translocation events; however, we observe that two overlapping inversion events can result in a translocation. The lengths of inversions are sampled from an exponential distribution with mean value 50 kb. Locations for inversion and horizontal transfer events are sampled uniformly throughout the genome, and all events are simulated to have taken place at a point in time given by a marked Poisson process over the phylogenetic tree. Finally, genome size is expected to stay relatively constant over time; thus, deletion events are sampled with the same size and frequency as events that introduce new sequence. Our implementation of the evolutionary model described above is referred to as the simple genome evolver, or just sgEvolver.
Experiments
Using the simple genome evolver, we designed and executed several experiments to compare the ability of Mauve and other alignment systems to align our target data set. Multiple alignment experiments used the phylogenetic guide tree estimated for the nine enterobacteria (see Figure 2), midpoint-rooted to provide an entry point for the ancestral sequence. Rather than generate a random ancestral sequence, 1 Mb of enterobacterial DNA was used to preserve the distribution of sequence motifs and repetitive subsequences found in our data set. An additional 1 Mb of enterobacterial DNA was used as a donor sequence pool for insertion and horizontal transfer events.
Figure 2.

An unrooted phylogenetic tree relating the nine enterobacterial genomes in Table 1. The tree is a phylogenetic guide tree calculated using Neighbor Joining by the Mauve alignment system.
Three experiments were performed, each of which consists of numerous simulations. The first experiment evaluates the robustness of Mauve and Multi-LAGAN, a cross-species genome comparison tool, to genomes with high nucleotide substitution and indel rates. A second experiment compares Mauve to Shuffle-LAGAN when aligning pairs of genomes with rearrangements. At the time these experiments were performed, Shuffle-LAGAN was the only publicly available genome aligner capable of aligning genomes in the presence of rearrangement. Our final experiment evaluates the ability of Mauve to align simulated genomes that resemble the nine target enterobacteria.
For each simulated data set, alignments were calculated using the Condor high-throughput computing environment at the University of Wisconsin. The Wisconsin Condor cluster contains >1000 nodes and allowed us to rapidly align thousands of simulated data sets. The calculated alignments were scored against correct alignments generated during the evolution process. We used the sum-of-pairs scoring procedure also used by BaliBASE (Thompson et al. 1999). In sum-of-pairs scoring, each pair of aligned residues in the calculated alignment that are aligned to each other in the correct alignment tallies a point. The total alignment score is then the ratio of points to total possible points.
Mauve Versus Multi-LAGAN
Our first experiment compared the ability of Mauve and Multi-LAGAN version 1.2 to align collinear sequences that had undergone increasing amounts of nucleotide substitution and indels. This experiment is designed to test the sensitivity of the anchoring methods used by each aligner. We evolved nine genomes at 20 levels of nucleotide substitution and 20 levels of indels, performing two replicate experiments of each combination of substitution rate and indel rate. The average Mauve and Multi-LAGAN alignment accuracy for each simulation is displayed in Figure 3. From the figure, it is obvious that Mauve's alignment score drops more rapidly than Multi-LAGAN's in the presence of an increasing substitution rate. We attribute this behavior to Mauve's use of multi-MUMs as alignment anchors. Multi-LAGAN's alignment anchors can contain substitutions and indels, making them much more sensitive than exactly matching subsequences. At lower levels of nucleotide substitution, Mauve appears to handle indels about as well as Multi-LAGAN. For the nucleotide substitution and indel rates previously reported in the enterobacterial data set, Mauve aligns the simulated genomes with a high degree of accuracy.
Figure 3.

The performance of Mauve (left) and Multi-LAGAN (right) when aligning sequences evolved with increasing amounts of nucleotide substitution and indels. The multi-MUM anchoring technique used by Mauve limits its ability to align distantly related sequences. Multi-LAGAN version 1.2 did not complete the alignments of genomes without indels, resulting in the black row at the bottom. The substitution and indel rate observed in the enterobacteria is denoted by an asterisk (*).
