

Abstract
Estrogens are disease modifiers in PAH. Even though female patients exhibit better right ventricular (RV) function than men, estrogen effects on RV function (a major determinant of survival in PAH) are incompletely characterized. We sought to determine whether sex differences exist in RV function in the SuHx model of PAH, whether hormone depletion in females worsens RV function, and whether E2 repletion improves RV adaptation. Furthermore, we studied the contribution of ERs in mediating E2’s RV effects. SuHx-induced pulmonary hypertension (SuHx-PH) was induced in male and female Sprague-Dawley rats as well as OVX females with or without concomitant E2 repletion (75 μg·kg−1·day−1). Female SuHx rats exhibited superior CI than SuHx males. OVX worsened SuHx-induced decreases in CI and SuHx-induced increases in RVH and inflammation (MCP-1 and IL-6). E2 repletion in OVX rats attenuated SuHx-induced increases in RV systolic pressure (RVSP), RVH, and pulmonary artery remodeling and improved CI and exercise capacity (V̇o2max). Furthermore, E2 repletion ameliorated SuHx-induced alterations in RV glutathione activation, proapoptotic signaling, cytoplasmic glycolysis, and proinflammatory cytokine expression. Expression of ERα in RV was decreased in SuHx-OVX but was restored upon E2 repletion. RV ERα expression was inversely correlated with RVSP and RVH and positively correlated with CO and apelin RNA levels. RV-protective E2 effects observed in females were recapitulated in male SuHx rats treated with E2 or with pharmacological ERα or ERβ agonists. Our data suggest significant RV-protective ER-mediated effects of E2 in a model of severe PH.
Keywords: sex differences, right ventricular failure, exercise capacity, apoptosis, estrogen receptor
pulmonary arterial hypertension (PAH; WHO group 1 pulmonary hypertension) is a sexually dimorphic disease of multifactorial etiology that, if left untreated, results in severe pulmonary vascular remodeling and profound hemodynamic alterations, eventually leading to right ventricular (RV) failure and death (44, 51, 52). Despite the development of several new drugs over the last 20 years, morbidity and mortality of PAH remain high, and the 3-year survival of incidence patients in the modern treatment era is only 54.9% (19).
Despite the striking female predominance with female-to-male ratios of 1.4–4.1:1 in modern registries (reviewed in Ref. 24), women with PAH exhibit better survival than men (3, 19, 41). This has been referred to as the “estrogen paradox” of PAH (9). Interestingly, female PAH patients have a higher RV ejection fraction (RVEF) than their male counterparts (22), and recent studies demonstrate that the more preserved RVEF in women is a significant contributor to the female survival advantage in PAH (20). These sex differences in RV function are likely mediated at least in part by biologically relevant effects of sex hormones, evidenced by studies demonstrating correlations of estrogen levels with RV function in healthy postmenopausal hormone replacement therapy users (55) and by studies demonstrating higher CIs in female PAH patients compared with male patients that are not observed in patients older than 45 years (56). These data suggest that to understand sex differences in mortality of PAH patients, it is imperative to understand the effects of sex hormones on the RV. A better understanding of these mechanisms may lead to novel therapeutic strategies for PAH patients of either sex. In addition, such studies are in line with the recent NIH initiative to reduce sex bias in research (8).
Although previous studies focused on evaluating estrogens effects in the pulmonary vasculature (reviewed in Refs. 1, 24), these studies were performed in milder forms of pulmonary hypertension (PH) and did not specifically investigate the effects of estrogens on the RV. However, since RV function is a critical determinant of survival in PAH (54), specific studies of E2 effects in the RV are needed. To date, only one study specifically investigated effects of E2 on RV function (29). However, this study was performed using the SuHx mouse model, a model that only incompletely recapitulates the human phenotype owing to its relatively mild degree of RV dysfunction (13, 29, 50). Furthermore, the study was limited to female mice. We therefore investigated the effects of sex hormones on RV function in male and female animals with severe angioproliferative PH and well-established RV dysfunction, using the rat SuHx model of PAH and RV failure. This model is characterized by several maladaptive processes also observed in human RV failure [e.g., cardiomyocyte apoptosis, inflammation, oxidative stress, capillary rarefication, and mitochondrial dysfunction (6, 7)] and thus provides a clinically relevant platform to study sex hormone signaling in the RV.
The aims of this study were to determine whether sex differences exist in SuHx-PH and whether E2 exerts beneficial effects on RV function and RV maladaptive remodeling in the failing RV. Since we previously demonstrated that E2 mediates its effects in PH via activation of ERs (23), and since ERs are present in cardiomyocytes (38), we also sought to determine the contribution of the 2 ER subtypes (ERα and ERβ) in mediating E2 effects on the RV. We demonstrate that sex hormone depletion worsens RV function and RV maladaptive remodeling in SuHx-PH, whereas treatment with E2 has protective effects on these parameters in both sexes. Studies of ER expression and activation suggest a role of both ERα and ERβ in mediating protective E2 effects on the RV, with a more prominent contribution of ERα.
MATERIALS AND METHODS
Animal care.
Studies were performed in age-matched adult male and female Sprague-Dawley rats (175–200 and 150–175 g, respectively; Charles River, Wilmington, MA). Animals were allowed ad libitum access to food and water for the duration of the experimentation. All animals received care in compliance with the Guide for the Care and Use of Laboratory Animals. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Indiana University School of Medicine.
Su5416/hypoxia exposure and experimental groups.
Su5416 (20 mg/kg subcutaneously; dissolved in DMSO; Sigma Aldrich, St. Louis, MO) was administered immediately prior to hypoxia exposure. Hypoxia exposure occurred in a hypobaric chamber (Patm = 362 mmHg; equivalent to 10% FiO2) as described previously (23). After 3 wk of hypoxia exposure, animals were returned to room air for another 4 wk, followed by exercise testing, echocardiography, and hemodynamic assessment (all described below). Three different sets of experiments were performed: In the main experiment, we tested the role of sex and endogenous sex hormones in male and female SuHx rats, OVX females, and OVX females replete with E2. In complementary experiments, we evaluated whether E2 effects demonstrated in female OVX SuHx rats can be replicated in male SuHx rats and whether these effects can be recapitulated with administration of selective ERα- or ERβ-agonists. A timeline and overview of the described experiments and experimental groups is provided in Fig. 1.
Fig. 1.

Experimental groups and timeline of the described experiments. Experimental groups for the main experiment and the 2 complementary experiments are shown in the boxes at the top of the figure. Timeline, experimental procedures and end points are shown at bottom. SuHx-PH was generated in male or female age-matched Sprague-Dawley rats by administration of Su5416 (Sugen) followed by 3 wk of hypobaric hypoxia [Patm = 362 mmHg; equivalent to 10% FiO2] and 4 wk of reexposure to room air. Subsets of female SuHx rats underwent OVX ± repletion of E2. OVX was performed 1 wk prior to Su5416 administration. E2 pellets were implanted at the time of OVX. Subsets of male rats were treated with E2, PPT (ERα agonist), DPN (ERβ agonist), or vehicle pellets (implanted during a dedicated session 1 wk prior to SuHx induction). Male and female rats not exposed to Su5416 or hypoxia served as controls. Maximal oxygen consumption (V̇o2max) was determined at least 48 h prior to echocardiography (Echo), hemodynamic assessment, and euthanasia. RVSP, right ventricular systolic pressure; sq, subcutaneously.
Drug administration.
All drugs were delivered via subcutaneous (sq) pellets (Innovative Research of America, Sarasota, FL). Drugs used were E2 p75 μg·kg−1·day−1 (23)], ERα-selective agonist PPT [850 μg·kg−1·day−1 (53)], ERβ-selective agonist DPN [850 μg·kg−1·day−1 (53)], as well as their vehicles (100% ethanol). The E2 dose used results in levels in the physiological range for adult female Sprague-Dawley rats, as reported before (45) and as confirmed by measurement of plasma E2 concentrations (see results). Pellets were implanted under isoflurane anesthesia (2%) by sterile technique via a 5-mm skin incision over the nape of the neck. The skin was closed with sterile stainless steel wound clips. Duration of the procedure was ∼5 min.
Ovariectomy.
In selected SuHx females, OVX was performed under isoflurane anesthesia by using sterile technique. Ovaries were resected through small bilateral flank incisions. Through the skin incision, the muscle wall was incised 1 cm lateral to the midline just below the last rib to enter the abdominal cavity. The periovarian fat pad was located and gently grasped and exteriorized. The ovary was identified, and care was taken to not directly handle the ovary to avoid abdominal implantation of ovarian tissue. A single absorbable suture ligature was placed on the ovarian pedicle. While the periovarian fat pad was held with forceps, the fallopian tube between the fat pad and uterus was clamped and crushed by use of mosquito hemostats. Following release of the hemostats, the crushed area was cut with scissors and the fat pad with the ovary was removed. The abdominal wall muscle layer was then closed with absorbable suture. Stainless steel wound clips were used for skin closure. Duration of the surgical procedure was ∼10–15 min. OVX animals that also underwent E2 replacement received estrogen pellets during the same session than OVX. Peri- and postoperative pain control was achieved with scheduled doses of ketoprofen (5 mg/kg sq at the time of surgery) and buprenorphine (0.05 mg/kg sq at the time of surgery and 8 and 16 h after surgery, respectively).
Exercise testing.
To determine exercise capacity [a clinically relevant marker of RV function and PH severity (26)], subsets of animals underwent treadmill familiarization running starting at 2 wk prior to the scheduled exercise test date. This consisted of running 5 min/day and 4×/wk at progressively faster pace (6–20 m/min) and incline (0–20°). This workload was selected to promote familiarization without promoting a training effect (25). After the completion of the 4-wk reexposure to room air, animals completed exercise testing for determination of V̇o2max. Metabolic measurements were obtained by use of an indirect open-circuit calorimetric system (Oxymax, Columbus Instruments, Columbus, OH). A gas analyzer was calibrated with standardized gas mixtures (Praxair, Danbury, CT), and baseline measurements were collected until V̇o2 consumption stabilized. A graded exercise test was performed, consisting of a stepwise increase in treadmill speed and incline stages every 3 min as follows: Stage I 10 m/min at 0°, Stage II 10 m/min at 5°, followed by an increase by 5 m/min and 5° for each consecutive stage. The test was terminated when V̇o2 plateaued despite increasing workload, or if the rat was unable to maintain position on the treadmill belt despite three consecutive electrical shocks to the tail without recovery (2). All V̇o2max values are expressed relative to body weight, and the highest achieved V̇o2 was recorded as the V̇o2max. To avoid confounding acute effects of exercise testing on echocardiographic, hemodynamic, and biochemical end points, at least 2 days were allowed between exercise testing and further assessments.
