

Abstract
We report the case of a paediatric patient with steroid-resistant nephrotic syndrome due to a novel dominant Wilms’ tumour 1 mutation. The nucleotide change C1184A, identified in exon 9, results in amino acid substitution Ser395Tyr. Genotyping of parents and healthy controls indicated that this is a de novo mutation not present in healthy individuals. The affected amino acid is evolutionarily conserved and is located in a functionally important domain of the protein involved in DNA binding. Molecular modelling based on crystallography data indicated that the substitution would have a deleterious effect on the protein function.
Keywords: nephrotic syndrome, SRNS, Wt1
Background
Nephrotic syndrome is a heterogeneous group of childhood disorders associated with proteinuria, hypoalbuminaemia, oedema and failure to thrive, and is usually managed by glucocorticoid treatment. Between 10% and 20% of the patients fail to respond to this therapy and are diagnosed with steroid-resistant nephrotic syndrome (SRNS).The prognosis for SRNS is usually poor, due to the increased risk of developing end-stage renal disease. Defects in several genes result in SRNS: Nphs1, Nphs2, Wt1, Actn4 and Cd2ap [1–5].
Mutations in the Wt1 gene are the second most common cause of sporadic SRNS following defects in Nphs2. Wt1 encodes a zinc-finger transcription factor with multiple isoforms, which has critical role for development of the genitourinary tract and maintenance of podocyte differentiation [6]. Dominant Wt1 mutations have been implicated in a large number of disorders such as Frasier, Denys–Drash and WAGR syndromes. SRNS alone is found to be associated with defects in exons 8 and 9 of the gene [5,7].
Here, we report on the case of a female SRNS patient with onset of the disease at 22 months of age and frequent relapses. The renal pathology in this child is due to a novel missense mutation affecting the DNA-binding zinc-finger domain of Wt1.
Case report
The patient was born to parents with no known family history of kidney disease, at the end of 28th gestational week, weighing 950 g with signs of respiratory distress syndrome and anaemia. She was diagnosed with nephrotic syndrome with oedema, erythrocyturia and albuminuria (4 g/L per 24 h) at 22 months of age. Ultrasound examination revealed hyperechogenic parenchyma. The female sex of the child was confirmed by STR analysis. Due to lack of response to steroid treatment, cyclophosphamide was introduced but failed to contain proteinuria (2.83 g/L per 24 h, blood pressure 115–200 mmHg systolic and 70–100 mmHg diastolic). An alternate regimen of immunosuppressant (cyclosporine and mycophenolate), ACE inhibitor and corticosteroid was established. At present, the child is 6 years old, with levels of proteinuria 1.11 g/L per 24 h.
Genetic testing for mutations in the most commonly affected genes, Nphs2 and Wt1, was carried out. While no defects were found in the podocin gene, a previously unknown nucleotide substitution, C > A in position 1184, was identified in Wt1. On the amino acid level, this results in replacement of serine with tyrosine in position 395. The substitution was found neither in the parents of the patient nor in any of the 120 unrelated control samples from individuals with no known kidney disorder.
A ClustalW2 multiple species alignment of the sequence surrounding serine 395 indicated that the affected residue is conserved among organisms as diverse as Xenopus, mouse, chimpanzee and human (Figure 1) [8].
Fig. 1.

ClustalW2 multiple species alignment of the area surrounding serine 395 residue. Ser395 is indicated with an arrowhead. All other conserved residues are designated with asterisks.
According to the Wt1 3D structure, Ser395 is located at the first turn of the α-helix in zinc-finger 3 (zf3) [9]. To investigate the effect of S395Y on Wt1, we used the mutation tool in COOT [10]. Analysis of the predicted mutant model revealed that the tyrosine’s bulky side chain would clash with DNA in two of three possible rotamers, while in the third rotamer, there would be a collision with Phe383 of the protein (Figure 2).
Fig. 2.
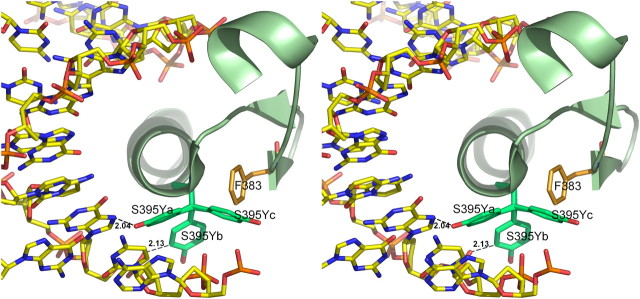
Stereo diagram of the effect of the Ser395Tyr substitution on zf3. The α-helix of Wt1 zinc-finger 3 is shown as secondary structure model with the three possible rotamers of Ser395 mutated to Tyr (designated as S395Ya, S395Yb and S395Yc, respectively) as well as the neighbouring residue Phe383. DNA is represented with stick model.
Discussion
We identified a novel heterozygous nucleotide change, C1184A, in exon 9 of Wt1 in a SRNS patient. On the protein level, this genetic variant leads to substitution of serine with tyrosine at position 395. The absence of the mutation in both parents indicates a de novo origin.
According to the data in the Human Gene Mutation Database (HGMD), missense mutations are the most common cause of Wt1-associated diseases. This poses difficulties in elucidating the genotype–phenotype correlations since the effect of a substitution on the protein level may be difficult to predict. The absence of the respective variant in healthy individuals and the evolutionary conservation of the affected residues are often reliable indications for the pathogenic character. This is the case with the C1184A substitution we report here. It was found to be absent among healthy controls and affects an evolutionarily conserved residue in a functionally important domain of the protein. However, in order to understand the correlation between genetic defect and pathology, one could use molecular models based on the crystal structure data for predicting the effect on the protein level. This approach has been used successfully for different genetic disorders.
A study of the molecular structure of Wt1 reveals the important role of serine 395 for correct folding of the zf3 and localization of the α-helix in the major groove of DNA [9]. The residue is conserved in all Wt1 zfs and also in the homologous domain of the ZIF268 protein. The structure of the Wt1 complex with the 14-bp DNA duplex showed that zf3 along with zf2 and zf4 makes base-specific contacts in the major groove. By introducing Ser395Tyr in the model, we could show that the bulky Tyr side chain would either prevent the proper folding of the protein or interfere with the DNA binding; hence, the mutation would have a negative impact on the function of Wt1 (Figure 2).
In summary, we have identified a new nucleotide change in Wt1, which results in serine to tyrosine substitution in position 395. This is a de novo mutation, not present in healthy individuals. Ser395Tyr affects an important, evolutionarily conserved part of the transcription factor responsible for DNA binding. Molecular modelling indicated that the serine residue is essential for the proper functioning of Wt1. Taken together, these results allow us to conclude that C1184A is pathological by nature.
Acknowledgments
This study was funded under Medical University-Sofia grant (MU-14/2008).
Conflict of interest statement. None declared.
References
- 1.Boute N, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan JM, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 3.Kestila M, et al. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 4.Kim JM, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300:1298–1300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 5.Mucha B, et al. Mutations in the Wilms’ tumor 1 gene cause isolated steroid resistant nephrotic syndrome and occur in exons 8 and 9. Pediatr Res. 2006;59:325–331. doi: 10.1203/01.pdr.0000196717.94518.f0. [DOI] [PubMed] [Google Scholar]
- 6.Wagner KD, Wagner N, Schedl A. The complex life of WT1. J Cell Sci. 2003;116:1653–1658. doi: 10.1242/jcs.00405. [DOI] [PubMed] [Google Scholar]
- 7.Ruf RG, et al. Prevalence of WT1 mutations in a large cohort of patients with steroid-resistant and steroid-sensitive nephrotic syndrome. Kidney Int. 2004;66:564–570. doi: 10.1111/j.1523-1755.2004.00775.x. [DOI] [PubMed] [Google Scholar]
- 8.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 9.Stoll R, et al. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J Mol Biol. 2007;372:1227–1245. doi: 10.1016/j.jmb.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]