Mauve Versus Shuffle-LAGAN
We proceeded to gauge the ability of Mauve and Shuffle-LAGAN version 1.2 to align sequences that had undergone increasing amounts of inversion and nucleotide substitution. Because Shuffle-LAGAN is a pairwise aligner, we reduced the number of taxa in our simulation from nine to two. Three simulations were performed for each of 110 combinations of nucleotide substitution rate and inversion rate. The average accuracies of Mauve and Shuffle-LAGAN for each experiment are shown in Figure 4. Special considerations must be taken when scoring Shuffle-LAGAN. Because Shuffle-LAGAN attempts to identify and align paralogous regions, a single residue in the first genome can be aligned to multiple residues in the second genome. For the purpose of scoring Shuffle-LAGAN, we awarded points for a given residue in the first genome if any of the residues in the second genome it was aligned to were correct.
Figure 4.

The performance of Mauve (left) and Shuffle-LAGAN (right) when aligning two sequences evolved with increasing amounts of nucleotide substitution and inversions. Mauve is clearly more accurate than Shuffle-LAGAN at lower substitution rates. Shuffle-LAGAN version 1.2 did not complete some alignments without rearrangements, resulting in black entries. The observed substitution and inversion rate in the enterobacteria is denoted by an asterisk (*).
The experiment shows that Mauve clearly excels at aligning rearranged sequences under lower substitution rates that do not hamper its multi-MUM anchoring process. Interestingly, Shuffle-LAGAN appears to perform better as the substitution rate increases. Based on our experience, we conjecture that this counterintuitive result is related to the repetitive nature of the ancestral enterobacterial sequence. Shuffle-LAGAN appears to have difficulty selecting anchors in repetitive sequences. Because our simulation does not model selective pressure or gene conversion for repetitive regions, they are independently randomly mutated, and as the nucleotide substitution rate increases, they become decreasingly repetitive. Shuffle-LAGAN's improved performance on more divergent genomes appears to be an artifact of our simulation method and is not likely to be observed on real data. Anchoring its alignment in unique subsequences provides Mauve with immunity to this phenomena.
An Enterobacteria-Like Simulation
Our final set of experiments sought to evaluate the ability of Mauve to align genomes similar to the enterobacteria. Evolutionary rates for the simulation were extrapolated from previously published observations of the differences between E. coli K-12 MG1655 and O157:H7 EDL933. For these two E. coli, there are ∼75,000 observed nucleotide substitutions, ∼4000 observed indels, 40 large horizontal transfer events, 400 small horizontal transfers, and one inversion. The observed frequencies were converted to rates used to assign event frequencies to branches of the phylogenetic guide tree. It is known that among the group of enterobacteria, the Salmonella have higher rates of inversion and rearrangement than the E. coli. To compensate, the inversion rate was adjusted to result in ∼30–40 inversion events. When varying the substitution and indel rates between 0% and 125% while keeping horizontal transfer and inversion rates constant, Mauve alignments consistently average 80% accurate, ±5% (data not shown). The quality of alignment does not appear to drop as the substitution and indel rates are increased in this range. Rather, it appears that horizontal transfer rates have a more significant impact on alignment quality. As horizontal transfer rates increase, the ratio of lineage-specific sequence to backbone sequence increases and Mauve's alignment algorithm aligns decreasing amounts of the total sequence. Figure 5 shows how Mauve's ability to align enterobacteria-like genomes changes as horizontal transfer rates increase. When scored only against regions of the simulated genomes considered as conserved backbone, Mauve consistently aligns with >98% accuracy. For the purpose of scoring the alignment, we define backbone as a region in the correct alignment containing >50 gap-free columns without stretches of 50 or more consecutive gaps in any single genome sequence. Based on our simulations, we believe our method accurately aligns the backbone of the nine enterobacteria; however, significant lineage-specific regions remain unaligned.
Figure 5.

The performance of Mauve when aligning sequences evolved with rates similar to those observed among the group of nine enterobacteria. In this experiment, the substitution, indel, and inversion frequencies were held constant at rates similar to those observed in the enterobacteria. The asterisk (*) denotes the combination of large and small horizontal transfer rates observed in the enterobacteria. As the rate of large horizontal transfer increases, the amount of lineage-specific sequence relative to backbone grows. Because Mauve cannot align large lineage-specific regions, the alignment score drops. When scored only on regions considered backbone sequence, the accuracy is consistently above 98%.
