

Abstract
Transforming growth factor-β induces a smooth muscle cell phenotype in undifferentiated mesenchymal cells. To elucidate the mechanism(s) of this phenotypic induction, we focused on the molecular regulation of smooth muscle-γ-actin, whose expression is induced at late stages of smooth muscle differentiation and developmentally restricted to this lineage. Transforming growth factor-β induced smooth muscle-γ-actin protein, cytoskeletal localization, and mRNA expression in mesenchymal cells. Smooth muscle-γ-actin promoter-luciferase reporter activity was enhanced by transforming growth factor-β, and deletion analysis revealed that CArG box 2 in the promoter was necessary for this transcriptional activation. CArG motifs bind transcriptional activator serum response factor; gel shift analyses revealed increased binding of serum response factor-containing complexes to this site in response to transforming growth factor-β, paralleled by increased serum response factor protein expression. Serum response factor expression was found to be up-regulated by transforming growth factor-β via transcriptional activation of the gene and post-transcriptional regulation. Using mesenchymal cells stably transfected with wild type or dominant-negative serum response factor, we demonstrated that its expression is sufficient for induction of a smooth muscle phenotype in mesenchymal cells and is necessary for transforming growth factor-β-mediated smooth muscle induction.
Transforming growth factor-β (TGF-β)1 is a member of a large family of cytokines that includes activins and bone morphogenic proteins (1). TGF-β, specifically, is known to play an important role during embryonic development in cellular growth and differentiation in a number of organ systems, including the gastrointestinal tract (2–4) and the vasculature (5). TGF-β is produced in a latent form in mesenchymal and epithelial cell types (6) and is thought to be activated in a plasmin-mediated process (7) that requires cell-cell interaction (8). Activated TGF-β binds to specific membrane receptors that possess Ser-Thr kinase activity (1). Intracellular TGF-β signaling occurs primarily via intermediate effector proteins, SMADS, which modulate the interactions of transcription factors with their cognate cis-elements to promote changes in gene expression, ultimately leading to changes in cellular phenotype.
TGF-β signaling is thought to direct, in part, the differentiation of smooth muscle (SM) cells from mesenchymal and neural crest precursors during vascular and gut development (4, 9). SM cells play important physiological roles in these tissues via modulation of vascular tone and resistance and control of gastrointestinal motility and fluid movement. The SM cell phenotype is characterized by coordinated expression of contractile proteins that include SM-α-actin (10–13), SM-γ-actin (14–18), SM myosin heavy chain (19–21), calponin (22, 40), SM 22α (23–25), and telokin (26, 27). Furthermore, these cells are thought to be capable of reversibly modulating their phenotype during postnatal development (28). This phenotypic modulation includes an alteration in the expression of proteins characteristic of the differentiated SM cell and has been implicated in the pathogenesis of cardiovascular (29) and gastrointestinal (30, 31) disease states. Paradoxically, TGF-β signaling may play a role in directing SM cell responses to disease. TGF-β expression is increased in SM components of the arterial wall following injury (32) and of the intestinal mesenchyme in patients with inflammatory bowel diseases (31). Furthermore, it has been shown that administration of neutralizing anti-TGF-β antibodies reduces the severity of lesions in carotid injury (33) and experimentally induced ulcerative colitis (34).
The exact mechanism(s) through which TGF-β signaling affects both SM cell differentiation and response to injury or disease is largely unknown. However, it has been demonstrated that TGF-β up-regulates SM-α-actin expression in human and rat SM via direct transcriptional regulation (13). Interestingly, although SM-α-actin is among the first genes expressed during SM differentiation, it is also expressed during development in a variety of other cell types, including skeletal and cardiac muscle, corneal fibroblasts, and astroglial cells (35). Furthermore, SM-α-actin expression is modulated by TGF-β in skin and corneal fibroblast during wound repair (35); hence, the transcriptional control of this gene by TGF-β may not represent a SM-specific event.
To gain a better understanding of the molecular mechanisms that regulate genes during SM differentiation, we have analyzed the expression of SM-γ-actin. Although a closely related isoform of SM-α-actin, SM-γ-actin expression is induced at late stages of SM differentiation (36) and developmentally restricted to SM in the vasculature and gastrointestinal tract (15, 47), with the exception of the post-meiotic spermatocyte (37). Thus, SM-γ-actin provides an excellent marker for the differentiated SM cell phenotype.
Transcriptional regulation of the SM-γ-actin gene appears to require the interaction of positive- and negative-acting cis-elements within the promoter. Two separate regions displaying positive transcriptional activity have been mapped in the SM-γ-actin gene promoter, and are referred to as the specifier and modulator domains (14). A key cis-element for SM-specific transcription present in both of the positive-acting transcriptional regulatory domains is the CArG/SRE motif. The positive-acting transcriptional activity of the specifier and modulator domains is derived from the binding of SRF to the CArG/SRE motif (CC(A/T)6GG), and we have previously shown that SRF complexes are key regulators of the developmental activation of the SM-γ-actin gene in vivo (15). Given that CArG/SRE elements are important for TGF-β inducibility of the skeletal and SM-α-actin genes, it is possible that SRF mediates TGF-β induction of SM-γ-actin during SM development. The CArG/ SRE motif has been implicated in the developmental regulation of other SM-specific genes as well, including SM-α-actin (38), SM22α (23, 39), calponin (22, 40), SM myosin heavy chain (21), and telokin (41). Moreover, SRF expression and function has been developmentally correlated with vascular and visceral SM differentiation in vivo (15, 36, 42, 43).
In the studies presented here, we examined the regulation of SM-γ-actin expression by TGF-β in 10T1/2 mesenchymal cells, which we have previously shown to serve as SM progenitors (44). We demonstrate that TGF-β increased steady-state levels of SM-γ-actin mRNA and induced production and cytoskeletal localization of SM-γ-actin protein. Increased mRNA levels resulted from TGF-β-induced transcriptional activation of the SM-γ-actin gene. This response was mediated through a specific CArG/SRE motif (CArG box 2) within the specifier segment of the promoter, which was found to be necessary for TGF-β-induced transcriptional activation. Furthermore, we show an increase in SRF binding activity upon the SM-γ-actin promoter in response to TGF-β, which was paralleled by increased SRF protein expression. SRF expression was up-regulated by TGF-β via transcriptional and post-transcriptional control mechanisms. Using SM progenitors stably transfected with wild type or dominant-negative SRF, we demonstrated that SRF protein expression is sufficient for induction of a SM phenotype in mesenchymal cells and is necessary for TGF-β-mediated SM induction.
Materials and Methods
Cell Culture
10T1/2 cells (ATCC CCL 226) were grown and maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and 4.5 g/liter glucose and supplemented with penicillin and streptomycin. All cells were maintained at 37 °C in a humidified atmosphere. For all cell culture experiments, cells were switched to DMEM containing 2% calf serum (DMEM, 2% CS) upon initial cell plating (To for each experiment); each experiment was repeated at least three times. Representative experiments are shown.
Immunocytochemistry
10T1/2 cells were cultured in four-chamber culture slides (Lab-Tek, Naperville, IL) in DMEM, 2% CS ± 1 ng/ml TGF-β1. After incubation for 24 h, the cells were fixed with 4% paraformaldehyde and immunostained for SM-γ-actin, as described previously (44). Anti-SM-γ-actin (mouse monoclonal antibody, ICN) was used at 1:1000 in blocking buffer consisting of 4% normal goat serum, 3% bovine serum albumin, 0.1% Triton X-100 in phosphate-buffered saline. At this concentration, this antibody is specific for the SM-γ-actin isoform and does not cross-react with other prevalent actin isoforms, including SM-α-actin.2 Antibody-antigen complexes were visualized using the Vectastain Elite ABC Kit (Vector, Burlingame, CA) and the biotinylated anti-mouse secondary antibody (1:250) provided by the manufacturer.
Western Blot Analyses
10T1/2 cells were cultured (6 × 105 cells/100 mm dish) with 0–5 ng/ml TGF-β1 for up to 3 days. Protein was isolated from cells as described previously (45). Ten μg of protein were electrophoresed on 10% SDS-PAGE minigels (Bio-Rad) and then electrophoretically transferred to Immobilon-P membranes (Millipore). Membranes were blocked with 5% nonfat dry milk, 1% bovine serum albumin in phosphate-buffered saline and then incubated 1–2 h with primary antibody (anti-SM-γ-actin, anti-SRF, and anti-HA, all at 1:1000 dilution). Antibody-antigen complexes were revealed using the ECL detection system (Amersham Biosciences, Inc.).
Northern Blot Analyses
10T1/2 cells were cultured (6 × 105 cells/ 100 mm dish) with 0–5 ng/ml TGF-β1 for up to 3 days. RNA was isolated from the cells using RNAzol B (Tel-Test, Friendswood, TX). Total RNA (10 μg per sample) was electrophorectically separated and transferred onto GeneScreen Plus nylon membrane (PerkinElmer Life Sciences). cDNA probes (murine SM-γ-actin, cytoplasmic -γ-actin, and SRF) were labeled using Ready-To-Go DNA labeling beads (Pharmacia Biosciences, Inc.), purified by centrifugation through MicroSpin S-200 HR columns (Pharmacia Biosciences, Inc.), and hybridized at 1.5 × 106 cpm/ml. After washing, membranes were exposed to either X-Omat AR film (Eastman Kodak Co.) or a screen designed for imaging using a PhosphorImager (Molecular Dynamics).
Plasmid Constructs
The avian SM-γ-actin gene and ∼2.3 kb of DNA comprising the promoter have been previously cloned and sequenced (GenBank™ accession number AFO12348 (14)). Deletions of the promoter were cloned into the PGL-3 Basic luciferase vector forming chimeric SM-γ-actin promoter-reporter genes (15) and utilized for the transfection studies described here. Mutagenesis of each of the six CArG/SRE motifs was accomplished by oligonucleotide mutagenesis using the Exsite™ PCR mutagenesis kit (Stratagene, La Jolla, CA), as described by the manufacturer. cDNA clones for mouse actin isoforms were obtained from Genome Systems (St. Louis, MO) and were enhanced sequence tag clones 1431913 (cytoplasmic γ-actin, GenBank™ accession number AA986689), 348425 (cytoplasmic β-actin, GenBank™ accession number W35717), and 314246 (SM-γ-actin, GenBank™ accession number W09992). Isoform-specific probes were generated from these cDNAs by PCR using oligonucleotide primers that amplify the 3′-UTR segments of the mRNAs.
Transient Transfections and Reporter Gene Assays
Various chimeric SM-γ-actin-luciferase reporter gene constructs were transiently transfected into cultured 10T1/2 cells using LipofectAMINE reagent (Invitrogen), according to manufacturer's protocol. Cells were plated at 70,000 cells/well in 12-well dishes in DMEM, 2% CS, incubated overnight at 37 °C, then rinsed with DMEM and treated with 1 μg/well of DNA. After an overnight incubation, the cells were rinsed in phosphate-buffered saline and treated with DMEM, 2% CS ± 1 ng/ml TGF-β for 48 h. Positive (luciferase gene under the control of the SV40 promoter/ enhancer) and negative (promoterless pGL-3 vector DNA) controls were included in each experiment. All promoter constructs were evaluated in a minimum of three separate experiments, three separate wells per experiments, and at least two separate plasmid preparations per construct.
Promoter activity was evaluated by measurement of firefly luciferase activity according to manufacturer's protocol (Promega, Madison, WI), using a Turner model 20 luminometer. Each lysate was analyzed in triplicate and luciferase activity was normalized to protein content. Normalized luciferase activity in TGF-β-treated cells was compared with measured activity in cells in control conditions transfected with the same promoter-reporter DNA. The calculated values (mean ± S.D.) were plotted as fold increase in response to TGF-β treatment.
DNA Fragments and Gel Shift Analyses
Double-stranded oligonucleotides corresponding to the CArG/SRE2 motif (nucleotides −135 to −105 of the promoter) or the α-skeletal actin gene SRE (15, 52) were end-labeled using the polynucleotide kinase reaction, and gel shift assays were preformed as described previously (15). Reaction mixtures were assembled in 50-μl volume containing 1 μg of poly(dI-dC) nonspecific competitor and 3 or 5 μg of nuclear lysate and preincubated of 10 min at room temperature. Appropriate labeled probe was then added and the mixture incubated for an additional 30 min. For control reactions, cold competitor probes were added at 50 times molar excess of labeled probe or an SRF-specific antibody added during the 10-min preincubation. Binding complexes were then resolved on 5% polyacrylamide gels, dried onto Whatman paper, and exposed to x-ray film (Kodak, Biomax BMR1). The nuclear lysates used in these assays were derived from TGF-β-treated, and nontreated, 10T1/2 cells and from SM tissue (avian gizzard) (15). For quantitative assessment of binding complexes, the dried gels were exposed to PhosphorImager screens and analyzed using the Molecular Analysis Software package (Bio-Rad). In each experiment, binding activity observed in control cells was normalized to a value of 1.0 and directly compared with binding in lysates from the corresponding TGF-β treated cells. These values represent the mean ± S.D.
Creation and Characterization of 10T1/2 Stable Transfectants
10T1/2 cells were transfected, via electroporation, with 5 μg of linearized HA-tagged expression plasmid containing no cDNA (vector control), 5 μg of linearized wild type (wt) SRF cDNA, or 5 μg of linearized SRF cDNA that contains a 3-bp substitution in the region known to mediate DNA binding, yielding a dominant negative SRF protein (dnSRF; described previously in Ref. 36). All cells were cotransfected with a plasmid containing the neomycin resistance gene (pCI-neo; Promega) at 1/10 the concentration of experimental vector. Twelve stable transfectant clones were generated from each experimental group (vector, wtSRF, dnSRF) via selection in normal growth media containing 1000 μg/ml G418; thereafter, stable clones were maintained in 500 μg/ml G418. Total protein was isolated from each clone and screened via Western analyses to assess the expression of SRF protein and the HA tag. Two or three positive clones from each transfectant group were cultured in experimental media (2% CS, DMEM) in the presence or absence of 1 ng/ml TGF-β1 for 24 h; protein was isolated from each and analyzed for expression of SM-γ-actin.
Results
Multipotent 10T1/2 mesenchymal cells are induced toward a SM phenotype upon direct coculture with endothelial cells and in response to direct treatment with TGF-β (44). This phenotypic transformation is characterized by up-regulation of SM-α-actin protein expression, induction of other SM-specific proteins including calponin, SM22α, and SM myosin heavy chain, and a dramatic shape change from flat and spread to elongated with pseudopodia (44). Recent reports indicate that SM-γ-actin is coordinately regulated with calponin and SM22α in coronary artery SM progenitors (36). Furthermore, the expression of SM-γ-actin is induced in 10T1/2 cells that are cocultured with endothelial cells.2 Therefore, we aimed to determine whether TGF-β induces the expression of SM-γ-actin, a late marker of SM phenotype, in SM precursors and, if so, by what mechanism.
TGF-β Regulation of SM-γ-Actin Expression
To address this issue, multipotent 10T1/2 mesenchymal cells were incubated with 1 ng/ml TGF-β1 for 24 h and then assayed for SM-γ-actin expression. We found that although 10T1/2 mesenchymal cells did not express SM-γ-actin protein in control conditions (Fig. 1C), its expression and cytoskeletal localization was induced by TGF-β1 (Fig. 1D) to a level similar to that of primary cultures of SM cells (Fig. 1B). As expected, endothelial cells did not express SM-γ-actin (Fig. 1A). The time course of TGF-β induction of SM-γ-actin protein expression in 10T1/2 cells was examined by Western blot analyses. TGF-β induction of SM-γ-actin protein occurred after 12–24-h exposure to 1 ng/ml TGF-β1 (Fig. 2A); this effect was dose-dependent from 0.1 to 5 ng/ml TGF-β1 (not shown). SM-γ-actin protein was not detected in 10T1/2 cells in control conditions at any time point throughout the experimental period (Fig. 2A; representative time points shown).
Fig. 1. TGF-β induction of SM-γ-actin protein expression and cytoskeletal localization.

10T1/2 mesenchymal cells were incubated with or without 1 ng/ml TGF-β1 for 24 h; bovine aortic endothelial cells and smooth muscle cells were incubated in control conditions for 24 h, as described under “Materials and Methods.” All cells were fixed in 4% paraformaldehyde and immunostained for SM-γ-actin (ICN antibody; 1:1000). 10T1/2 cells (C), as well as endothelial cells (A), did not express SM-γ-actin protein; however, TGF-β induced expression and cytoskeletal localization of SM-γ-actin in 10T1/2 cells (D) to a level similar to that of primary cultures of smooth muscle cells (B). Bar = 10 μm.
Fig. 2. Time course of TGF-β induction of SM-γ-actin protein and mRNA expression.
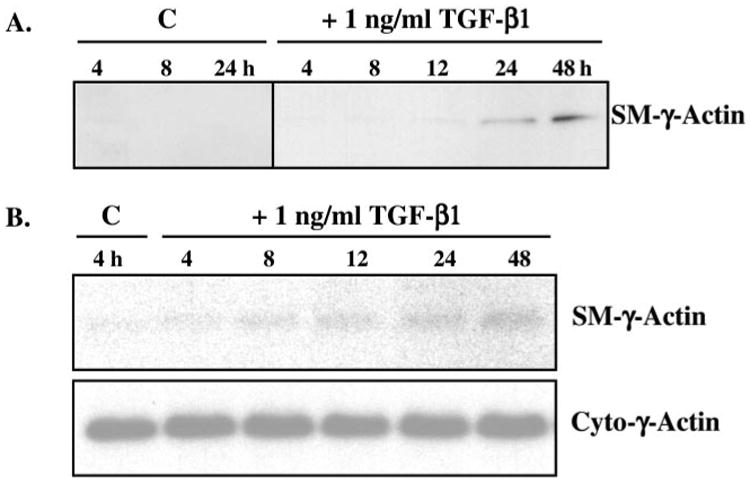
A, 10T1/2 mesenchymal cells were incubated with or without 1 ng/ml TGF-β1 for 0, 4, 8, 12, 24, and 48 h, after which total protein lysates were isolated and subjected to Western blot analyses (10 μg of total protein/lane). SM-γ-actin protein (∼43 kDa) expression was induced after 12–24 h of TGF-β treatment compared with control (C) conditions. B, 10T1/2 mesenchymal cells were treated with 1 ng/ml TGF-β1 (as in A), after which total RNA was isolated and subjected to Northern blot analyses (5 μg of total RNA/lane). SM-γ-actin mRNA (∼1.5 kb) expression was induced after ∼8 h. Blots were re-hybridized with a specific probe to cytoplasmic -γ-actin as a loading control. These data, taken together, suggest that SM-γ-actin gene expression is transcriptionally regulated in 10T1/2 cells by TGF-β.
To elucidate the mechanism by which TGF-β up-regulated SM-γ-actin production, we examined its effect on SM-γ-actin mRNA. We performed Northern blot analyses on total RNA isolated from 10T1/2 cells treated with or without 1 ng/ml TGF-β1 for up to 48 h. SM-γ-actin mRNA was up-regulated after ∼8 h exposure to TGF-β1 (Fig. 2B), prior to induction of protein expression, suggesting that SM-γ-actin gene expression may be transcriptionally regulated by TGF-β in 10T1/2 mesenchymal cells.
Transcriptional Regulation of SM-γ-Actin Promoter
To more directly determine whether TGF-β transcriptionally activates SM-γ-actin gene expression, we performed promoter-reporter analyses using various regions of the SM-γ-actin promoter linked to the firefly luciferase reporter gene. Initially, chimeric deletion constructs, containing different lengths of SM-γ-actin promoter, ranging from full-length (2294 bp) to 65 bp, were transiently transfected into 10T1/2 cells that were then incubated in the presence or absence of 1 ng/ml TGF-β1 for 48 h (Fig. 3). A chimeric reporter gene construct, containing the initial 65 bp of the SM γ-actin promoter (SMGA −65), was capable of promoting transcription over that observed for the promoterless control plasmid, pGL3-Basic in 10T1/2 cells. However, this reporter gene was not differentially activated in TGF-β-treated and untreated (control) cells (Fig. 3). These data indicate that although the SM-γ-actin TATA sequence motif contained within the SMGA −65 clone was capable of promoting basal transcription in 10T1/2 cells, there were no sequence motifs within this region of DNA capable of responding to TGFβ signaling. An additional 40 bp (SMGA −108) or 50 bp (SMGA −116) of DNA that contains the first CArG/SRE sequence alone (SMGA −108), or in combination with a well conserved E-box motif within the SM-γ-actin promoter (14), were also incapable of demonstrating a TGF-β-dependent transcriptional response. However, the addition of the 20 bp between −130 and −116 of the SM-γ-actin promoter produced a 2.5-fold response to TGF-β treatment, as compared with control conditions (SMGA −136, Fig. 3). The most prominent cis-element within this region is the CArG/SRE2 motif at −120 (14). The TGF-β response was maintained at similar levels (1.5–2.6-fold) with the addition of DNA out to ∼2.3 kb flanking the SM γ-actin gene. These experiments indicate that CArG/SRE motifs within the SM γ-actin promoter are involved in the TGF-β-induced transcriptional activity, with DNA sequences between −136 and −116 containing the CArG/SRE2 element at −120 being essential for this response.
Fig. 3. TGF-β-induced transcriptional activation of the SM-γ-actin promoter.

Chimeric deletion constructs, containing different lengths of SM-γ-actin promoter, ranging from full-length (2294 bp) to 65 bp, were transiently transfected (1 μg DNA/transfection) into 10T1/2 cells (70,000 cells/well) that were then incubated in the presence or absence of 1 ng/ml TGF-β1 for 48 h. Promoter activity was assessed in transfected cells by measurement of the firefly luciferase reporter. Each lysate was analyzed in triplicate, and luciferase activity was normalized to protein content. To determine the responsiveness of a promoter to TGF-β treatment, the luciferase activity generated in lysates of TGF-β-treated cells was directly compared with activity obtained with the same construct in untreated cells and is plotted as a fold increase response to TGF-β. The promoter constructs used in the experiments are diagrammed in A, with their response to TGF-β in 10T1/2 cells plotted (mean + S.D.) in B. There was a TGF-β-dependent response of 2–3-fold with promoter DNA containing the first 136 bp (SMGA −136). The TGF-β response was maintained at similar levels with the addition of DNA out to −2.3 kb flanking the gene.
SRF Binding at CArG/SRE2 Is Required for TGF-β Transcriptional Regulation of SM-γ-Actin Promoter
CArG/SRE motifs, which bind the transcription factor SRF, have been implicated in the transcriptional regulation of genes expressed by smooth muscle cells. Therefore, we aimed to determine whether TGF-β-induced transcriptional activity was mediated via binding to these cis-elements in the SM-γ-actin promoter. To do so, plasmids containing full-length SM-γ-actin promoter regions in which each of its six CArG boxes was individually mutated were used in transfection experiments. Site-directed mutants were made by a PCR-based, oligonucleotide-mediated mutagenesis technique, and each contained changes in the CArG motif at the GG residues (CC(A/T)6GG → CC(A/T)6CC). The G residues are critical for SRF binding (46), and we have previously demonstrated that such changes abolish SRF binding to the CArG motifs of the SM-γ-actin gene (15, 42, 43). TGF-β-induced transcriptional activity was measured in 10T1/2 mesenchymal cells that were transiently transfected with plasmids containing each of the mutated promoters and then incubated in the presence or absence of 1 ng/ml TGF-β for 48 h. Mutations in CArG/SRE sequences 1, 3, 4, 5, and 6 did not abolish, but consistently reduced, the TGF-β response by 40–60% (Fig. 4). In contrast, mutations in the CArG/SRE2, which disrupt SRF binding to this cis-element, totally abolished TGF-β-induced transcriptional activation (Fig. 4). Thus, these experiments, combined with our deletion analyses, implicate the CArG/SRE-binding motifs as the transducer(s) of TGF-β transcriptional activation of the SM-γ-actin gene and indicate that CArG/SRE2 is critical for this response.
Fig. 4. CArG/SRE2 is required for TGF-β transcriptional regulation of SM-γ-actin promoter.
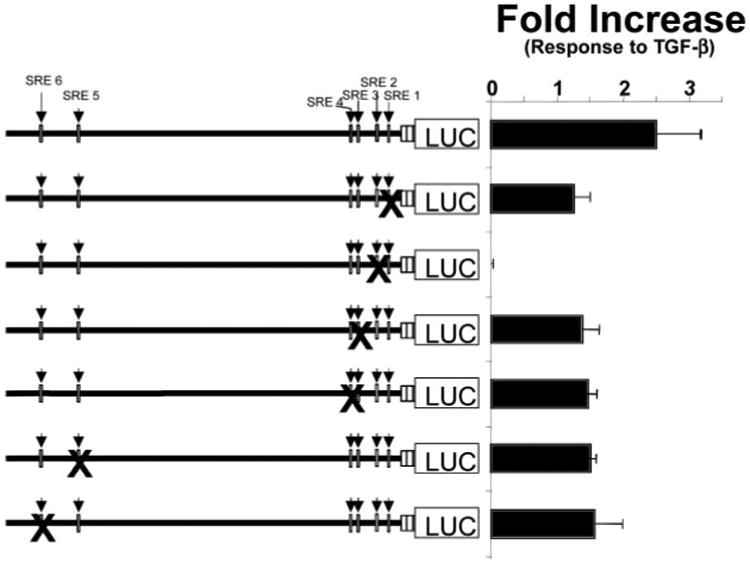
Plasmids containing full-length SM-γ-actin promoter regions in which each of its six CArG/SRE motifs were individually mutated were transiently transfected into 10T1/2 cells. The TGF-β responsiveness of these mutated DNAs was evaluated by incubating cells in the presence or absence of 1 ng/ml TGF-β for 48 h and measuring the resultant luciferase activity in cell lysates. Mutations in CArG/SRE sequences 1, 3, 4, 5, and 6 did not abolish, but consistently reduced, TGF-β transcriptional activation by 40–60%. In contrast, mutations in the CArG/SRE2, which disrupt SRF binding to this cis-element, totally abolished TGF-β-induced transcriptional activation.
We have previously demonstrated that while appropriate developmental expression of the SM-γ-actin gene is dependent upon the binding activities of multiple trans-acting factors to sequences flanking the 5′ region of the gene, SRF binding at the CArG/SRE motifs plays a key role in the transcriptional activation of this gene (14, 47). To determine whether there was a change in SRF binding upon the SM-γ-actin promoter in TGF-β-treated mesenchymal cells, we performed gel shift analyses with nuclear lysates derived from 10T1/2 cells incubated with 0 or 1 ng/ml TGF-β for 48 h. We focused upon the contribution of SRF binding to the CArG/SRE2 motif as it was found to be essential for the TGF-β response.
As shown in Fig. 5, multiple complexes were formed with a probe containing the CArG/SRE2 motif and surrounding sequences (lanes 2 and 3, Fig. 5B) using 5 μg of nuclear lysate from 10T1/2 cells. The probe used in this experiment (135CAATAAAACACCTTATATGGCCATATGGCT −106) is a highly conserved segment of the SM-γ-actin promoter (Fig. 5A) and contains a number of cis-acting elements that have been shown to be critical for smooth muscle-specific SM-γ-actin transcription (14, 15, 18, 49, 75). Thus the formation of multiple complexes is likely due to several DNA-protein interactions. A 50-fold excess of unlabeled, native DNA fragment abolished all complex formation (lanes 4 and 5), whereas a competitor probe containing an SRF-binding site derived from the α-skeletal actin gene (lanes 6 and 7, Fig. 5B) specifically abolished the formation of the middle migrating complex (denoted by the arrow). Conformation that the middle complex contained SRF as its principal DNA-binding component was demonstrated by control experiments, which included antibody supershift experiments (data not shown) as we have performed in previous studies (15, 38, 42, 43, 47, 52). Competition experiments with an oligonucleotide in which the CArG/SRE2 motif was mutated to prevent SRF binding (CArG/SRE2-mut, Fig. 5A) did not inhibit the formation of the SRF-containing complex with the native sequence; however, other DNA-binding complexes were prevented from forming (lanes 8 and 9). This experiment clearly demonstrates that there is SRF binding activity in control, untreated cells and that there was enhanced SRF binding of the CArG/SRE2 element probe in nuclear lysates derived from TGF-β-treated 10T1/2 cells.
Fig. 5. SRF binding activity at CArG/SRE2 is enhanced in response to TGF-β.
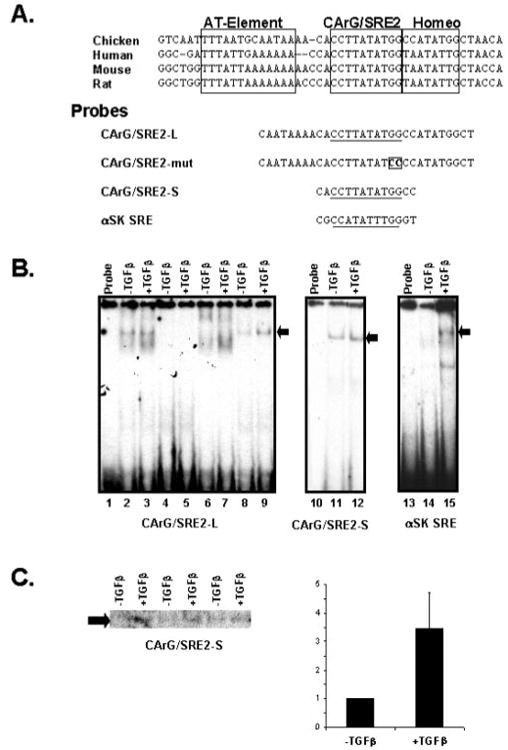
Nuclear lysates were derived from 10T1/2 mesenchymal cells incubated with or without 1 ng/ml TGF-β for 48 h and utilized for gel shift analyses. A, an alignment of SM-γ-actin promoter sequences from chicken (14), human (18), mouse (49), and rat (75) containing the region surrounding the CARG/SRE2 motif is shown. The sequence from this highly conserved segment of the SM-γ-actin promoter contains an A-T-rich and homeodomain binding motifs in addition to the CArG/SRE2 element that function in SMGA transcription. Below the sequence alignment is the sequence of the oligonucleotide probes used for the gel shift experiments shown here. The CArG/ SRE2-L probe contains significant sequence 5′ and 3′ to the SRE element, whereas the CArG/SRE2-S probe contains the minimal SRF binding motif. The CArG/SRE2-mut probe contains a mutated SRE binding site and the α-SK SRE is a strong SRF binding site derived from the α-skeletal actin gene. B, multiple complexes were observed with 5 μg of nuclear lysate derived from treated and control cells using the CArG/SRE2-L probe (lanes 2 and 3). A 50× molar excess of unlabeled probe prevented the formation of binding complexes with the SM-γ-actin CArG/SRE2 probe (lanes 4 and 5), whereas a 50× excess of a probe that contained an SRF binding site derived from the α-skeletal actin gene efficiently inhibited the formation of one prominent complex (denoted with the arrow), indicating that this complex contained SRF as its principal binding component (lanes 6 and 7). A competitor oligonucleotide in which the CArG/SRE motif was mutated to prevent SRF binding did not inhibit the formation of the SRF complex with the native probe sequence; however the other complexes were efficiently prevented from forming (lanes 8 and 9). The arrow to the right of the autoradiograph denotes the position of the SRF containing complex. Using 5 μg of nuclear lysate the CArG/SRE2-S (lanes 11 and 12) and α-SK SRE (lanes 14 and 15) showed some varible nonspecific bands with a major band (denoted by the arrow) of SRF binding activity. C, quantitative assessment of SRF binding activity from 10T1/2 cells was performed by probing lysates from multiple, separate experiments with the CArG/ SRE2-S oligonucleotide probe and separating the resultant binding complexes on a single gel. SRF-binding complex formation with this probe using 3 μg of nuclear lysate from three separate experiments are shown. The radioactivity associated with the SRF complex band was then quantitated using a Bio-Rad PhosphorImager and the Molecular Analysis Software package (Bio-Rad). The amount of binding derived from treated cells was compared with that observed in control cells, which was designated as a value of 1. The relative binding activity was averaged and plotted ± the S.D.
To specifically examine the SRF binding activity, we preformed control experiments utilizing oligonucleotides containing minimal binding sites from the SM-γ-actin (CArG/SRE2-S, Fig. 5A) and α-skeletal (α-SK SRE, Fig. 5A) actin genes. Both the SM-γ-actin and α-skeletal SRE-binding motifs exhibited a singular major SRF-binding complex that was enhanced in lysates from TGF-β-treated cells (lanes 10–15, Fig. 5B). Quantitative analysis of SRF binding using the SRE probes demonstrated that lysates from treated cells exhibited ∼3-fold (3.2 ± 1.5; Fig. 5C) more SRF complex formation than found in control, untreated cells even when minimal amounts of nuclear lysate is probed (3 μg of total lysate, Fig. 5C). Thus, these data show that there are multiple cis-trans interactions near the CArG/SRE2 motif of the SM-γ-actin promoter in 10T1/2 cells and that the complex containing SRF as a principal binding component is enhanced in lysates derived from TGF-β-treated cells.
TGF-β Up-regulates SRF Expression in SM Precursors
SRF gene expression was examined in TGF-β-treated 10T1/2 cells to determine whether the changes observed in SRF binding activity resulted from alterations in SRF expression levels. Western blot analyses revealed a measurable increase in SRF protein after 8–12 h of TGF-β incubation (Fig. 6A), which precedes the appearance of SM-γ-actin protein (∼12–24 h) and coincides with increases in SM-γ-actin mRNA in response to TGF-β (Fig. 2). SRF protein was not detected in 10T1/2 cells cultured in control conditions at any time point throughout the experimental period.
Fig. 6. TGF-β regulation of SRF expression.
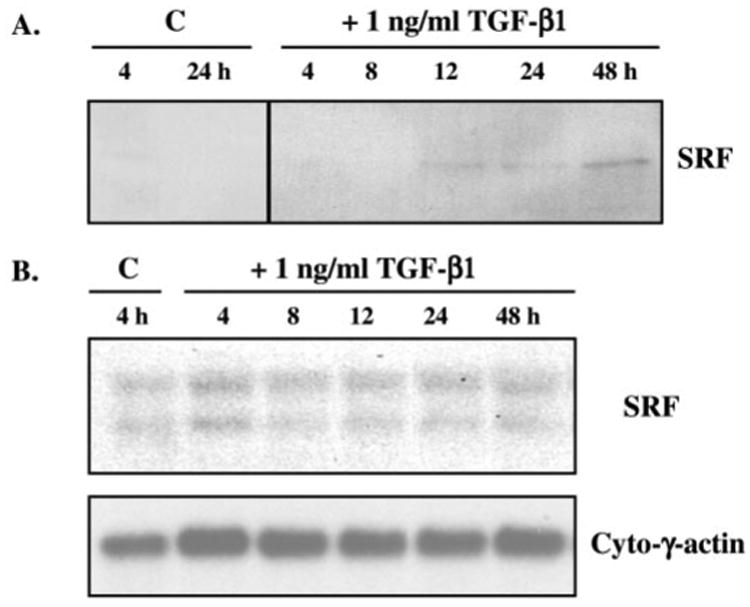
A, 10T1/2 cells were treated with 1 ng/ml TGF-β for up to 48 h, after which time total protein was isolated and subjected to Western blot analyses (10 μg of total protein/lane) using anti-SRF (ICN; 1:500). SRF protein (∼65 kDa) expression was induced after 8–12 h of TGF-β exposure compared with control (C) conditions. B, total RNA was isolated from similarly treated cells and probed, via Northern blot analysis, with a murine SRF cDNA. This probe recognized two mRNA species (∼4.5 and 2.5 kb) in the RNA populations derived from the 10T1/2 cells, which are the products of both differential polyadenylation and alternate splicing of the SRF gene transcript (42). Blots were re-hybridized with a specific probe to cytoplasmic γ-actin as a loading control. While there was a consistent 25–30% increase, in multiple experiments, in SRF mRNA in TGF-β-treated cells as compared with controls, which was detectable after 4 h, the mRNA levels did not exhibit a steady increase over the time of incubation as did the appearance of SRF protein.
The levels of SRF mRNA in treated and control cells were examined to determine whether increased SRF protein was due to increased steady-state mRNA levels (Fig. 6B). Total RNA was isolated from cells and probed by hybridization with a murine SRF cDNA (42). This probe recognized two mRNA species in the RNA populations derived from the 10T1/2 cells, which are the products of both differential polyadenylation (42) and alternate splicing (42) of the SRF gene transcript. Observations derived from multiple experiments showed that there was a consistent 25–30% increase in normalized SRF mRNA within cells treated with TGF-β compared with controls, evidenced as early as 4 h. However, the mRNA levels did not exhibit a steady increase over time as did SRF protein. Thus, there was a discordant increase in SRF protein relative to the subtle up-regulation of its mRNA in response to TGF-β, unlike the coordinate induction of SM-γ-actin protein and mRNA. The observed increase in SRF protein expression was paralleled by increased SRF binding activity upon CArG/SRE2 of the SM-γ-actin promoter (Fig. 5) and consistent with the timing of TGF-β activation of SM-γ-actin gene expression in SM precursors (Fig. 2).
Our analyses revealed that SRF mRNA is expressed in 10T1/2 cells in control conditions; however, SRF protein is only detectable by Western blotting analysis in response to TGF-β. There is SRF-directed binding activity, and therefore SRF protein, in control 10T1/2 cells (Fig. 5), indicating that SRF content in these cells is below the sensitivity of our Western assays. Thus, we are measuring significant increases in SRF protein content in TGF-β-treated cells in our experiments relative to slight increases in mRNA. These observations, combined with previous studies in vivo (43, 15) and in vitro (42, 46)3 suggest that TGF-β regulation of SRF expression also involves post-transcritpional mechanism(s) of control.
SRF Is Necessary and Sufficient for TGF-β Induction of a SM Phenotype
To determine whether SRF is necessary and sufficient for TGF-β-induced SM phenotype in mesenchymal cells, we created stable 10T1/2 transfectant cell lines that express either wild type (wtSRF) or a dominant negative form of the SRF protein (dnSRF), as described above. To create stable transfectants, 10T1/2 cells were electroporated with an HA-tagged expression plasmid containing wtSRF, dnSRF, or no cDNA (vector control) and cotransfected with an expression plasmid containing the neomycin resistance gene (pCI-neo). Stable transfectant clones were generated via neomycin selection and screened via Western analyses to assess the expression of the SRF protein and HA tag. Western analyses revealed that the wtSRF and dnSRF clones expressed HA-tagged SRF; whereas, vector control transfectants did not (representative clones shown in Fig. 7A).
Fig. 7. SRF is necessary and sufficient for TGF-β induction of a SM phenotype.

10T1/2 cells were transfected, via electroporation, with 5 μg of linearized HA-tagged expression plasmid containing no cDNA (vector control), 5 μg of linearized wtSRF cDNA, or 5 μg of linearized dnSRF cDNA. All cells were cotransfected with 0.5 μg of linearized pCI-neo plasmid, and stable transfectant clones were generated from each experimental group via selection in 1000 μg/ml G418-containing media. Total protein was isolated from each clone and screened via Western analyses (10 μg of total protein/lane) to assess the expression of SRF protein and the HA tag. A, transfectants expressing wtSRF (10T-wt SRF) and dnSRF (10T-dnSRF) exhibited SRF protein expression, and concomitant HA tag, via Western analyses; stable clones containing only the plasmid vectors (10T-v) did not exhibit SRF or HA expression. One representative clone of the 12 generated for each experimental group is shown. B, two or three clones from each group were cultured in the presence or absence of 1 ng/ml TGF-β1 for 24 h; protein was isolated from each and analyzed for expression of SM-γ-actin. Stable transfectants containing only vector (10T-v) exhibited a dose-dependent increase in SM-γ-actin protein expression in response to TGF-β. Transfectants expressing wtSRF (10T-wtSRF) exhibited elevated levels of SM-γ-actin in control conditions, which was further increased in response to TGF-β. Expression of dnSRF in mesenchymal cells prevented TGF-β induction of SM-γ-actin protein expression. Results generated from a representative clone for each experimental group are shown.
Multiple clones from each transfectant group (10T-v, 10T-wtSRF, and 10T-dnSRF) were cultured in the presence or absence of 1 ng/ml TGF-β1. Total protein was isolated from each and analyzed for the expression of SM-γ-actin protein; representative results from one clone of each experimental group are shown in Fig. 7B. 10T-vector transfectant clones exhibited an expected up-regulation of SM-γ-actin protein expression in response to TGF-β (Fig. 7B); this response was dose-dependent from 0 to 5 ng/ml TGF-β1 (data not shown). Transfectant cell lines expressing wtSRF exhibited elevated levels of SM-γ-actin protein in control conditions, compared with untreated 10T1/2 cells (see Fig. 2) or vector control clones; TGF-β treatment further increased SM-γ-actin protein levels in these clones (Fig. 7B). The wtSRF clones also exhibited a change in morphology from flat and spread to elongated with many pseudopodia, reminiscent of SM cells in culture (not shown). In contrast, transfectants expressing dnSRF exhibited no SM-γ-actin protein expression in control conditions or exhibited no increase in SM-γ-actin protein expression in response to TGF-β treatment (Fig. 7B).
Discussion
Previously (44, 48), we demonstrated that multipotent 10T1/2 mesenchymal cells serve as SM progenitors and, as such, provide an excellent in vitro model to examine molecular details of SM cell differentiation. We have shown that the expression of SM-specific proteins, including SM-α-actin, cal-ponin, SM22α, and SM myosin heavy chain, is up-regulated in 10T1/2 mesenchymal cells upon contact with endothelial cells and that this phenotypic induction is mediated, at least in part, by TGF-β (44). Recent studies revealed that SM-γ-actin is also up-regulated in 10T1/2 mesenchymal cells directly cocultured with endothelial cells, in a TGF-β-dependent manner.2 These results are consistent with recent findings from other laboratories demonstrating that SM-γ-actin is coordinately expressed with the above mentioned cytoskeletal and contractile proteins that collectively characterize the differentiated SM cell phenotype (14, 36).
Our present studies focus on the molecular mechanisms underlying TGF-β-induction of a SM phenotype; specifically, we investigated TGF-β regulation of SM-specific γ-actin gene expression. We observed that, along with previously documented morphological changes (44), TGF-β induced the production and cytoskeletal localization of SM-γ-actin in 10T1/2 mesenchymal cells. Increased SM-γ-actin protein was paralleled by an increase in its mRNA, indicating that, as in normal SM developmental processes (14,36), TGF-β regulation of SM-specific gene products is mediated by mechanisms that provide increased steady-state levels of mRNA within cells. We further demonstrated that TGF-β enhanced transcriptional activation of the SM-γ-actin gene. Deletion and mutagenesis experiments demonstrated that the CArG/SRE motif located at −120 bp (CArG/ SRE2) of the SM-γ-actin promoter played a critical role in the observed TGF-β transcriptional activation, suggesting that this response occurred via an SRF-mediated pathway.
Consistent with this theory, we found that TGF-β up-regulated SRF protein expression via transcriptional and post-transcriptional controls, and we observed an increase in SRF-binding complexes in nuclear lysates derived from TGF-β-treated 10T1/2 cells. Furthermore, we found that stable expression of wtSRF in mesenchymal cells was sufficient to induce a SM phenotype and that stable expression of dnSRF suppressed the observed TGF-β-induced SM phenotype in mesenchymal cells. Thus, our present results demonstrate that TGF-β induction of SM differentiation is mediated via the regulation of SRF expression.
While TGF-β has been shown to up-regulate the expression of multiple SM-specific proteins, our data suggest that the mechanisms by which this response is mediated may be gene-specific. For example, although SM-γ-actin and SM-α-actin are closely related isoforms of the same gene family, modulation of their expression via TGF-β appears to be uniquely controlled. In the case of SM-α-actin, TGF-β responsiveness involves two separate cis-elements: 1) the CArG/SRE motif, which we found to mediate TGF-β-induced SM-γ-actin expression and 2) a G-A-rich TGF-β control element or TCE (11), which is recognized in the promoter region of various SM-specific genes, but whose involvement in TGF-β responsiveness of these genes has not been investigated (11). The segment of the SM-γ-actin gene promoter that we demonstrate here to be necessary for TGF-β activation of the gene (−135 bp) is highly conserved across species (14, 16, 18, 49) and does not contain any obvious TCE motifs. However, we did observe multiple protein-DNA complexes formed with oligonucleotide containing the SM-γ-actin CArG/SRE and surrounding sequences. Thus, it is possible that SRF works in conjunction with other factors to regulate SM-γ-actin transcription in response to TGF-β. Synergistic interactions of SRF with other factors that activate gene transcription have been demonstrated for several muscle-restricted genes (43, 50, 51), including SM-γ-actin (52). Moreover, these interactions may not necessarily require DNA binding by the accessory factor, such as in the facilitated binding of SRF to CArG B of the SM-α-actin gene promoter (50). Nonetheless, it is clear that TGF-β induction of SM-γ-actin transcription does not require secondary protein interactions at a TCE or TCE-like motif.
The apparent differences in TGF-β transcriptional regulation between such SM-specific genes may dictate observed differences in developmental expression. During embryogenesis (53) and in developmental models (36), SM-α-actin is among the first genes expressed in the SM lineage, although not restricted to this lineage. Given that TGF-β is required for SM development, it seems logical that the first SM gene induced would require unique regulation via a specialized TGF-β control element. Along these same lines of thought, perhaps other genes that are up-regulated at later stages of SM differentiation, and are required for differentiated function, are more dependent on mediators downstream of TGF-β signaling, such as SRF. If so, it would be possible that TGF-β temporally induces the production of key positive and negative factors during the progressive stages of SM development and, in so doing, directs the coordinated expression of the genes that collectively characterize the SM cell phenotype. Alternatively, other factors, regulated independently from TGF-β, may act in conjunction with TGF-β-inducible factors. In support of this idea, comparison of the promoter regions of various SM-specific genes reveals unique cis-elements (such as retinoic acid response elements, RAREs) that may be utilized for the sequential, stepwise expression of genes needed for SM cell function.
Also, since TGF-β signaling is mediated via intracellular transducer SMAD proteins, which assemble multisubunit complexes that move to the nucleus and regulate transcription (54), perhaps differential regulation of these proteins yields multilayered SM development. The SMAD regulatory proteins make DNA contact at the core sequence 5′-CAGAC-3′ (55), which is not found in the SM-γ-actin promoter sequences needed for TGF-β responsiveness (−135 bp, Fig. 3). SMAD proteins are able to interact with a variety of cofactors, some of which have no apparent intrinsic transactivating activity (56, 57) and many that do contain such activity (58 – 60). Thus, appropriate SM-γ-actin transcriptional activation requires SRF binding, and because SRF is also capable of forming multiple complexes with transcriptional activators and co-factors, it may require SMAD complex interactions with SRF or SRF accessory factors.
There are potential SMAD binding sites within the SRF promoter that may account for the rapid increase in mRNA observed with TGF-β treatment. One of these sites overlaps with a potential AP-1 site (at −436) of the SRF core promoter (42), which is a combination of cis-elements found to be operative in other genes that respond to TGF-β regulation via the SMAD proteins (59). It has been shown that two CArG/SRE motifs and Sp1 binding sites within the SRF gene promoter are necessary for both serum-stimulated (61) and muscle-restricted (41) transcription of the gene. Although this proximal segment of the SRF promoter (∼136 bp) was needed for specific transcription, this segment was not sufficient for muscle-restricted SRF transcription. Indeed, sequences adjacent to the SRF proximal promoter were needed to establish full activation of this promoter in skeletal muscle cells and for an increased transcriptional capacity of the SRF promoter during myogenesis (41). Our studies revealed induction of SRF gene transcription leading to a rapid 25–50% increase in mRNA in mesechymal cells in response to TGF-β. Although the 5′ segment of the SRF promoter contains multiple SMAD-like core binding sequences, the contribution of these elements to the TGF-β-stimulated transcriptional activation of the gene remains to be evaluated.
In contrast to the rapid increase in SRF mRNA content, we observed a delayed accumulation of SRF protein in mesenchymal cells, in response to TGF-β. These data suggest that post-transcriptional regulation plays an important role in the control of SRF expression during SM differentiation. Post-transcriptional control of SRF in response to TGF-β may involve regulation of protein stability and/or translational control of mRNA expression. TGF-β has been implicated in the translational control of several gene products including collagenase 3 (62), elastin (63, 64), growth hormone releasing factor (65), ribonucleotide reductase R2 (66), and receptor for hyaluronan-mediated motility (67). For the regulation of ribonucleotide reductase R2 and receptor for hyaluronan-mediated motility, it appears that TGF-β acts to stabilize their respective mRNAs in the cytoplasm of treated cells, and this occurs via specific cis-trans interactions directed by the 3′-UTR mRNA segments (66, 67). Related studies from our laboratories suggest that SRF expression may be translationally controlled in response to TGF-β, in a process mediated through the 5′-UTR of the SRF mRNA (15, 42, 43, 46).3 The SRF 5′-UTR is complex, enriched in G and C bases (268 out of 348 bases or ∼77% G + C) (41) and, thus, shares characteristics of mRNAs encoding a number of developmental regulatory proteins (Antp Ubx homeodomain, RAR-β, c-Mos, c-Myc, FGF2, PDGF2, VEGF), including TGF-β1 (68, 69), that are regulated in a spatio-temporal manner via 5′-UTR translational control.
A number of factors, both inter- and intracellular, may influence TGF-β-mediated differentiation responses upon SM development observed in our studies. During vascular development, SM differentiation occurs only in mesenchymal precursors that have contacted mature endothelial cells (53). We have observed a similar phenomenon in our coculture model system and have determined that TGF-β mediates the endothelial cell-induced SM differentiation (44). TGF-β in this system, and during development, is presumably activated in response to cell-cell contact (8). In the developing GI tract, mesenchymal cells are found to respond to soluble signals from epithelial cells, such as sonic hedgehog, by up-regulating the production of the TGF-β family (70). Here, TGF-β signaling may not only influence SM development directly by affecting SRF expression, but likely stimulates the production of basement membrane components that stimulate the SM developmental pathway (14). Moreover, other transcriptional factors may play a role in SM development. Mice deficient for the factor COUP-TFII exhibit defective vascular development secondary to the lack of SM cell and pericyte investment of endothelial tubes (71). It would be of great interest to determine whether factors such as COUP-TFII are up-regulated by locally produced and activated TGF-β or perhaps by other soluble effectors, such as retinoids, that are known to play a role in SM development (72).2 Interestingly, retinoid signaling is associated with increased production (73) and activation (74) of TGF-β, and may be an initiating signal for SM development during embryogenesis.
In summary, the in vivo microenvironment that dictates the differentiation of SM from mesenchymal and neural crest precursors is complex, and the regulation of this process is undoubtedly multilayered. The studies detailed here reveal the biological connection between two factors independently shown to play critical roles in the regulation of smooth muscle development, TGF-β and SRF. Thus, these findings significantly contribute to our understanding of the biological control of smooth muscle development and provide insights into aberrant smooth muscle differentiation that occurs in prevalent pathological conditions.
Acknowledgments
We thank Charnae Williams for technical assistance.
Footnotes
The abbreviations used are: TGF-β, transforming growth factor-β; SM, smooth muscle; DMEM, Dulbecco's modified Eagle's medium; SRF, serum response factor; HA, hemagglutinin; UTR, untranslated region; wt, wild type; dn, dominant-negative; CS, calf serum.
L. Lai and K. K. Hirschi, unpublished observation.
N. S. Belaguli, unpublished data.
References
- 1.Massaque J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 2.Staehling-Hampton K, Hoffmann FM, Baylies MK, Rushton E, Bate M. Nature. 1994;372:783–786. doi: 10.1038/372783a0. [DOI] [PubMed] [Google Scholar]
- 3.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts A. Wound Repair Regen. 1995;3:408–418. doi: 10.1046/j.1524-475X.1995.30405.x. [DOI] [PubMed] [Google Scholar]
- 5.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Development (Camb) 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 6.Nakajima Y, Miyazono K, Nakamura H. Cell Tissue Res. 1999;295:257–267. doi: 10.1007/s004410051232. [DOI] [PubMed] [Google Scholar]
- 7.Nunes I, Munger JS, Harpel JG, Nagano Y, Shapiro RL, Gleizes PE, Rifkin DB. Int J Obes Rel Metab Dis. 1996;3:S4–S8. [PubMed] [Google Scholar]
- 8.Antonelli-Orlidge A, Saunders KB, Smith SR, D'Amore PA. Proc Natl Acad Sci U S A. 1989;86:4544–4548. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah NM, