

Abstract
Familial lecithin:cholesterol acyltransferase deficiency (FLD) is an autosomal recessive disorder characterized by corneal opacity, hemolytic anemia, low high-density lipoprotein cholesterol (HDL-C) and proteinuria. Two novel lecithin:cholesterol acyltransferase (LCAT) mutations[c.278 C>T (p.Pro69Leu); c.950 T>C (p.Met293Thr)] were identified in a 27-year-old man and in a 30-year-old woman, respectively. Both patients manifested corneal opacity, hemolytic anemia, low low-density lipoprotein cholesterol and HDL-C and proteinuria. Lipid deposits with vacuolar lucent appearance in glomerular basement membranes were observed in both cases. APOE genotype was also investigated: the first case results ϵ4/ϵ3, the second ϵ2/ϵ2; however, they shared a similar phenotype characterized by the presence of intermediate-density lipoproteins (IDL) remnant and the absence of lipoprotein-X. In conclusion, our findings suggest that APOE ϵ2/ϵ2 may not be the major determinant gene for the appearance of IDL in FLD patients.
Keywords: APOE genotype, familial LCAT deficiency (FLD), IDL remnant, lipoprotein-X
Background
Mutations in the lecithin:cholesterol acyltransferase (LCAT) gene cause either familial lecithin:cholesterol acyltransferase deficiency (FLD) (OMIM# 245900) or fish-eye disease (FED, OMIM# 136120) [1]. FLD patients are characterized by a significant reduction in serum high-density lipoprotein cholesterol (HDL-C) level, corneal opacity, normochromic anemia and proteinuria with progression to end-stage renal disease [1, 2]. Plasma low-density lipoprotein cholesterol (LDL-C) levels are widely variable among FLD patients although plasma LCAT activity catalyzing HDL (LCATα activity) and catalyzing apolipoprotein B containing lipoproteins (LCATβ activity) is absent or nearly absent. The loss of LCAT activity leads to increased levels of phosphatidylcholine and free cholesterol (FC) in the blood and to the formation of an abnormal lipoprotein called lipoprotein-X (Lp-X). As a result, accumulation of FC occurs in various tissues, including the cornea, erythrocyte membrane and the kidney [3]. Recent report suggests an etiological role for Lp-X in the development of glomerulosclerosis [4].
Here, we describe two novel mutations of LCAT gene, a C to T transition in Exon 2 converting proline 69 to leucine (Pro69Leu) and a T to C transition in Exon 6 converting methionine 293 to threonine (Met293Thr). Since Baass et al. [5] hypothesized that the presence of the APOE ε2 allele contributed to the accumulation of intermediate-density lipoprotein (IDL) in the patients with LCAT deficiency, we investigated their APOE genotypes and phenotypic manifestations of the patients with FLD.
Case reports
Case presentation
Case 1.
A 27-year-old Japanese man was referred to Okayama University Hospital because he had extremely low HDL-C and LDL-C concentrations. On physical examination, he showed bilateral corneal opacity, but no other abnormal findings. Laboratory findings demonstrated a mild degree of normochoromic-normocytotic anemia (11.3 g/dL). Although his renal function was normal, (+1) proteinuria was revealed via the dipstick method (0.21 g/day). The consanguinity was found in his parents as shown in Figure 1A.
Fig. 1.
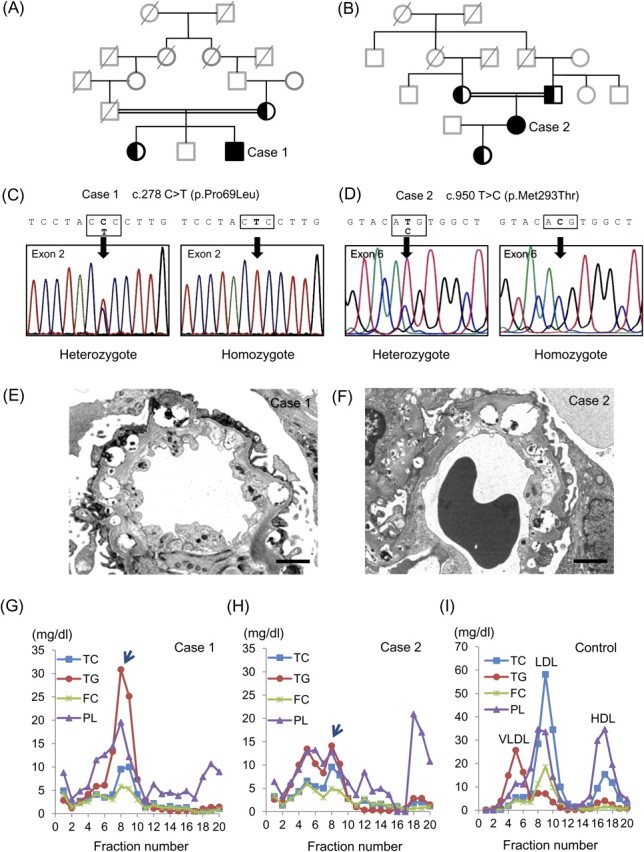
Presented cases with FLD. (A, B) Family trees of Cases 1 and 2. Squares are male, circles are female. Filled symbols indicate homozygous carriers and left filled symbols indicate heterozygous carriers. The deceased subjects are indicated with the oblique lines. Members without genetic analysis are presented in gray tone. The consanguinity of patients' parents are indicated by doubled line. (C, D) Determination of the LCAT gene sequences of proband and their families. Heterozygous mutation of mother and elder sister and homozygous mutation of proband are shown in Cases 1 (C) and 2 (D). (E, F) Renal biopsy findings of Cases 1 and 2. Electron micrograph shows lipid deposits with a vacuolar lucent appearance in the GBM (bar = 2 μm). (G–I) High-performance liquid chromatography patterns of Cases 1 (G), 2 (H) and normal healthy control (I).