Alignment of Nine Enterobacterial Genomes
We applied Mauve to align the nine enterobacterial genomes listed in Table 1. Previous studies of these genomes indicates they underwent significant genome rearrangement, horizontal transfer, and other recombination (Perna et al. 2001; Deng et al. 2003). Mauve consumed 3 h to align the nine taxa on a 2.4-GHz computer with 1 GB of RAM. The alignment of the nine taxa reveals 45 LCBs with a minimum weight of 69. Figure 2 shows the guide tree generated for these species. The visualization of the genome rearrangement structure generated by the Mauve viewer is shown in Figure 6. We can quickly visually confirm several known inversions such as the O157:H7 EDL933 inversion relative to K-12 (Perna et al. 2001) and the large inversion about the origin of replication among the S. enterica serovars Typhi CT18 and Ty2 (Deng et al. 2003).
Figure 6.

Locally collinear blocks identified among the nine enterobacterial genomes listed in Table 1. Each contiguously colored region is a locally collinear block, a region without rearrangement of homologous backbone sequence. LCBs below a genome's center line are in the reverse complement orientation relative to the reference genome. Lines between genomes trace each orthologous LCB through every genome. Large gray regions within an LCB signify the presence of lineage-specific sequence at that site. Each of the 45 blocks has a minimum weight of 69. The Shigella and Salmonella genomes have undergone more genome rearrangements than the E. coli, possibly because of the presence of specific mobile genetic elements. The computation consumed ∼3 h on a 2.4-GHz workstation with 1 GB of memory. The figure was generated by the Mauve rearrangement viewer.
We proceeded to extract conserved backbone sequence from the alignment. Again, backbone is defined as regions of the alignment containing >50 gap-free columns without stretches of 50 or more consecutive gaps in any single genome sequence. Under this definition, the nine enterobacteria have 2.86 Mb of conserved backbone sequence broken into 1252 backbone segments. Across the backbone the level of nucleotide identity is high, as shown by the identity matrix in Table 2.
Table 2.
Identity Matrix for 2.86 Mb of Shared Backbone Regions Among the Nine Enterobacteria Listed in Table 1
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | E. coli K12 MG1655 | 1.000 | — | — | — | — | — | — | — | — |
| 2 | E. coli EDL933 | 0.977 | 1.000 | — | — | — | — | — | — | — |
| 3 | E. coli VT-2 Sakai | 0.978 | 1.000 | 1.000 | — | — | — | — | — | — |
| 4 | E. coli CFT073 | 0.965 | 0.966 | 0.967 | 1.000 | — | — | — | — | — |
| 5 | S. flexneri 2a | 0.976 | 0.975 | 0.975 | 0.963 | 1.000 | — | — | — | — |
| 6 | S. flexneri 2a 2457T | 0.976 | 0.975 | 0.975 | 0.962 | 0.999 | 1.000 | — | — | — |
| 7 | S. Typhimurium | 0.794 | 0.793 | 0.793 | 0.793 | 0.791 | 0.791 | 1.000 | — | — |
| 8 | S. typhi CT18 | 0.792 | 0.791 | 0.791 | 0.792 | 0.790 | 0.789 | 0.981 | 1.000 | — |
| 9 | S. typhi Ty2 | 0.793 | 0.793 | 0.793 | 0.793 | 0.791 | 0.791 | 0.984 | 0.996 | 1.000 |
Although an average of only 58% of the genomes is conserved across species, the level of sequence identity is remarkably high, suggesting that horizontal transfer and differential gene loss may account for the majority of phenotypic diversity among bacteria in this group.