Echocardiography.
Rats were lightly anesthetized with 1–2% isoflurane via nose cone. Two-dimensional short axis and long axis images were acquired by using a high-resolution ultrasound system (Vevo 2100, VisualSonics, Toronto, ON, Canada) equipped with an 18–38 MHz scan probe. Short axis M mode was utilized to measure RV thickness. Long axis images were used to measure the RVOT at the level of the tricuspid valve at midsystole. Doppler was used to identify maximal velocities within the proximal pulmonary artery and to record VTI; the latter was calculated with Vevo Analysis software on three serial cardiac cycles. Stroke volume (SV) was determined by using the formula SV = VTI × 3.142 × (½ RVOT)2, with CO being calculated as CO = SV × heart rate (HR). Upon determination of all echocardiographic end points, rats were recovered from anesthesia.
Hemodynamic assessment and euthanasia.
After a recovery period of at least 4 h following echocardiography, RVSP was measured via right subclavian vein catheterization under isoflurane anesthesia (2%) with a 2-F Millar catheter (Millar Instruments, Houston, TX) as described previously (23). Animals were orotracheally intubated and mechanically ventilated (100% FiO2) during measurements; airway pressures were monitored and kept <5 cmH2O. All measurements were obtained during euthermia and at normal pH. Mean arterial pressure was measured by left carotid artery cannulation with PE-50 tubing. Once all hemodynamic measurements were obtained, animals were exsanguinated via the arterial line while anesthesia was maintained. Blood was collected in heparinized tubes and then centrifuged at 2,400 rpm for 30 min at 4°C. The supernatant was then centrifuged at 14,000 rpm at 4°C for another 10 min. Plasma was collected and immediately frozen at −80 degrees for later analysis.
Other in vivo end points.
Hematocrit (Hct) levels were determined from arterial blood by use of i-STAT analyzer (Abbott Point of Care, Princeton, NJ). After Su5416 administration, animals were weighed once a week, on the day of exercise testing, and upon euthanasia. Weight gain during PH development is expressed as percentage change from weight upon PH induction [(euthanasia weight − weight upon Su5416 administration)/weight upon Su5416 administration].
RVH was assessed by measuring the Fulton index (weight of RV divided by weight of the LV plus septum; RV/[LV+S]) as described previously (23). Immediately after determination of RV and LV+S weights, sections of the RV were snap-frozen for further biochemical analyses or immersed in 10% buffered formalin for immunohistochemistry studies.
Pulmonary vascular remodeling.
Lungs were flushed with normal saline through a catheter in the PA until clear return was obtained from the left atrium. The left lung was then inflated via the trachea with 10% buffered formalin under constant pressure (20 mmHg), immersed in 10% buffered formalin, and embedded in paraffin. Verhoeff-Van Gieson staining was performed on lung sections. Pulmonary vascular wall area was then determined in a blinded fashion by tracking the inner and outer border of the pulmonary arterial wall by use of Kodak software (×20 objective) and by dividing the measured area (pulmonary wall area) by the entire vessel area (entire area surrounded by outer pulmonary arterial wall border; thus representing pulmonary wall area + lumen area). Pulmonary vascular remodeling was expressed as the ratio of the pulmonary wall area to the entire vessel area. Only vessels <200 μm were included in the analysis. At least 10 vessels per animal were assessed. PAs were identified by proximity to terminal bronchioles or alveolar ducts as described previously (23). Images were obtained by using a Nikon Eclipse 80i microscope with camera and NIS-Elements 4.0 software (Nikon Instruments, Melville, NY).
Western blotting was performed with the following antibodies: mouse monoclonal anti-vinculin (clone cp74, CalBiochem/EMD Millipore, Billerica, MA); rabbit monoclonal anti-aromatase (Abcam, Cambridge, MA); and rabbit polyclonal anti-Bax (Cell Signaling, Danvers, MA), anti-Bcl2 (Cell Signaling), anti-GPR30 (clone N-15, Santa Cruz, Dallas, TX), anti-ERα (clone HC-20, Santa Cruz), and anti-ERβ (clone H-150, Santa Cruz). Secondary antibodies were anti-mouse horseradish peroxidase (HRP) and anti-rabbit-HRP from GE Healthcare (Little Chalfont, Buckinghamshire, UK) and Cell Signaling, respectively. Tissue was homogenized in ice-cold RIPA buffer (ThermoFisher Scientific, Waltham, MA) containing proteinase inhibitor cocktail (Sigma) and PhosSTOP phosphatase inhibitor cocktail (Roche, Indianapolis, IN). Protein concentration was measured by the BCA protein assay (ThermoFisher Scientific). Western blots were performed as previously described (23) in whole lung homogenates and in tissue homogenates from the RVOT. All primary antibodies were used at a dilution of 1:1,000 in 5% BSA in TBST (25 mM Tris, 1 M NaCl, 1% Tween 20), except for anti-Bax and anti-GPR30 antibodies, which were diluted 1:250 and 1:500, respectively, in 5% BSA in TBST. Secondary antibodies were diluted 1:5,000 in 5% BSA in TBST, except for GPR30 blots, which was used at a dilution of 1:2,000.
Real-time RT-PCR.
Total RNA from 10–30 mg snap-frozen RVOT tissue was extracted using the RNeasy Fibrous Tissue kit (Qiagen, Germantown, MD) and quantified with a NanoDrop 2000 Spectrophotometer (ThermoScientific, Wilmington, DE). To generate cDNA for real-time PCR analysis, 1 μg total RNA was reverse transcribed using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen Life Technologies, Carlsbad, CA). Real-time PCR reactions were performed using the Applied Biosystems 7500 Real-time PCR system. TaqMan gene expression assay primers that crossed exon boundaries were selected to avoid contamination of genomic DNA (Table 1). Changes in mRNA expression were determined by the comparative CT (ΔΔCt) method. Data were normalized to Hprt1 and expressed as fold change to male normoxia controls.
Table 1.
Overview of primers used
| Gene | TaqMan Assay | Protein |
|---|---|---|
| Hprt1 | Rn01527840_m1 | Hypoxanthine phosphoribosyltransferase 1 |
| Apln | Rn00581093_m1 | Apelin 1 |
| Ccl2 | Rn00580555_m1 | Monocyte chemotactic protein1 |
| Il6 | Rn0140330_m1 | Interleukin 6 |
Caspase-3 activity was determined in RVOT homogenates using a fluorescence-based quantitative caspase-3/7 activity assay (Promega, Madison, WI) as described previously (43).
RV glutathione (GSH) concentration was determined by using a GSH assay kit (Cayman Chemical, Ann Arbor, MI) on RVOT homogenates according to the manufacturer's instructions. The assay is based on the reaction of GSH with DTNB [5,5′-dithio-bis-2-(nitrobenzoic acid); Ellman's reagent] and utilizes glutathione reductase for the quantification of GSH. GSH concentration was determined by the end point method.
RV GLUT1 expression.
GLUT1, a marker of mitochondrial dysfunction and cytoplasmic glycolysis (47), was measured in cryosectioned RV tissue from the RVOT. After application of 3% BSA blocking solution, anti-rabbit GLUT1 (Abcam no. ab652) was applied at 1:150 dilution overnight at 4°C. Alexa Fluor 568 anti-rabbit secondary (Life Technologies no. A10042, Grand Island, NY) was applied the following day at 1:150 dilution for 45 min in a dark room. Wheat germ agglutinin (a cell surface marker) and 4′-6-diamidino-2-phenylindole (DAPI) were then applied in sequence. Slides were imaged by immunofluorescence microscopy at ×40. Imaging thresholds were set to detect staining above the degree of autofluorescence as determined by negative controls. Images were analyzed by a blinded investigator using ImageJ software (NIH, Baltimore, MD). Five images per animal were analyzed.
RV estrogen receptor immunolocalization.
A piece of the RV free wall apex was immersed in 10% buffered formalin, embedded in paraffin, and sectioned in to 4-μm-thick slices. RV tissue was then stained with antibodies for ERα (1:200; Abcam) or ERβ (1:10; Dako, Carpinteria, CA). Dako EnVision+ polyclonal rabbit kit and EnVision+ monoclonal rabbit kit, respectively, were used for visualization. Images were obtained by using a Nikon Eclipse 80i microscope with camera and NIS-Elements 4.0 software.
E2 plasma concentration measurements.
Plasma samples were thawed on ice and E2 levels were measured by using Calbiotech Mouse/Rat E2 ELISA (Calbiotech, Spring Valley, CO) according to the manufacturer's instructions. The sensitivity of the assay is <3 pg/ml.
Data and statistical analysis.
Results are expressed as means ± SE. Experimental groups were compared by one-way analysis of variance (ANOVA) with post hoc Tukey's or Dunnett's multiple-comparisons test (GraphPad Prism version 6.0b, La Jolla, CA). Where appropriate, 2-way ANOVA was performed. Kruskal-Wallis ANOVA by ranks was performed on nonparametric data. Student's t-test was performed if analysis was limited to two experimental groups. Correlation analyses were performed by determining Pearson's correlation coefficient. Differences at an alpha level of 0.05 (P < 0.05) were considered statistically significant. Data outliers were identified with GraphPad Prism. If a large number of animals was included in an experimental group, selected biochemical and structural analyses were performed in subgroups of animals only. Tissues for these analyses were randomly selected in a blinded manner by a research technician not familiar with the experimental results.