Case 2.
A 30-year-old Japanese woman was referred because of very low HDL-C and LDL-C associated with (1+) proteinuria via dipstick (0.08 g/day). She also showed bilateral corneal opacity and mild hemolytic anemia (11.2 g/dL), demonstrating target cells on her peripheral blood smear. Her renal function was within normal range. She was a daughter of consanguineous parents (Figure 1B).
Sequencing of LCAT gene, renal biopsy findings and LCAT activity
Direct sequencing of LCAT gene and comparison with reference sequence (NM_000229) revealed that Case 1 had a C to T nucleotide substitution in Exon 2 resulting in Pro69Leu [c.278 C>T (p.Pro69Leu)] (Figure 1C) and Case 2 had a T to C substitution in Exon 6 resulting in Met293Thr mutations [c.950 T>C (p.Met293Thr)] (Figure 1D). Both cases showed homozygous mutation and their parents showed heterozygous mutation. We further examined the APOE genotype of the presented cases. Case 1 carried APOE ε4/ε3 genotype, whereas Case 2 carried APOE ε2/ε2 genotype (Table 1). The mother of Case 1 carried APOE ε3/ε3 genotype and genomic DNAs were not available from the deceased father. The parents of Case 2 carried APOE ε3/ε2 genotype. Renal biopsy in both cases revealed similar histological findings; diffuse vacuolization of the glomerular basement membranes (GBM) by periodic acid-methenamine silver stain and lipid deposits with a vacuolar lucent appearance in the mesangial matrix and the GBM by electron microscopy (Figure 1E and F). In both cases, half normal LCAT activity was observed by using exogenous substrate, suggesting the diagnosis of FED. However, clinical manifestations with hemolytic anemia and renal involvement coincided with the features of FLD.
Table 1.
Characteristics and lipoprotein profiles of presented casesa
| Normal values | Case 1 | Case 2 | Mother of Case 2 | |
| Sex | Male | Female | Female | |
| Age (years) | 27 | 30 | 55 | |
| c.278 C>T (p.Pro69Leu) | Homozygous | none | none | |
| c.950 T>C (p.Met293Thr) | none | Heterozygous | Heterozygous | |
| Total cholesterol (mg/dL) | 130–220 | 84 | 73 | 193 |
| Triglycerides (mg/dL) | 40–150 | 90 | 71 | 97 |
| LDL-cholesterol (mg/dL) (direct homogenous assay) | 70–139 | 28 | 24 | 126 |
| HDL-cholesterol (mg/dL) | 41–85 | 3 | 5 | 43 |
| FC (mg/dL) | 25–60 | 61 | 50 | 55 |
| Cholesteryl ester (mg/dL) | 90–200 | 23 | 23 | 138 |
| FC/TC (%) | <28 | 72.6 | 68.5 | 28.4 |
| RemL-C (mg/dL) | 0.0–7.5 | 34.4 | 23.2 | 5.4 |
| Phospholipid (mg/dL) | 150–250 | 157.8 | 178.3 | ND |
| Apo A-I (mg/dL) | 119–155 | 46 | 69 | 124 |
| Apo A-II (mg/dL) | 25.9–35.7 | 5.3 | 5.1 | 28.3 |
| Apo B (mg/dL) | 73–109 | 60 | 40 | 113 |
| LCAT activity (nmol/mL/h/37°C) | M 67.3–108.2 | 31.7 | 29.4 | ND |
| F 53.3–95.5 | ||||
| APOE genotype | ϵ4/ϵ3 | ϵ2/ϵ2 | ϵ3/ϵ2 |
RemL-C, remnant-like particles cholesterol measured with MetaboLead RemL-C; ND, not determined.
Plasma lipids and lipoproteins
Both Cases 1 and 2 revealed similar lipid profile; LDL-C and HDL-C were extremely low (Table 1). Furthermore, FC/total cholesterol (TC) ratio was much higher than normal value, suggesting reduced activity of esterification of plasma cholesterol due to LCAT deficiency. In both cases, the lipoproteins in fraction 7–10, corresponding to IDL particle size, were characterized by triglycerides (TG)- and phospholipid (PL)-rich. Since Lp-X is FC- and PL-rich lipoprotein particle, the peaks appeared in fraction 7–10 seem to be remnant lipoproteins containing IDL remnant (Figure 1G and H; arrows). In addition, Lp-X was not detected by agarose gel electrophoresis in both cases. The remnant-like particles cholesterol was measured with MetaboLead RemL-C well-reflects IDL remnant compared with immunoseparation assay, the high values of RemL-C in both cases also support the elevation of IDL remnant.
Discussion
Lynn et al. [6] recently reported that accumulation of Lp-X in renal lesion can stimulate monocyte chemoattractant protein-1 expression in mesangial cells via nuclear factor-κB. Nishiwaki et al. [7] reported Lp-X is larger than normal LDL-C in size, and the percentages for TG, cholesteryl ester, FC and PL were 9.5, 6.2, 27.6 and 56.8% in Lp-X, respectively. In our cases, we found TG- and PL-rich remnant IDL particles, where TC and FC contents are very low. Previous research has observed that APOE ε2 allele carriers reveal decreased LDL-C and increased TG and lipoprotein remnants, whereas APOE ε4 carriers have increased LDL-C and TG in comparison to ε3/ε3 subjects [8, 9]. It has been suggested that lipoproteins of ε2 carriers have a low affinity for lipoprotein receptors, which induces decreased cholesterol delivery to the hepatocytes and a following upregulation of hepatic sterol synthesis and LDL receptors [10]. Therefore, a decreased conversion of very low-density lipoprotein into LDL and appearance of IDL is observed in ε2 carriers. Baass et al. [5] hypothesized that the presence of the APOE ε2 allele contributed to the accumulation of IDL instead of Lp-X in the patients with LCAT deficiency, but IDL remnant is even higher in Case 1 carrying APOE ε4/ε3 genotype compared with Case 2 with APOE ε2/ε2 genotype (Figure 1G and H). The discrepancy of these observations may attribute to the difference in mutations of LCAT gene rather than APOE genotype and further characterization of enzymatic activities of mutated gene products is required in future investigations.
In conclusion, we describe two unrelated FLD patients with corneal opacity, hemolytic anemia, abnormalities in serum lipoprotein profile and lipid accumulation in GBM. Instead of Lp-X, we detected IDL remnant in both cases and similar pathological findings in renal biopsy. Genotyping APOE gene revealed APOE ε4/ε3 in Case 1 and a rare association of APOE ε2/ε2 in Case 2, thus APOE ε2/ε2 may not be the major modifier gene for the appearance of IDL in FLD patients. More cases need to be investigated to draw the definitive conclusions on the relation among LCAT mutation, APOE genotype and lipoprotein profile in FLD patients.
References
- 1.Kuivenhoven JA, Pritchard H, Hill J, et al. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res. 1997;38:191–205. [PubMed] [Google Scholar]
- 2.Funke H, von Eckardstein A, Pritchard PH, et al. Genetic and phenotypic heterogeneity in familial lecithin: cholesterol acyltransferase (LCAT) deficiency. Six newly identified defective alleles further contribute to the structural heterogeneity in this disease. J Clin Invest. 1993;91:677–683. doi: 10.1172/JCI116248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerin M, Dolphin PJ, Chapman MJ. Familial lecithin:cholesterol acyltransferase deficiency: further resolution of lipoprotein particle heterogeneity in the low density interval. Atherosclerosis. 1993;104:195–212. doi: 10.1016/0021-9150(93)90191-v. [DOI] [PubMed] [Google Scholar]
- 4.Lambert G, Sakai N, Vaisman BL, et al. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem. 2001;276:15090–15098. doi: 10.1074/jbc.M008466200. [DOI] [PubMed] [Google Scholar]
- 5.Baass A, Wassef H, Tremblay M, et al. Characterization of a new LCAT mutation causing familial LCAT deficiency (FLD) and the role of APOE as a modifier gene of the FLD phenotype. Atherosclerosis. 2009;207:452–457. doi: 10.1016/j.atherosclerosis.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 6.Lynn EG, Siow YL, Frohlich J, et al. Lipoprotein-X stimulates monocyte chemoattractant protein-1 expression in mesangial cells via nuclear factor-kappa B. Kidney Int. 2001;60:520–532. doi: 10.1046/j.1523-1755.2001.060002520.x. [DOI] [PubMed] [Google Scholar]
- 7.Nishiwaki M, Ikewaki K, Bader G, et al. Human lecithin:cholesterol acyltransferase deficiency: in vivo kinetics of low-density lipoprotein and lipoprotein-X. Arterioscler Thromb Vasc Biol. 2006;26:1370–1375. doi: 10.1161/01.ATV.0000217910.90210.99. [DOI] [PubMed] [Google Scholar]
- 8.Heeren J, Beisiegel U, Grewal T. Apolipoprotein E recycling: implications for dyslipidemia and atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:442–448. doi: 10.1161/01.ATV.0000201282.64751.47. [DOI] [PubMed] [Google Scholar]
- 9.Topic A, Spasojevic Kalimanovska V, Zeljkovic A, et al. Gender-related effect of apo E polymorphism on lipoprotein particle sizes in the middle-aged subjects. Clin Biochem. 2008;41:361–367. doi: 10.1016/j.clinbiochem.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 10.Mahley RW, Rall SC., Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]