Rearrangements in Three Mammalian Genomes
Although we designed our methods with the intent of aligning bacterial genomes, we applied Mauve to the entire mouse, rat, and human genomes to assess the scalability of our methods. For this experiment, we used the “finished” human genome build 34, mouse genome build 32, and rat genome RGSC build 3.1. Rather than complete a full alignment, Mauve was used to determine the global rearrangement structure and LCBs in the three genomes. Finding an initial set of anchors with minimum length 31 bp consumed ∼12 h on a 1.6-GHz desktop workstation. Computing the anchors consumes roughly 3 GB RAM; however, the workstation was equipped with only 2.5 GB of true memory, and disk-based virtual memory was used to supply the remaining need. Figure 7 shows the complex rearrangement structure of these three mammalian genomes. In this data set it is difficult to determine the “correct” number of LCBs: depending on the minimum weight parameter used, the number of LCBs ranges from about 1000 to 2000. Furthermore, the large minimum anchor size (31 bp) precludes identification of small, local rearrangements of the type previously reported by Brudno et al. (2003b). A full mouse–rat–human alignment using Mauve may help resolve the true number of collinear blocks and facilitate identification of local rearrangements, but has not yet been performed.
Figure 7.

Mauve visualization of locally collinear blocks identified between concatenated chromosomes of the mouse, rat, and human genomes. Each of the 1251 blocks has a minimum weight of 90. Red vertical bars demarcate interchromosomal boundaries. The Mauve rearrangement viewer enables users to interactively zoom in on regions of interest and examine the local rearrangement structure. The computation consumed ∼12 h on a 1.6-GHz workstation with 2.5 GB of memory.
DISCUSSION
Since their first application to molecular biology some 30 years ago, sequence alignment techniques have progressed considerably. With the advent of genome sequencing, a new type of sequence alignment problem, that of whole-genome comparison, has emerged. Early approaches to genome alignment were designed to tackle dramatically increased sequence lengths, but did not consider the additional types of evolutionary events observed on the genome scale. Genome rearrangements, horizontal transfer, and duplication obfuscate orthology. As genomes continue to be sequenced, automatic and accurate identification of genome rearrangements becomes increasingly important, especially as high levels of rearrangement have been observed among both eukaryotes and prokaryotes (Lefebvre et al. 2003b; Pevzner and Tesler 2003a,b).
Our genome alignment method represents a first step toward multiple genome comparison in the presence of large-scale evolutionary events. It is capable of aligning conserved regions in the presence of genome rearrangement, and appears to scale efficiently to long genomes. Furthermore, Mauve aligns genomes identically irrespective of their input order by identifying multi-MUMs in subsets of the genomes and calculating a guide tree for progressive alignment. The remaining unaligned regions are often either repetitive, or lineage-specific regions acquired through horizontal transfer or other means. Repeat analysis using tools such as Repeat Masker, REPuter (Kurtz et al. 2000), and FORRepeats (Lefebvre et al. 2003a) can help to further classify unaligned regions.
Much research has been devoted to inference of rearrangement history that could lead to observed permutations in gene order (Bader et al. 2001; Bourque and Pevzner 2002; Larget et al. 2002; Eichler and Sankoff 2003). The locally collinear blocks identified during the alignment process serve as a foundation for such methods. LCBs can naturally be reduced to the signed permutation matrix typically used as input by these inference tools.
Our evaluation of alignment quality using simulated genome evolution yields several insights that will inform researchers seeking an appropriate alignment tool. The comparison of Mauve to Multi-LAGAN empirically confirms the sensitivity of the CHAOS anchoring technique and LAGAN alignment method. Multi-LAGAN successfully aligns much more divergent genomes than Mauve and is better suited to cross-species comparison when the genomes are collinear.
Similarly, the comparison of Mauve to Shuffle-LAGAN highlights important differences in each alignment method. Mauve excels at aligning closely related sequences that have undergone modest amounts of nucleotide substitution or inversion, consistently achieving scores above 90% when either rate is low relative to the other. Conversely, Shuffle-LAGAN does best when the inversion rate is low and nucleotide substitutions are frequent, topping out at 77.8% accuracy with ∼500,000 nucleotide substitutions and 400 inversions among the two genomes. As previously mentioned, Shuffle-LAGAN's difficulty anchoring in the presence of repetitive subsequences appears to cause the anomalous result. When conducting this comparative experiment, we executed Shuffle-LAGAN as per the instructions distributed with the software; however, in the Shuffle-LAGAN paper, the authors apply RepeatMasker to the genomes prior to alignment. Repeat-Masker is not applied to the genomes by the Shuffle-LAGAN software as distributed, and the addition of such a step may improve the accuracy of Shuffle-LAGAN alignments.
The design of our genome simulation system was motivated in part by our desire to evaluate the method's ability to align genomes similar to the nine enterobacteria. Of course, our model simplifies or ignores many aspects of the actual evolutionary forces at work. Nucleotide substitution rates vary widely throughout the genome. Our simulation incorporated general rate heterogeneity using a γ distribution, α = 1, but did not consider observed patterns of site-specific rate heterogeneity such as third base pair substitutions in coding regions. Furthermore, our model does not reflect the phenomena of gene duplication and subsequent loss that are known to occur frequently in the enterobacteria. Factors such as strand bias and site-specific rate heterogeneity for insertion, deletion, or inversion events that may significantly alter patterns of genome evolution are not incorporated into the model. Despite these shortcomings, the simple genome evolver has allowed us to demonstrate the accuracy of our alignment system when presented with certain well-defined patterns of evolution. The evaluation of alignment quality in the presence of increasing amounts of horizontal transfer suggests that Mauve's ability to completely align genomes declines in the presence of large lineage-specific sequence elements. Because our method requires homologous sequence in all genomes to anchor the alignment, lineage-specific regions larger than the maximum permitted size for progressive alignment (10 kb by default) remain unaligned. Small lineage-specific regions do not have as great an impact on alignment quality.
Our experience with Mauve clearly indicates that many challenges remain in genome alignment. A sensitive anchoring technique that recognizes and ignores repetitive subsequences would permit our method to be applied to more distantly related organisms. A method for determining breakpoints with anchors existing in a subset of the genomes would facilitate anchored alignment of the large lineage-specific regions currently missed. Some organisms are known to have small, local sequence rearrangements such as reordering of protein domains in coding regions. In such cases, the proximity of the rearrangement to neighboring homologous sequence should clearly be considered. Other types of rearrangement do not exhibit locality bias: symmetric inversions about the origin and terminus of replication and rearrangements mediated by mobile elements are common in prokaryotes and can move sequence to distant parts of the genome. Although Shuffle-LAGAN's scoring metric accounts for locality, it is clear that not all recombination mechanisms are subject to such a constraint. A more sophisticated rearrangement scoring method may attempt to infer the recombination mechanism suggested by a particular pattern of anchors and then score the rearrangement based on parameters tuned to that mechanism of recombination.
The availability and analysis of genome sequences has revealed the importance of large-scale evolutionary events. In light of these large-scale events, the genome comparison problem fundamentally differs from the traditional sequence alignment task. By considering such large-scale events, the methods presented here represent a significant advance toward the goal of automatic multiple genome comparison.
Acknowledgments
We thank Mark Craven for insightful comments and suggestions. Funding for this research was provided by NIH Grant GM62994-02. In addition, A.D. was supported in part by NLM Training Grant 1T15LM007359-01.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2289704.
Footnotes
[Supplemental material is available online at www.genome.org. The source code and binaries are freely available for academic and nonprofit research. Commercial licenses are also available. See http://gel.ahabs.wisc.edu/mauve for more details.]
Under this definition of an LCB, multi-MEMs on nontandem repetitive elements would break LCBs. Each multi-MEM would become its own independent LCB with identical weight, leaving the greedy breakpoint elimination algorithm with no means for discrimination.
References
- Bader, D.A., Moret, B.M., and Yan, M. 2001. A linear-time algorithm for computing inversion distance between signed permutations with an experimental study. J. Comput. Biol. 8: 483–491. [DOI] [PubMed] [Google Scholar]
- Batzoglou, S., Pachter, L., Mesirov, J.P., Berger, B., and Lander, E.S. 2000. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 10: 950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette, M., Bourque, G., and Sankoff, D. 1997. Breakpoint phylogenies. Genome Inform Ser. Workshop Genome Inform. 8: 25–34. [PubMed] [Google Scholar]
- Blattner, F.R., Plunkett III, G., Bloch, C.A., Perna, N.T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J.D., Rode, C.K., Mayhew, G.F., et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1474. [DOI] [PubMed] [Google Scholar]
- Bourque, G. and Pevzner, P.A. 2002. Genome-scale evolution: Reconstructing gene orders in the ancestral species. Genome Res. 12: 26–36. [PMC free article] [PubMed] [Google Scholar]
- Bray, N. and Pachter, L. 2003. MAVID multiple alignment server. Nucleic Acids Res. 31: 3525–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, N., Dubchak, I., and Pachter, L. 2003. AVID: A global alignment program. Genome Res. 13: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno, M., Do, C.B., Cooper, G.M., Kim, M.F., Davydov, E., Green, E.D., Sidow, A., and Batzoglou, S., NISC Comparative Sequencing Program. 2003a. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13: 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno, M., Malde, S., Poliakov, A., Do, C.B., Couronne, O., Dubchak, I., and Batzoglou, S. 2003b. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 19 Suppl 1: I54–I62. [DOI] [PubMed] [Google Scholar]
- Calabrese, P.P., Chakravarty, S., and Vision, T.J. 2003. Fast identification and statistical evaluation of segmental homologies in comparative maps. Bioinformatics 19 Suppl 1: i74–i80. [DOI] [PubMed] [Google Scholar]
- Darling, A.E., Mau, B., Blattner, F.R., and Perna, N.T. 2004. GRIL: Genome rearrangement and inversion locator. Bioinformatics 20: 122–124. [DOI] [PubMed] [Google Scholar]
- Delcher, A.L., Kasif, S., Fleischmann, R.D., Peterson, J., White, O., and Salzberg, S.L. 1999. Alignment of whole genomes. Nucleic Acids Res. 27: 2369–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, W., Liou, S.-R., Plunkett III, G., Mayhew, G.F., Rose, D.J., Burland, V., Kodoyianni, V., Schwartz, D.C., and Blattner, F.R. 2003. Comparative genomics of Salmonella enterica serovar Typhi strains Ty2 and CT18. J. Bacteriol. 185: 2330–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler, E.E. and Sankoff, D. 2003. Structural dynamics of eukaryotic chromosome evolution. Science 301: 793–797. [DOI] [PubMed] [Google Scholar]
- Hacker, J. and Carniel, E. 2001. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2: 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, T., Makino, K., Ohnishi, M., Kurokawa, K., Ishii, K., Yokoyama, K., Han, C.G., Ohtsubo, E., Nakayama, K., Murata, T., et al. 2001. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8: 11–22. [DOI] [PubMed] [Google Scholar]
- Hohl, M., Kurtz, S., and Ohlebusch, E. 2002. Efficient multiple genome alignment. Bioinformatics 18 Suppl 1: S312–S320. [DOI] [PubMed] [Google Scholar]
- Jin, Q., Yuan, Z., Xu, J., Wang, Y., Shen, Y., Lu, W., Wang, J., Liu, H., Yang, J., Yang, F., et al. 2002. Genome sequence of Shigella flexneri 2a: Insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 30: 4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. and Zahler, A.M. 2000. Conservation, regulation, synteny, and introns in a large-scale C. briggsae–C. elegans genomic alignment. Genome Res. 10: 1115–1125. [DOI] [PubMed] [Google Scholar]
- Kurtz, S., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. 2000. Computation and visualization of degenerate repeats in complete genomes. Proc. Int. Conf. Intell. Syst. Mol. Biol. 8: 228–238. [PubMed] [Google Scholar]
- Larget, B., Simon, D.L., and Kadane, J. 2002. On a Bayesian approach to phylogenetic inference from animal mitochondrial genome arrangements. J. Roy. Stat. Soc. B 64: 681–693. [Google Scholar]
- Lee, C., Grasso, C., and Sharlow, M.F. 2002. Multiple sequence alignment using partial order graphs. Bioinformatics 18: 452–464. [DOI] [PubMed] [Google Scholar]
- Lefebvre, A., Lecroq, T., Dauchel, H., and Alexandre, J. 2003a. FORRepeats: Detects repeats on entire chromosomes and between genomes. Bioinformatics 19: 319–326. [DOI] [PubMed] [Google Scholar]
- Lefebvre, J.F., El-Mabrouk, N., Tillier, E., and Sankoff, D. 2003b. Detection and validation of single gene inversions. Bioinformatics 19 Suppl 1: I190–I196. [DOI] [PubMed] [Google Scholar]
- Martins, W.S., del Cuvillo, J., Cui, W., and Gao, G.R. 2001. Whole genome alignment using a multithreaded parallel implementation. Symposium on Computer Architecture and High Performance Computing, pp. 1–8.
- McClelland, M., Sanderson, K.E., Spieth, J., Clifton, S.W., Latreille, P., Courtney, L., Porwollik, S., Ali, J., Dante, M., Du, F., et al. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413: 852–826. [DOI] [PubMed] [Google Scholar]
- Morgenstern, B. 1999. DIALIGN 2: Improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 15: 211–218. [DOI] [PubMed] [Google Scholar]
- Morgenstern, B. 2000. A space-efficient algorithm for aligning large genomic sequences. Bioinformatics 16: 948–949. [DOI] [PubMed] [Google Scholar]
- Morgenstern, B., Dress, A., and Werner, T. 1996. Multiple DNA and protein sequence alignment based on segment-to-segment comparison. Proc. Natl. Acad. Sci. 93: 12098–12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302: 205–217. [DOI] [PubMed] [Google Scholar]
- Parkhill, J., Dougan, G., James, K.D., Thomson, N.R., Pickard, D., Wain, J., Churcher, C., Mungall, K.L., Bentley, S.D., Holden, M.T., et al. 2001. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413: 848–852. [DOI] [PubMed] [Google Scholar]
- Perna, N.T., Plunkett III, G., Burland, V., Mau, B., Glasner, J.D., Rose, D.J., Mayhew, G.F., Evans, P.S., Gregor, J., Kirkpatrick, H.A., et al. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409: 529–533. [DOI] [PubMed] [Google Scholar]
- Pevzner, P. and Tesler, G. 2003a. Genome rearrangements in mammalian evolution: Lessons from human and mouse genomes. Genome Res. 13: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner, P. and Tesler, G. 2003b. Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proc. Natl. Acad. Sci. 100: 7672–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. and Grassly, N.C. 1997. Seq-Gen: An application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 13: 235–238. [DOI] [PubMed] [Google Scholar]
- Rokas, A., Williams, B.L., King, N., and Carroll, S.B. 2003. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425: 798–804. [DOI] [PubMed] [Google Scholar]
- Saitou, N. and Nei, M. 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Schwartz, S., Elnitski, L., Li, M., Weirauch, M., Riemer, C., Smit, A., Green, E.D., Hardison, R.C., and Miller, W., NISC Comparative Sequencing Consortium. 2003a. MultiPipMaker and supporting tools: Alignments and analysis of multiple genomic DNA sequences. Nucleic Acids Res. 31: 3518–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, S., Kent, W.J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003b. Human–mouse alignments with BLASTZ. Genome Res. 13: 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTALW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Plewniak, F., and Poch, O. 1999. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 27: 2682–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillier, E.R. and Collins, R.A. 2000. Genome rearrangement by replication-directed translocation. Nat. Genet. 26: 195–197. [DOI] [PubMed] [Google Scholar]
- Ureta-Vidal, A., Ettwiller, L., and Birney, E. 2003. Comparative genomics: Genome-wide analysis in metazoan eukaryotes. Nat. Rev. Genet. 4: 251–262. [DOI] [PubMed] [Google Scholar]
- Wei, J., Goldberg, M.B., Burland, V., Venkatesan, M.M., Deng, W., Fournier, G., Mayhew, G.F., Plunkett III, G., Rose, D.J., Darling, A., et al. 2003. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect. Immun. 71: 2775–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, R.A., Burland, V., Plunkett III, G., Redford, P., Roesch, P., Rasko, D., Buckles, E.L., Liou, S.-R., Boutin, A., Hackett, J., et al. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. 99: 17020–17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://gel.ahabs.wisc.edu/mauve; the Mauve alignment system and visualization environment.