RESULTS
OVX worsens SuHx-induced increases in RV mass, whereas E2 replacement in OVX rats attenuates SuHx-induced PH. We first investigated whether sex differences exist in response to SuHx. Surprisingly, we found no significant differences between male and female rats with regards to SuHx-induced alterations in RVSP, RVH, or PA remodeling (Fig. 2, A–C). However, female SuHx rats had less weight gain during PH development (Fig. 2D) and lower Hct levels (Fig. 2E). Removal of endogenous estrogens via OVX, on the other hand, significantly worsened SuHx-induced increases in RV mass and (to a lesser degree) RVSP (Fig. 2, A and B). E2 repletion in OVX rats had dramatic effects on PH end points and led to a 50–60% decrease in RV mass and RVSP, respectively (Fig. 2, A and B). Although SuHx-induced pulmonary vascular remodeling was not affected by OVX, a significant decrease in this parameter was noted in E2-repleted OVX rats (Fig. 2C). Taken together, these data suggest that endogenous E2 has protective effects with regards to PH development in females and that E2 repletion in OVX females robustly attenuates SuHx-induced PH.
Fig. 2.

OVX worsens Su5416/hypoxia (SuHx)-induced increases in right ventricular (RV) mass, whereas E2 replacement attenuates SuHx-induced pulmonary hypertension (SuHx-PH) in OVX rats. Effects of sex, OVX, and E2 repletion on RV mass, expressed as RV weight divided by combined weight of left ventricle plus septum [RV/(LV+S)] (A), RV systolic pressure (RVSP; B), PA remodeling (assessed as %PA wall area; C), weight gain since Su5416 administration (expressed as % change from pre-Su5416 baseline; D), and hematocrit (Hct; E) in SuHx-PH rats are shown. SuHx-PH induction, OVX, and E2 repletion were as outlined in Fig. 1. Male and female rats exposed to neither Su5416 nor hypoxia served as controls. PA wall area in C was determined in a blinded fashion by tracking the inner and outer border of the PA wall after Verhoeff-Van Gieson staining, and by dividing the measured area (PA wall area) by the entire vessel area (PA wall area + lumen area). Representative immunohistochemical images are shown at left; bar graphs at right show quantified expression. Size bars = 50 μm. normox, Untreated normoxia control; SuHx, untreated SuHx groups; female OVX SuHx, ovariectomized SuHx rats; female OVX+E2 SuHx, ovariectomized female SuHx rats with concomitant E2 repletion. Values are means ± SE. *,**,***,****P < 0.05, P < 0.01, P < 0.001, P < 0.0001 vs. same-sex normoxia control; #,###P < 0.05, P < 0.001 vs. female SuHx; °,°°,°°°,°°°°P < 0.05, P < 0.01, P < 0.001, P < 0.0001 vs. female OVX SuHx; ^,^^^^P < 0.05, P < 0.0001 vs. male SuHx or normoxia control, respectively (1-way ANOVA with post hoc Tukey's test).
OVX worsens RV remodeling, whereas E2 replacement in OVX rats dramatically improves RV morphology and function. We next sought to evaluate the effects of sex, sex hormone withdrawal, and E2 repletion on RV function. Although 2D echocardiography did not demonstrate sex differences in RV free wall thickness (Fig. 3A), we noted that female SuHx-PH rats demonstrated better RV function than their male SuHx counterparts, as evidenced by a lower percentage of animals with RV notching (75% vs. 100%; Fig. 3B) and a 90% higher CI (Fig. 3C). It should be noted, however, that females also had higher CI at baseline (Fig. 3C); we therefore performed additional testing using two-way ANOVA, which demonstrated that sex did not differentially alter SuHx-induced declines in CI (evidenced by a nonsignificant interaction P value). Similar to its effects on Fulton index, OVX led to a more pronounced increase in RV free wall thickness (Fig. 3A). OVX also led to a more pronounced decrease in CI (Fig. 3C; evidenced by a statistically significant difference vs. female normoxia controls that was not observed in female SuHx). Similar to its effects on RV mass, RVSP, and pulmonary vascular remodeling, E2 repletion in OVX led to a robust improvement in RV morphology and function, evidenced by a 40% decrease in RV free wall thickness (Fig. 3A), a dramatic decrease in the percentage of animals with RV notching (Fig. 3B), and a 30% more preserved CI (Fig. 3C). To investigate the functional consequences of these findings, we investigated effects of OVX and OVX+E2 on exercise capacity, as measured by V̇o2max determination during treadmill running. Although OVX did not further worsen SuHx-induced decreases in V̇o2max, we found that E2 repletion in OVX rats significantly increased exercise capacity and restored this to values similar to those in female normoxia controls (Fig. 3D). In conglomerate, these data suggest beneficial effects of E2 on RV morphology, RV function, and exercise capacity in SuHx-PH.
Fig. 3.

OVX worsens RV remodeling in SuHx-PH, whereas E2 replacement in OVX rats improves RV morphology, RV function, and exercise capacity. Effects of sex, OVX, and E2 repletion in OVX rats on RV wall thickness (A), notching of Doppler signal in RV outflow tract (B), CI (C), and exercise capacity (determined by V̇o2max measurement via treadmill running; D) in SuHx rats are shown. RV wall thickness, RV outflow tract notching, and CI were determined by echocardiography. SuHx exposure, OVX, and E2 repletion were as outlined in Fig. 1. Male and female rats exposed to neither Su5416 nor hypoxia served as controls. Values are means ± SE, except in B, where values are expressed as percentages. Numbers listed above bars in B indicate number of animals with RV outflow tract notching/total number of animals in that group. Groups are as outlined in legend for Fig. 2. *,***P < 0.05, P < 0.001 vs. same-sex normoxia control; °,°°°P < 0.05, P < 0.001 vs. female OVX SuHx (1-way ANOVA with post hoc Tukey's test); ^P < 0.05 vs. male normoxia control, ^^P < 0.01 vs. male SuHx (2-way ANOVA with Sidak's multiple comparison test).
E2 attenuates RV oxidative stress, proapoptotic signaling, metabolic dysfunction, and proinflammatory cytokine expression in OVX rats with SuHx-PH. In an effort to identify underlying mechanisms of E2’s protective effects in the RV, we investigated E2 effects on RV oxidative stress, proapoptotic signaling, mitochondrial dysfunction, angiogenesis regulation, and proinflammatory cytokine activation, all of which have been implicated in the pathogenesis of RV dysfunction in PH (5, 15). We noticed strong trends for OVX to increase total GSH activation (indicating oxidative stress; Fig. 4A), to increase caspase-3 activation, and to decrease bcl-2/bax ratios (with the latter two indicating increased proapoptotic signaling; Fig. 4, B and C). Although OVX effects on apoptotic signaling were not statistically significant, it is noteworthy that the OVX group exhibited the highest caspase-3 activity levels and the lowest bcl-2/bax ratios of all experimental groups. Similarly, in real-time RT-PCR studies of angiogenesis and inflammation markers, OVX rats exhibited the most severe decreases in the proangiogenic and inotropic peptide apelin (4, 27) and the most severe increases in the proinflammatory cytokines MCP-1 and IL-6 (Fig. 4, E–G). In contrast, E2 repletion in OVX resulted in profound and statistically significant 30–40% decreases in GSH and caspase-3 activation, as well as 100% increase in the bcl-2/bax ratio and 50% decrease in GLUT1 expression [a marker of mitochondrial dysfunction and cytoplasmic glycolysis (47); Fig. 4, A–D]. Though not statistically significant compared with OVX, E2-replete OVX rats exhibited a 30% increase in apelin RNA (Fig. 4E). Furthermore, E2 repletion in OVX females led to a 50% decrease in MCP-1 RNA and prevented the SuHx-induced increase in this parameter that was observed in the OVX group (Fig. 4F). Lastly, E2-replete OVX rats exhibited a significant 90% decrease in IL-6 RNA levels (Fig. 4G). These data indicate potential salutary effects of E2 on RV oxidative stress, proapoptotic signaling, mitochondrial f unction, regulation of angiogenesis and/or inotropic signaling, and proinflammatory cytokine activation.
Fig. 4.

E2 replacement in OVX SuHx rats attenuates RV oxidative stress, proapoptotic signaling, cytoplasmic glycolysis and proinflammatory cytokine activation. Effects of sex, OVX, and E2 repletion in OVX rats on total glutathione (GSH) levels (A), caspase-3 activity (B), bcl-2/bax ratio (C), GLUT1 expression (D), and mRNA levels of apelin (E), MCP-1 (F), and IL-6 (G) in RV homogenates are shown. SuHx exposure, OVX, and E2 repletion were as outlined in Fig. 1. Male and female rats exposed to neither Su5416 nor hypoxia served as controls. All samples were derived from RV outflow tract. Glutathione activation was determined by glutathione assay. Caspase-3 activity was determined by fluorescence-based quantitative caspase-3/7 activity assay and is expressed as fold change from untreated male normoxia control. Bcl-2/bax ratio was determined by Western blotting and densitometric analysis; a representative Western blot is shown in C, top, with densitometry results from all samples depicted at bottom. GLUT1 expression was assessed by immunofluorescence with quantification by ImageJ; representative images are shown at left of D, with results from ImageJ analysis depicted in bar graph on right (AU, arbitrary units). Size bars = 50 μm. Apelin, MCP-1, and IL-6 were determined by real-time RT-PCR and are expressed as fold change ΔΔCt vs. male normoxia control. Groups are as outlined in legend for Fig. 2. Values are means ± SE. *,***P < 0.05, P < 0.001 vs. same-sex normoxia control; #P < 0.05 vs. female SuHx; °P < 0.05 vs. female OVX SuHx (1-way ANOVA with post hoc Tukey's or Dunnett's test).
E2 levels are inversely correlated with increases in RVH and RVSP. Contrary to our original hypothesis, we found no significant sex differences in RVH, RVSP, and pulmonary vascular remodeling in this model of PH. We thus investigated the sex hormone axis in our experimental groups. Interestingly, E2 serum levels in female SuHx rats were decreased compared with healthy controls, resulting in levels similar to those in male animals and OVX rats (Fig. 5A). Furthermore, when evaluating differences between the lowest and the highest E2 level in each group, we noted that E2 levels in SuHx females showed less fluctuation compared with healthy females (Fig. 5B). These data suggest that female SuHx rats may be relatively E2 deficient. On the other hand, and as expected, E2 levels in the OVX group were similar to those in male rats, and levels in OVX+E2 were similar to healthy female rats (Fig. 5A). Interestingly, female rats with the highest E2 levels tended to have the least pronounced increases in RVH and RVSP, resulting in moderate inverse correlations between serum E2 levels and RVH and RVSP, respectively (Fig. 5, C and D), and suggesting a relationship between higher serum E2 levels and attenuated PH end points.
Fig. 5.

Plasma E2 levels in SuHx-PH. Effects of sex, OVX, and E2 repletion in OVX rats on plasma E2 levels in SuHx-PH rats are shown in A. Plasma E2 levels were measured by Calbiotech E2 ELISA. SuHx exposure, OVX, and E2 repletion were as outlined in Fig. 1. Male and female rats exposed to neither Su5416 nor hypoxia served as controls. Values are means ± SE. °P < 0.05 vs. female OVX SuHx (1-way ANOVA with post hoc Tukey's test). B: box-and-whiskers plots of distribution of plasma E2 levels in healthy control females and in intact SuHx rats. In each group (n, 6/group), the difference (Δ) between each measured E2 level and the lowest E2 level in that group was calculated and plotted on the y-axis. Boxes represent 25th, 50th, and 75th percentiles; whiskers represent minimum and maximum E2 levels for that particular group. Note strong tendency for higher Δ in control rats compared with SuHx rats. C and D: correlations between plasma E2 levels and RV hypertrophy (determined by Fulton index; see Fig. 2A) and RVSP (see Fig. 2B) in female animals. Data points represent individual animals. Rats from the 4 female treatment groups were included in the analyses. Pearson correlation coefficients and levels of significance are shown in the boxes at top right of each panel. Note inverse relationship between plasma E2 and RV hypertrophy and RVSP, respectively.
RV ERα expression is affected by hormonal status and correlates with decreased RVSP and RVH as well as increased CO and RV apelin expression. To explain the significant effects of E2 on RV function, we next sought to determine the expression of the two main ERs [ERα and ERβ (24, 37)] in the RV. We noted that normal female controls exhibited increased abundance of ERα in the RV than their male counterparts (Fig. 6A). There was a nonsignificant trend for SuHx to decrease ERα in females, whereas OVX SuHx rats exhibited a significant 30% decrease in ERα expression (as compared with female normoxia controls; Fig. 6A). E2 administration to OVX SuHx rats restored ERα levels to values found in untreated female controls (Fig. 6A). Correlation analyses in female control and untreated, OVX, and OVX+E2 SuHx rats demonstrated that higher RV ERα expression was tightly correlated with decreased RVSP and RVH as well as increased CO and increased RV apelin RNA levels (Fig. 6B), suggesting a prominent role of ERα in mediating E2 effects on the RV. To determine the location of ERα within the RV, we performed immunohistochemistry on sections from the apex of the RV free wall. We found ERα to be strongly expressed in cardiomyocytes and in coronary artery endothelial cells (Fig. 6, C–F). ERα expression within cardiomyocytes occurred in a punctate pattern in transverse section and in a striated pattern in longitudinal sections [as has been described for ERα in the LV (32, 40)]. Interestingly, a striking absence of ERα was noted in the coronary vascular smooth muscle layer (Fig. 6, E and F). RV ERβ expression, on the other hand, was not affected by sex, PH, or hormone withdrawal or repletion (Fig. 6G), and consequentially no correlations with PH end points were found (data not shown). As with ERα, ERβ was expressed in a striated or punctate pattern in RV cardiomyocytes as well as in coronary endothelial cells (data not shown).
Fig. 6.

E2 repletion in OVX rats attenuates SuHx/OVX-induced decreases in RV ERα expression, and RV ERα expression correlates negatively with RV hypertrophy and RVSP, and positively with CO and proangiogenic and procontractile signaling. A: effects of sex, OVX, and E2 repletion in OVX rats on ERα in RV homogenates from SuHx-PH rats. SuHx exposure, OVX, and E2 repletion were as outlined in Fig. 1. Male and female rats exposed to neither Su5416 nor hypoxia served as controls. RV samples were derived from RV outflow tract. ERα expression was determined by Western blotting and densitometric analysis (normalized for vinculin control and expressed as fold change vs. male normoxia control). Representative Western blot is shown at left, with densitometry results from all samples depicted in the bar graph at right. Note that OVX in female SuHx-PH rats was associated with a decrease in RV ERα expression, which was attenuated by E2 repletion. Groups are as outlined in legend for Fig. 2. Values are means ± SE. *P < 0.05 vs. female normoxia control; °P < 0.05 vs. female OVX SuHx; ^^P < 0.01 vs. male normoxia control (1-way ANOVA with post hoc Dunnett's test). B: correlations between RV ERα expression (determined by Western blotting and densitometry normalized for vinculin; see A) and RV hypertrophy (determined by Fulton index; see Fig. 2A), RVSP (see Fig. 2B), CO (determined by echocardiography), and RV apelin RNA expression (by real-time RT-PCR; see Fig. 4E) in female rats. Data points represent individual animals. Rats from the 4 female treatment groups were included in the analyses. Pearson correlation coefficients and levels of significance are indicated in the boxes at top right of each panel. C–F: representative images of ERα expression in RV sections derived from apex of free wall of normal females (D and E) and SuHx females replete with E2 (F), the groups with the strongest RV ERα expression. Negative control is depicted in C. Note predominant ERα expression in cytoplasm of RV cardiomyocytes, which occurred in a striated pattern in longitudinal sections (long arrows in D) and in a punctate pattern in transverse sections (arrowheads in D). Note that no ERα expression was observed in smooth muscle layer of coronary vessels (E), whereas coronary endothelial cells stained positive for ERα (short arrows in E). Findings noted in normal females were recapitulated in SuHx OVX+E2 rats (F). All images are ×40, size bar = 10 μm. G–I: effects of sex, OVX, and E2 repletion in OVX rats on RV ERβ (G), GPR30 (H), and aromatase (I) expression. Representative Western blots are shown at top, with densitometry results (normalized for vinculin control and expressed as fold change vs. male normoxia control) from all samples depicted in bar graphs. No significant differences were noted by 1-way ANOVA with post hoc Tukey's or Dunnett's test.
Since recent data indicate that nongenomic E2 effects in nonreproductive systems may also be mediated by the G protein-coupled receptor GPR30 (24, 37), we also investigated the expression of this receptor in the RV. Although GPR30 was not affected by SuHx in males, we found that GPR30 tended to be decreased in female SuHx animals by 25–45%, with a particularly strong trend for a decrease in untreated SuHx rats (P = 0.06 vs. female normoxia controls; Fig. 6H). To evaluate whether there is evidence of local estrogen production in the RV, we measured aromatase [CYP19A; the major enzyme responsible for estrogen synthesis (33)] expression in RV homogenates. Although aromatase expression tended to be the lowest in OVX+E2 SuHx rats, we found no statistically significant differences between groups (Fig. 6I).
Since E2 repletion in OVX rats affected lung vascular remodeling (Fig. 2C), we also investigated ERα, ERβ, and GPR30 expression in the lung. Western blots on whole lung homogenates did not reveal any significant group differences for ERα (Fig. 7A). ERβ was higher in normal females than in males but was not affected by SuHx (Fig. 7A). OVX did not affect lung ERβ, whereas E2 replacement in OVX led to a nonsignificant increase in ERβ. Lung GPR30 expression was increased by SuHx-PH in males and by SuHx+OVX in females (Fig. 7B). E2 repletion, on the other hand, abolished the SuHx+OVX-induced increase in GPR30 abundance, suggesting an inhibitory effect of E2 on GPR30 expression in the lung.
Fig. 7.

Effects of sex, OVX, and E2 repletion in OVX rats on expression of ERα and ERβ (A) as well as GPR30 (B) in lung homogenates from SuHx-PH rats. SuHx exposure, OVX, and E2 repletion were as outlined in Fig. 1. Male and female rats exposed to neither Su5416 nor hypoxia served as controls. Lung samples were derived from right lung. ERα, ERβ, and GPR30 expression were determined by Western blotting and densitometric analysis (normalized for vinculin control). Representative Western blots are shown at top, with densitometry results from all samples depicted in bar graphs. Groups are as outlined in legend for Fig. 2. Values are means ± SE. ^P < 0.05 vs. male normoxia control; *,**P < 0.05, P < 0.01 vs. same-sex normoxia control; °P < 0.05 vs. female OVX SuHx (1-way ANOVA with post hoc Tukey's test).
E2 attenuates SuHx-induced increases in RV mass in male rats. In light of our findings implicating E2 as a major modifier of PH end points, and given the dramatic effects of E2 administration on RV function in female rats, we sought to evaluate whether E2 treatment also attenuates RV dysfunction in male SuHx rats. E2 administration to males occurred in a manner identical to that in females (75 μg·kg−1·day−1 via subcutaneous pellets), resulting in levels physiological for adult female Sprague-Dawley rats (Table 2). E2 administration led to a robust decrease in SuHx-induced increases in RV mass (Fig. 8A). Interestingly, E2 decreased neither RVSP (Fig. 8B) nor pulmonary vascular remodeling (Fig. 8C), suggesting a predominant RV-directed effect. As described before in our studies in HPH (23), E2 had negative effects on weight gain after PH development (Fig. 8D). Lastly, E2 treatment was associated with a small but significant decrease in Hct levels (Fig. 8E). Taken together, these data suggest that E2, while potentially having undesired effects on weight gain and Hct levels in male SuHx rats, and while not affecting PA remodeling, has beneficial effects on RVH.
Table 2.
E2 repletion in male SuHx-PH rats results in plasma levels similar to those in healthy females
| Group | Plasma E2 level, pg/ml |
|---|---|
| Male SuHx | 8.14 ± 0.50 |
| Male SuHx E2 | 14.34 ± 2.32* |
| Male SuHx veh | 6.35 ± 0.33 |
| Healthy female | 16.44 ± 4.95 |
Sugen/hypoxia (SuHx)-induced pulmonary hypertension (SuHx-PH) was generated by administration of Su5416 (20 mg/kg subcutaneously) followed by 3 wk of hypobaric hypoxia (atmospheric pressure Patm = 362 mmHg; equivalent to 10% inspired oxygen fraction) and 4 wk of reexposure to room air. 17β-Estradiol (E2) repletion (75 μg·kg−1·day−1 via subcutaneous pellets) was started 1 wk prior to SuHx. Plasma E2 levels were measured by Calbiotech E2 ELISA. Male SuHx, untreated male SuHx group; male SuHx E2, male SuHx rats with concomitant E2 repletion; male SuHx veh, male SuHx rats with E2 vehicle. Plasma E2 levels from healthy female rats from Fig. 5 are included for comparison. Values are means ± SE. N = 6–13/group.
P < 0.01 vs. male SuHx (1-way ANOVA with post hoc Dunnett's test).
Fig. 8.

E2 administration to male SuHx-PH rats attenuates RV hypertrophy but does not affect SuHx-induced increases in RVSP or pulmonary vascular remodeling. E2 effects on RV mass, expressed as RV weight divided by combined weight of left ventricle plus septum [RV/(LV+S)] (A), RV systolic pressure (RVSP; B), pulmonary vascular remodeling (expressed as % PA wall area; C), weight gain since Su5416 administration (expressed as % change from pre-Su5416 baseline; D), and hematocrit (Hct; E) in male SuHx-PH rats are shown. SuHx-PH induction and E2 repletion were as outlined in Fig. 1. PA wall area was determined as outlined in Fig. 2. Size bars = 50 μm. Normox, healthy male control rats (no Su5416, no hypoxia); SuHx, untreated male SuHx rats; SuHx E2, male SuHx rats with concomitant E2 repletion; SuHx veh, male SuHx rats receiving E2 vehicle. Values are means ± SE. *,**,***,****P < 0.05, P < 0.01, P < 0.001, P < 0.0001 vs. normoxia control; ##, ####P < 0.01, P < 0.0001 vs. SuHx (1-way ANOVA with post hoc Tukey's test).
E2 improves RV function and exercise capacity in male rats. Similar to female rats, E2 treatment was associated with a robust 30% decrease in RV free wall thickness (Fig. 9A), as well as a robust increase in RV function, as demonstrated by a 30% increase in VTI (a surrogate marker of RV stroke volume; Fig. 9B), a decrease in the prevalence of RVOT notching (Fig. 9C), and a doubling of CI (Fig. 9D). These changes translated into a significant improvement in exercise capacity, as evidenced by a robust 20% increase in V̇o2max (Fig. 9E). In summary, these data suggest beneficial effects of E2 on RV morphology, RV function, and exercise capacity in male rats with SuHx-PH. Viewed in light of the data indicating beneficial effects on RV mass in absence of effects on RVSP and pulmonary vascular remodeling (Fig. 8, A–C), these data further support an RV-directed mechanism of action of E2 in SuHx-PH.
Fig. 9.

E2 administration to male SuHx-PH rats improves RV morphology, RV function, and exercise capacity. Effects of E2 on RV wall thickness (A), VTI (B), notching of Doppler signal in RV outflow tract (C), CI (D), and exercise capacity (determined by V̇o2max measurement via treadmill running; E) in male SuHx rats are shown. RV wall thickness, VTI, RV outflow tract notching, and CI were determined by echocardiography. SuHx exposure and E2 administration were as outlined in Fig. 1; treatment groups are as defined in Fig. 8. Values are means ± SE, except in C, where values are expressed as percentages. Numbers listed above bars in C indicate number of animals with RV outflow tract notching/total number of animals in that group. *,**P < 0.05, P < 0.01 vs. normoxia control; #,##P < 0.05, P < 0.01 vs. SuHx and SuHx veh (1-way ANOVA with post hoc Tukey's test).
E2 attenuates maladaptive signaling in male RVs. Given the findings of improved RV function in E2-treated male rats, and given our findings of attenuated RV maladaptive signaling in E2-treated females, we hypothesized that E2 would positively affect maladaptive changes in male SuHx rats as well. Indeed, E2-treated males exhibited an attenuation of SuHx-induced alterations in proapoptotic signaling, as demonstrated by a robust increase in bcl-2/bax, as well as a decrease in caspase-3 activity (Fig. 10, A and B). In addition, E2-treated males demonstrated a significant 30% increase in apelin RNA, suggesting enhanced proangiogenic and inotropic signaling (Fig. 10C). E2 also tended to exert favorable effects on RV proinflammatory cytokine and GLUT1 expression; however, these effects were not as robust as those observed in female rats (data not shown).
Fig. 10.

E2 administration to male SuHx-PH rats E2 decreases RV proapoptotic signaling and increases RV apelin RNA levels. Effects of E2 on bcl-2/bax ratio (A), caspase-3 activity (B), and apelin RNA in RV homogenates are shown. All samples were derived from RV outflow tract. SuHx exposure and E2 administration were as outlined in Fig. 1; treatment groups are as defined in Fig. 8. Bcl-2/bax ratio was determined by Western blotting and densitometric analysis; a representative Western blot is shown in A, top, with densitometry results from all samples depicted at bottom. Caspase-3 activity was determined by fluorescence-based quantitative caspase-3/7 activity assay and is expressed as fold change from untreated male normoxia control. Apelin RNA was measured by real-time RT-PCR and is expressed as fold change ΔΔCt vs. male normoxia control. Values are means ± SE. *,***P < 0.05, P < 0.001 vs. normoxia control; #P < 0.05 vs. SuHx and SuHx veh (1-way ANOVA with post hoc Tukey's test).
RV-protective E2 effects can be recapitulated by treatment with selective ERα or ERβ agonists. Given our previous findings of ER-mediated protective effects of E2 in HPH (23), and given our data demonstrating ER expression in cardiomyocytes and implicating ERα in mediating protective E2 effects in female SuHx rats (Fig. 6), we tested whether the RV-protective effects of E2 could be recapitulated by treatment with selective ERα and ERβ agonists. We treated male rats with the ERα-selective agonist PPT or the ERβ-selective agonist DPN in doses previously shown to be effective in rat models of PH (53). Concomitant experiments were performed in E2-treated rats as well as untreated SuHx rats that were used as reference groups. PPT and DPN vehicle pellets are identical to E2 vehicle pellets found to be without effects in Figs. 8–10 and for that reason were not separately tested. Interestingly, both PPT and DPN recapitulated beneficial effects of E2 on RV function, as shown by similar increases in CI (Fig. 11A). Stroke volume index was affected in a similar pattern, even though here the ERβ agonist, despite a strong trend, did not achieve statistical significance vs. untreated SuHx (Fig. 11B). RVOT notching was observed in PPT-treated animals in a range similar to that of E2-treated rats (Fig. 11C). Effects of DPN on this parameter, on the other hand, were less pronounced (Fig. 11C). Salutary E2 effects on exercise capacity were recapitulated by both PPT and DPN, with PPT being even more potent that E2 or DPN (Fig. 11D). Both PPT and DPN tended to mimic E2 effects on RVH (Fig. 11E). Of note, potentially undesired E2 effects on Hct and weight gain were not replicated by PPT or DPN treatment (Fig. 11, F and G).
Fig. 11.

Treatment of male SuHx-PH rats with selective ERα-agonist PPT or selective ERβ-agonist DPN recapitulates beneficial effects of E2 on RV function and exercise capacity. Effects of E2, PPT, and DPN on CI (A), stroke volume index (SVI; B), notching of Doppler signal in RV outflow tract (C), exercise capacity (determined by V̇o2max measurement via treadmill running; D), RV hypertrophy (E), hematocrit (Hct; F), and weight gain since Su5416 administration (G) in male SuHx rats are shown. SuHx exposure as well as E2, PPT, and DPN administration were as outlined in Fig. 1. CI, SVI, and RV outflow tract notching were determined by echocardiography. RV hypertrophy was determined by Fulton index. Values are means ± SE, except in C, where values are expressed as percentages. Numbers listed above bars in C indicate number of animals with RV outflow tract notching/total number of animals in that group. *,****P < 0.05, P < 0.0001 vs. normoxia control; #,##,###,####P < 0.05, P < 0.01, P < 0.001, P < 0.0001 vs. SuHx; $$,$$$$P < 0.01, P < 0.0001 vs. SuHx PPT and SuHx DPN; ns, not significant (1-way ANOVA with post hoc Tukey's or Dunnett's test).
On a molecular level, whereas their effects were not as robust as those of E2, neither the PPT group nor the DPN exhibited significant SuHx-induced increases in GSH (P = not significant vs. normoxia control; Fig. 12A); administration of PPT or DPN was thus sufficient in attenuating SuHx effects on this parameter. PPT exhibited similar effects on bcl-2/bax as E2, whereas no such effects were noted for DPN (Fig. 12B).
Fig. 12.

Effects of treatment of male SuHx-PH rats with selective ERα-agonist PPT or selective ERβ-agonist DPN on glutathione (GSH) activation (A) and proapoptotic signaling (bcl-2/bax ratio; B). SuHx exposure, E2, PPT, and DPN administration were as outlined in Fig. 1. All samples were derived from RV outflow tract. Glutathione activation was determined by glutathione assay. Bcl-2/bax ratio was determined by Western blotting and densitometric analysis; a representative Western blot is shown in B, top, with densitometry results from all samples depicted in the bar graph at bottom. Values are means ± SE. *P < 0.05 vs. normoxia control; #,##P < 0.05, P < 0.01 vs. SuHx; $P < 0.05 vs. SuHx DPN (1-way ANOVA with post hoc Tukey's or Dunnett's test).
Taken together, these data suggest that both the ERα agonist and the ERβ agonist can replicate E2 effects on RV function and exercise capacity, suggesting that both ERs are involved in mediating E2 effects in males. However, the ERα agonist appeared to be more consistent and more robust in improving these end points.
DISCUSSION
To our knowledge, this is the first study to comprehensively investigate the effects of sex as well as endogenous and exogenous sex hormones in a model of severe, angioproliferative PH with significant RV dysfunction and failure. Our studies revealed several new findings. First, we demonstrate that whereas neither RVSP nor RV hypertrophy or PA remodeling differ between sexes, there are significant sex differences with regards to RV function, which was superior in females compared with males (demonstrated by a 90% higher CI). Second, we show that OVX tends to worsen relevant PAH end points related to RV hypertrophy and RV function, whereas there was a dramatic improvement in nearly all end points investigated when OVX rats were replete with E2. Third, we demonstrate that E2 attenuates pathogenetically relevant maladaptive RV processes, such as proapoptotic signaling, oxidative stress, cytoplasmic glycolysis, apelin repression, and activation of proinflammatory cytokines. Fourth, we demonstrate that OVX decreases RV ERα expression in SuHx rats and that E2 repletion increases expression of this receptor. The significant correlations between RV ERα and hemodynamic (RVSP, CO), structural (RVH), and biochemical alterations (apelin expression) strongly suggest a role of this ER in modulating RV function in PAH. Lastly, we show that protective RV effects of E2 can even be elucidated when this hormone is given to male SuHx rats (suggesting a robustness of effects across sexes), and that E2’s RV effects can be recapitulated at least in part by treatment with selective ERα and ERβ agonists, again implicating ERs as modulators of RV function in PH. The moderate correlations between ERα and PH end points in female rats and the more pronounced effects of the ERα agonist in male rats suggest that ERα may play a more prominent role in the SuHx-RV than ERβ. A summary of our results is provided in Fig. 13.
Fig. 13.
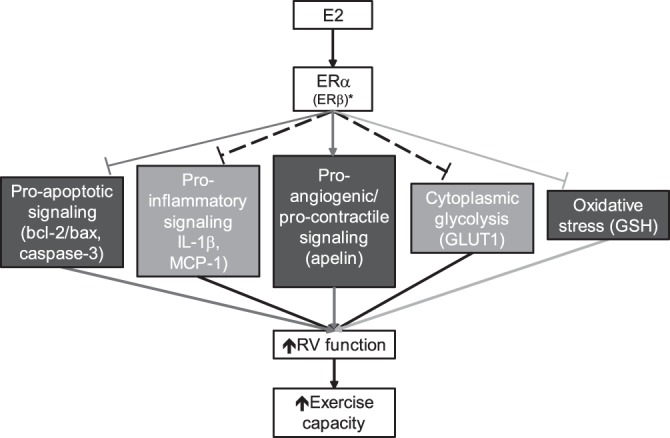
Schematic representation of experimental results. Endogenous or exogenous E2 favorably affects multiple end points linked to RV failure, thus improving RV function and exercise capacity in male and female rats with SuHx-PH. Straight arrows indicate activation by E2; T-shaped lines indicate inhibition. Dark gray lines indicate pathways primarily affected by ERα; light gray lines indicate pathways linked to both ERα and ERβ. Dashed lines indicate pathways affected by E2, but not specifically linked to ERα or ERβ. *ERβ may have a more prominent role in male animals. End points depicted in dark gray were positively affected by E2 in both female and male rats; end points depicted in light gray were significantly affected only in females.
Although RV effects of E2 have been studied in a limited number of prior investigations, these studies were limited by E2 administration to male rats only (23, 53) or by investigation of E2 effects in females with only a mild degree of RV dysfunction (29). Lastly, a recent study, although investigating sex differences in SuHx rats, did not investigate effects of OVX and/or E2 repletion and did not specifically focus on RV effects (33). Although these prior studies significantly contributed to the field, our experimental approach has several specific strengths. First, we investigate a wide range of intact and hormone-manipulated male and female animals that allows for a detailed assessment of the role of sex as well as endogenous and exogenously administered sex hormones in PH. To our knowledge, this is the most comprehensive assessment of these factors in severe angioproliferative PH to date. Second, we provide a detailed physiological, structural, and biochemical assessment of our experimental groups. A particular strength is the comprehensive echocardiographic assessment of RV function and the ability to perform V̇o2max testing. The latter, to our knowledge, has never been performed in SuHx-PH. As in humans, determination of exercise capacity in experimental PAH is of utmost importance, since it allows for an objective assessment of the functional consequences of the pulmonary vascular and RV abnormalities observed in PAH (26). Similarly, we investigate E2 effects across a wide range of relevant biochemical and molecular processes, all of which have been implicated in the pathogenesis of RV failure in PAH (5, 15). Third, we were able to achieve E2 levels in E2-treated animals that were similar to those in healthy females and thus well within the physiological range. This is in contrast to a previous study, in which E2 supplementation resulted in supraphysiological levels (29). Our results suggest that even treatment with physiological doses of E2 in either males or females is beneficial to RV function. We elected to use the Calbiotech assay because of its excellent sensitivity and excellent performance compared with the gold standard method of gas chromatography/tandem mass spectrometry (GC/MSMS) (17). High accuracy of our measurements is demonstrated by the fact that E2 levels in our samples were consistent with levels previously reported for Sprague-Dawley rats (10, 45). Furthermore, internal consistency of our samples was demonstrated by the observation that E2 levels in OVX females were similar to those measured in males, and by the finding that E2 levels in E2-repleted OVX rats were similar to those in healthy females.
Through this experimental approach, we were able to expand prior reports of beneficial effects of E2 on RV function in experimental PAH by demonstrating novel antiapoptotic and anti-inflammatory effects, as well as beneficial effects on glutathione activation, cytoplasmic glycolysis, and proangiogenic/inotropic signaling. These changes appear functionally relevant, since they were paralleled by beneficial effects on RV structure, function, and exercise capacity. Furthermore, we characterize the hormonal milieu and RV ER expression in SuHx rats, and we show that RV ERα abundance (and to a lesser degree plasma E2 levels) negatively correlates with SuHx-induced RV remodeling and hemodynamic alterations as well as with increases in CO. In addition, ERα expression also correlated with the proangiogenic and inotropic peptide apelin. Taken together, these data implicate ERα as a potential mediator of E2-mediated RV protection in females. Interestingly, RV-protective effects of E2 observed in female OVX SuHx rats could be replicated when E2 was given to male SuHx rats, albeit with the effects being somewhat less robust. This may be due to differences in E2 metabolism and/or differences in ER abundance, sensitivity, and downstream signaling (37, 38). RV effects of E2 in male SuHx rats could be replicated in part by treatment with selective ERα and ERβ agonists, suggesting that both of these ERs are implicated in mediating E2 effects on the RV. However, the respective contribution of the two main ERs to mediating E2 effects in males appears complex. Although both the ERα agonist as well as the ERβ agonist recapitulated E2’s effects, the effects of the ERα agonist were more consistent and more potent. Nevertheless, some of the effects of the selective ER subtype agonists tended to be less robust than those of E2 (e.g., effects on GSH activation or bcl-2/bax; Fig. 12), suggesting that both ERs may need to be activated simultaneously in order for E2 to achieve its maximum effects. Alternatively, non-ER-mediated mechanisms may be involved. This scenario is reminiscent of what has been described with regards to protective effects of estrogens in other organ systems. For example, neuroprotective effects of E2 in the central nervous system (CNS) are mediated through both ERα and ERβ, but ER involvement differs between cell types and employs different cellular mechanisms depending on which ER is involved (49). In addition, estrogen abundance may also affect ER involvement. For example, one paradigm proposed in the CNS is that ERα is more important at physiological E2 levels, whereas ERβ plays a stronger a role at therapeutic levels (36, 49). Consequentially, neuroprotective effects of estrogens, as well as of selective ERα- or ERβ-agonists, are currently being investigated for potential therapeutic application in various clinical conditions including multiple sclerosis, another sexually dimorphic disease that, like PAH, is characterized by female disease susceptibility yet better survival (8).
Similarly, both ERα and ERβ have been implicated in protective E2 effects in the LV, where their contribution appears to be context, cell type, and sex specific (37). Studies in various models of acute or chronic LV injury (including chronic LV pressure overload) demonstrated antifibrotic, antihypertrophic, anti-inflammatory, antiapoptotic/prosurvival, and antioxidant effects of E2, as well as beneficial effects on mitochondrial function (11, 21, 28, 39, 42, 59). Furthermore, E2 enhances nitric oxide signaling in the LV (40), and a recent study suggested that LV inotropic effects of phosphodiesterase (PDE)-5 inhibition in female rodents are estrogen-dependent (48). Both ERs appear to be involved in mediating E2 protection in the LV (28, 39, 42, 58, 60).
We found ERα to be predominantly expressed in the cytoplasm of cardiac myocytes and to a lesser degree in coronary endothelial cells. This pattern is consistent with what has been reported for ERα expression in the LV and correlates with expression in the contractile apparatus (32, 40). Interestingly, RV ERα abundance followed trends in plasma E2 levels, suggesting that E2 levels and the presence of ovaries regulate RV ERα expression, at least in part. The observation that RV ERα protein expression and E2 plasma levels tended to be decreased in female SuHx rats compared with their healthy counterparts supports the notion that a suppressed E2-ERα axis in female SuHx rats may explain the lack of more robust differences in end points between male and female SuHx rats. This observation, combined with the significant correlations of RV ERα abundance with RV function end points, suggests that enhancing ERα signaling may be a therapeutic strategy to attenuate PH-induced alterations in female RVs. RV ERβ expression, on the other hand, was not significantly affected by SuHx-PH or hormonal manipulation, suggesting that expression of this receptor is less susceptible to increases in RV afterload and/or endocrine changes.
Even though the RV is embryologically, anatomically, and physiologically different from the LV (16), our studies suggest that estrogens exert similar benefits in the pressure-overloaded RV as in the LV (38). Specifically, we observed antihypertrophic, anti-inflammatory, antiapoptotic, proangiogenic, and/or inotropic as well as potentially antioxidant effects consistent with those described in the LV. E2 effects on these end points are relevant, since all these pathophysiological processes have been implicated in the pathogenesis of RV failure (5, 47). The molecular targets of these actions remain to be determined and are the focus of mechanistic studies that are currently ongoing in our laboratory.
Like in the LV, the structural and biochemical changes evoked by E2 in the RV translate into improved cardiac function and improved exercise capacity, underlining their clinical relevance. The novel observation that RV-protective E2 effects are seen even in severe PH suggests that E2-mediated RV protection is not limited to mild PH and mild RV dysfunction [as shown previously (29)]. The clinical implication of these findings is that they may allow for the development of hormonal and potentially even nonhormonal RV directed therapies in early as well as advanced stages of PAH [a current treatment gap and clear area of need in PAH (57)].
However, protective actions of E2 on the RV cannot be discussed without considering its effects on the pulmonary vasculature. In contrast to the RV, where estrogens appear to be uniformly beneficial, the data on E2 effects on the pulmonary vasculature are discordant, with some studies showing beneficial effects while others demonstrate estrogens to be harmful (1, 24). Similarly, our data suggest that E2 in SuHx-PH has differential effects on pulmonary vascular remodeling. Although neither OVX nor E2 replacement in males affected this parameter, we did observe a decrease in PA remodeling when E2 was given to OVX SuHx-PH rats. The latter may have contributed at least in part to the improved RV function in E2-replete OVX rats. The biological correlate for these disparate findings of E2 on the PA is unclear, but we speculate that differences in ER expression, ER signaling, or E2 metabolism may play a role. Differential activation or repression of coactivators and corepressors as well as differential expression of target genes may also contribute. The discrepancy in published data as well as the complex relationship between E2 treatment, sex, and hormonal status in our studies demonstrate that E2 actions on the pulmonary vasculature are highly complex and may depend on the endogenous hormonal milieu, pattern of E2 release, and/or E2 concentration in the PA (discussed below). The fact that we did not notice worsening of pulmonary vascular remodeling with E2 in intact SuHx females (as compared with males), E2-treated SuHx+OVX females, or E2-treated male SuHx animals is reassuring, but in light of data demonstrating worsening PA remodeling with E2 in other models (33; reviewed in Refs. 1, 24), we recognize that the ultimate E2 effect on the pulmonary vasculature may be context dependent. In particular, it may differ based on factors such as sex of the experimental groups, animal model used, E2 concentration given, E2 release pattern (e.g., cyclical vs. continuous), and duration of E2 administration. In addition, genetic (e.g., BMPR2 mutations) and environmental factors (e.g., exposure to serotoninergic drugs) more common in women may affect E2 responses in the pulmonary vasculature. Nevertheless, even if E2 were to worsen pulmonary vascular remodeling, such an effect may be tolerable as long as the concomitant RV-protective effects allow for sufficient RV-PA coupling. Ultimately, it is RV adaptation rather than PA remodeling that determines survival in PAH (54, 57), and in this context E2 clearly seems to facilitate the capacity of the RV to adapt to the increased afterload. Along these lines, it is worth mentioning that E2 also seems to exert beneficial effects on RV remodeling even in studies in which it was shown to worsen the remodeling process in the pulmonary vasculature (61). Future studies in the pulmonary artery banding model of PAH and RV failure will help to further evaluate E2 effects on the RV in absence of any potentially confounding pulmonary vascular effects. Similarly, studies using pressure-volume loops in SuHx rats will help evaluate RV adaption to increases in afterload.
It is noteworthy that hemodynamic, structural, and biochemical alterations induced by SuHx were much more attenuated in E2-treated OVX rats compared with intact female SuHx rats. Intuitively, one would expect the magnitude of changes end points in these groups to be similar. There are two potential explanations for this observation. First, while achieving physiological E2 concentrations in the OVX+E2 group, E2 was consistently released via subcutaneously implanted pellets, thus not replicating the physiological cyclical variations in E2 release that are observed in animals with intact ovaries. This may be relevant, since a menstrual cycle-related “estrogen withdrawal phenomenon” has been implicated in the worsening symptoms observed in patients with vascular diseases like migraines (31), Raynaud's syndrome (14), or angina (30) when their E2 levels decrease during the menstrual cycle. Second, E2 levels in OVX rats, although similar to levels measured in healthy female control rats, tended to be higher than those observed in female SuHx rats. This increase in E2 levels may have been sufficient to elicit more favorable effects. The fact that E2 levels in female SuHx rats tended to be lower than those in normal female rats and similar to those measured in male rats suggests that the female SuHx rats may be relatively E2 deficient. This may explain why several of the end points measured were not different between male and female SuHx rats: E2 levels simply may not have been different enough to elicit significant differences between the sexes. Similarly, the decrease in E2 in intact SuHx females may have limited the effect of further hormone manipulation via OVX. This may explain why several markers of maladaptive RV remodeling in Fig. 4 were not significantly altered by OVX. The reason for the decrease in E2 levels in SuHx females is unclear; this may be a function of a suppressed pituitary-gonadal axis due to severe illness (62), or it may represent a direct effect of SuHx on E2 production or metabolism.
E2’s effects on weight gain appear perplexing. However, E2 has known effects on appetite, insulin sensitivity, and energy expenditure. For example, decreased E2 levels after menopause or OVX are associated with hyperphagia, reduced energy expenditure, and weight gain (35, 46), whereas E2 replacement, by decreasing food intake and increasing energy expenditure, prevents OVX-induced obesity (35, 46). Similarly, hormone replacement therapy reverses the metabolic dysfunctions and obesity noted in postmenopausal women (35, 63). In light of the emerging evidence linking metabolic abnormalities to PAH development (18), it is conceivable that metabolic effects of E2 may have contributed to the less severe PH phenotype noted in E2-treated animals. On the other hand, E2’s effects on weight gain may reflect an unwanted effect that may limit its clinical applicability. The fact that selective activation of ERα or ERβ was not sufficient to affect weight gain suggests that both ERs may need to be activated simultaneously for E2 to exert its effects on weight. E2 effects on insulin sensitivity and lipid metabolism in PAH require further study.
Our study has limitations. First, some of the analyses may have been underpowered, leading to a type 2 statistical error. For example, there were trends for differences in glutathione activation as well as in apelin, MCP-1, and IL-6 RNA that did not reach statistical significance. Studies in a larger number of animals may bring out statistical differences for these end points, but in light of the robust and significant differences in other parameters, we did not pursue additional animal studies. Second, use of RV pressure-volume loops is considered the gold standard for evaluating RV adaptation to increases in pulmonary vascular resistance (57); however, our comprehensive analysis of RV function by echo and exercise capacity (complemented by a detailed analysis of biochemical end points) allows for a robust and clinically relevant evaluation of RV function. Third, the exact mechanism of E2-mediated RV protection in PH remains unclear. Although our studies suggest that both ERα and ERβ are involved in these effects, future studies in genetically modified animals may further dissect the contribution of both ERs and the downstream signaling pathways they employ. On the other hand, given the known sex differences in response to PAH therapies (12, 20, 34), it would be of interest to determine whether established PAH therapies (e.g., PDE-5 inhibitors or endothelin receptor antagonists) affect E2 levels or mediate their effects at least in part through hormonal changes. Lastly, results obtained in the SuHx model may not be applicable to other models.
In conclusion, we identify estrogens and their receptors as beneficial modulators of RV function in experimental severe angioproliferative PH. These findings may allow for the development of novel RV-directed therapies for both male and female patients with PAH.
GRANTS
This work was supported by the Indiana University Health-Indiana University School of Medicine Strategic Research Initiative (T. Lahm), a Pfizer ASPIRE Award (T. Lahm), VA Merit 1I01BX002042-01A2 (T. Lahm), NIH 5T32HL091816-05 (A. L. Frump, K. N. Goss, A. R. Cucci), and AHA Midwest Affiliates Scientist Development Grant 13SDG17140066 (M. B. Brown).
DISCLOSURES
T. Lahm's institution has received funding for investigator-initiated basic science research from Gilead, Inc. and Pfizer. T. Lahm serves on the speaker bureau for Bayer.
AUTHOR CONTRIBUTIONS
A.L.F. and T.L. conception and design of research; A.L.F., K.N.G., A.V., M.A., A.J.F., J.W., A.R.C., M.B.B., and T.L. performed experiments; A.L.F., K.N.G., A.V., M.A., R.T., J.F., J.W., A.R.C., M.B.B., and T.L. analyzed data; A.L.F., M.B.B., and T.L. interpreted results of experiments; A.L.F., A.V., M.A., and T.L. prepared figures; A.L.F. and T.L. drafted manuscript; A.L.F., K.N.G., M.B.B., and T.L. edited and revised manuscript; A.L.F., K.N.G., A.V., M.A., A.J.F., R.T., J.F., J.W., A.R.C., M.B.B., and T.L. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Jordan Wood, Mary Van Demark, Constance Temm, Crystal Sorg, and Robert G. Presson Jr, MD, for expert technical assistance. The authors thank Irina Petrache, MD, for helpful editing of the manuscript.
Glossary
- CI
cardiac index
- CO
cardiac output
- DPN
diarylpropionitrile
- E2
17β-estradiol
- ER
estrogen receptor
- FiO2
inspired oxygen fraction
- GPR30
G protein-coupled receptor 30
- GSH
glutathione
- GLUT1
glucose transporter 1
- HPH
hypoxia-induced pulmonary hypertension
- IL-6
interleukin 6
- LV
left ventricle
- MCP-1
monocyte chemoattractant protein 1
- OVX
ovariectomy
- PA
pulmonary artery
- PAH
pulmonary arterial hypertension
- Patm
atmospheric pressure
- PH
pulmonary hypertension
- PPT
4,4′4″[4-Propyl-(1H)-pyrazole-1,3,5-triyl] trisphenol
- RV
right ventricle
- RVH
right ventricular hypertrophy
- RVOT
right ventricular outflow tract
- RVSP
right ventricular systolic pressure
- SuHx
Su5416/hypoxia
- V̇o2max
maximal oxygen consumption
- VTI
velocity-time integral
REFERENCES
- 1.Austin ED, Lahm T, West J, Tofovic SP, Johansen AK, Maclean MR, Alzoubi A, Oka M. Gender, sex hormones and pulmonary hypertension. Pulm Circ 3: 294–314, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedford TG, Tipton CM, Wilson NC, Oppliger RA, Gisolfi CV. Maximum oxygen consumption of rats and its changes with various experimental procedures. J Appl Physiol 47: 1278–1283, 1979. [DOI] [PubMed] [Google Scholar]
- 3.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 122: 164–172, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Berry MF, Pirolli TJ, Jayasankar V, Burdick J, Morine KJ, Gardner TJ, Woo YJ. Apelin has in vivo inotropic effects on normal and failing hearts. Circulation 110: II187–II193, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135: 794–804, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 120: 1951–1960, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med 182: 652–660, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature 509: 282–283, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Jesus Perez VA. Making sense of the estrogen paradox in pulmonary arterial hypertension. Am J Respir Crit Care Med 184: 629–630, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Dupon C, Kim MH. Peripheral plasma levels of testosterone, androstenedione, and oestradiol during the rat oestrous cycle. J Endocrinol 59: 653–654, 1973. [DOI] [PubMed] [Google Scholar]
- 11.Fliegner D, Schubert C, Penkalla A, Witt H, Kararigas G, Dworatzek E, Staub E, Martus P, Ruiz Noppinger P, Kintscher U, Gustafsson JA, Regitz-Zagrosek V. Female sex and estrogen receptor-β attenuate cardiac remodeling and apoptosis in pressure overload. Am J Physiol Regul Integr Comp Physiol 298: R1597–R1606, 2010. [DOI] [PubMed] [Google Scholar]
- 12.Gabler NB, French B, Strom BL, Liu Z, Palevsky HI, Taichman DB, Kawut SM, Halpern SD. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest 141: 20–26, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, Taraseviciene-Stewart L, Sung Y, Kraskauskas D, Farkas D, Conrad DH, Nicolls MR, Voelkel NF. A brief overview of mouse models of pulmonary arterial hypertension: problems and prospects. Am J Physiol Lung Cell Mol Physiol 302: L977–L991, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenstein D, Jeffcote N, Ilsley D, Kester RC. The menstrual cycle and Raynaud's phenomenon. Angiology 47: 427–436, 1996. [DOI] [PubMed] [Google Scholar]
- 15.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation 117: 1717–1731, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I. Anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation 117: 1436–1448, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Haisenleder DJ, Schoenfelder AH, Marcinko ES, Geddis LM, Marshall JC. Estimation of estradiol in mouse serum samples: evaluation of commercial estradiol immunoassays. Endocrinology 152: 4443–4447, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 115: 1275–1284, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jais X, Montani D, Souza R, Simonneau G. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 122: 153–163, 2010. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, Kawut SM, Bogaard HJ, Boonstra A, Vonk Noordegraaf A. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 145: 1230–1236, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kararigas G, Fliegner D, Gustafsson JA, Regitz-Zagrosek V. Role of the estrogen/estrogen-receptor-β axis in the genomic response to pressure overload-induced hypertrophy. Physiol Genomics 43: 438–446, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Kawut SM, Al-Naamani N, Agerstrand C, Rosenzweig EB, Rowan C, Barst RJ, Bergmann S, Horn EM. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest 135: 752–759, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, Justice MJ, Brown MB, Van Demark M, Trulock KM, Dieudonne D, Reddy JG, Presson RG, Petrache I. 17beta-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med 185: 965–980, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L7–L26, 2014. [DOI] [PubMed] [Google Scholar]
- 25.Lambert MI, Noakes TD. Dissociation of changes in V̇o2max, muscle Q̇o2, and performance with training in rats. J Appl Physiol 66: 1620–1625, 1989. [DOI] [PubMed] [Google Scholar]
- 26.Lewis GD, Bossone E, Naeije R, Grunig E, Saggar R, Lancellotti P, Ghio S, Varga J, Rajagopalan S, Oudiz R, Rubenfire M. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation 128: 1470–1479, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Zeng H, Chen JX. Apelin-13 increases myocardial progenitor cells and improves repair postmyocardial infarction. Am J Physiol Heart Circ Physiol 303: H605–H618, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation 120: 245–254, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu A, Schreier DA, Tian L, Eickhoff JC, Wang Z, Hacker TA, Chesler NC. Direct and indirect protection of right ventricular function by estrogen in an experimental model of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 307: H273–H283, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lloyd GW, Patel NR, McGing E, Cooper AF, Brennand-Roper D, Jackson G. Does angina vary with the menstrual cycle in women with premenopausal coronary artery disease? Heart 84: 189–192, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacGregor EA, Frith A, Ellis J, Aspinall L, Hackshaw A. Incidence of migraine relative to menstrual cycle phases of rising and falling estrogen. Neurology 67: 2154–2158, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Mahmoodzadeh S, Eder S, Nordmeyer J, Ehler E, Huber O, Martus P, Weiske J, Pregla R, Hetzer R, Regitz-Zagrosek V. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J 20: 926–934, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, Fullerton J, Nilsen M, Loughlin L, Thomas M, MacLean MR. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med 190: 456–467, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathai SC, Hassoun PM, Puhan MA, Zhou Y, Wise RA. Sex differences in response to tadalafil in pulmonary arterial hypertension. Chest 147: 188–197, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 34: 309–338, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morissette M, Jourdain S, Al Sweidi S, Menniti FS, Ramirez AD, Di Paolo T. Role of estrogen receptors in neuroprotection by estradiol against MPTP toxicity. Neuropharmacology 52: 1509–1520, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Murphy E. Estrogen signaling and cardiovascular disease. Circ Res 109: 687–696, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy E, Steenbergen C. Estrogen regulation of protein expression and signaling pathways in the heart. Biol Sex Differ 5: 6, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nikolic I, Liu D, Bell JA, Collins J, Steenbergen C, Murphy E. Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J Mol Cell Cardiol 42: 769–780, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Nuedling S, Kahlert S, Loebbert K, Doevendans PA, Meyer R, Vetter H, Grohe C. 17 Beta-estradiol stimulates expression of endothelial and inducible NO synthase in rat myocardium in-vitro and in-vivo. Cardiovasc Res 43: 666–674, 1999. [DOI] [PubMed] [Google Scholar]
- 41.Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, Grunig E, Staehler G, Rosenkranz S, Halank M, Held M, Lange TJ, Behr J, Klose H, Claussen M, Ewert R, Opitz CF, Vizza CD, Scelsi L, Vonk-Noordegraaf A, Kaemmerer H, Gibbs JS, Coghlan G, Pepke-Zaba J, Schulz U, Gorenflo M, Pittrow D, Hoeper MM. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 129: 57–65, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Pedram A, Razandi M, Narayanan R, Dalton JT, McKinsey TA, Levin ER. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy. Mol Biol Cell 24: 3805–3818, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, Petrache HI, Flotte TR, Tuder RM. alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol 169: 1155–1166, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Resta TC, Kanagy NL, Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol 280: L88–L97, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Rogers NH, Perfield JW 2nd, Strissel KJ, Obin MS, Greenberg AS. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 150: 2161–2168, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res 115: 176–188, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki H, Nagayama T, Blanton RM, Seo K, Zhang M, Zhu G, Lee DI, Bedja D, Hsu S, Tsukamoto O, Takashima S, Kitakaze M, Mendelsohn ME, Karas RH, Kass DA, Takimoto E. PDE5 inhibitor efficacy is estrogen dependent in female heart disease. J Clin Invest 124: 2464–2471, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spence RD, Wisdom AJ, Cao Y, Hill HM, Mongerson CR, Stapornkul B, Itoh N, Sofroniew MV, Voskuhl RR. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERalpha signaling on astrocytes but not through ERbeta signaling on astrocytes or neurons. J Neurosci 33: 10924–10933, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Tuder RM, Archer SL, Dorfmuller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62: D4–D12, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tuder RM, Stacher E, Robinson J, Kumar R, Graham BB. Pathology of pulmonary hypertension. Clin Chest Med 34: 639–650, 2013. [DOI] [PubMed] [Google Scholar]
- 53.Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, van der Laarse A, Eghbali M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med 184: 715–723, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KM, Westerhof N, Vonk-Noordegraaf A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 58: 2511–2519, 2011. [DOI] [PubMed] [Google Scholar]
- 55.Ventetuolo CE, Ouyang P, Bluemke DA, Tandri H, Barr RG, Bagiella E, Cappola AR, Bristow MR, Johnson C, Kronmal RA, Kizer JR, Lima JA, Kawut SM. Sex hormones are associated with right ventricular structure and function: the MESA-right ventricle study. Am J Respir Crit Care Med 183: 659–667, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ventetuolo CE, Praestgaard A, Palevsky HI, Klinger JR, Halpern SD, Kawut SM. Sex and hemodynamics in pulmonary arterial hypertension. Eur Respir J 43: 523–530, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, Hassoun PM. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol 62: D22–D33, 2013. [DOI] [PubMed] [Google Scholar]
- 58.Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-α mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol 290: H2204–H2209, 2006. [DOI] [PubMed] [Google Scholar]
- 59.Wang M, Tsai BM, Reiger KM, Brown JW, Meldrum DR. 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J Mol Cell Cardiol 40: 205–212, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, Kelly M, Meldrum DR. Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol 296: R972–R978, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17beta oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res 90: 373–382, 2011. [DOI] [PubMed] [Google Scholar]
- 62.Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 60: 444–450, 1985. [DOI] [PubMed] [Google Scholar]
- 63.Wren BG. The benefits of oestrogen following menopause: why hormone replacement therapy should be offered to postmenopausal women. Med J Aust 190: 321–325, 2009. [DOI] [PubMed] [Google Scholar]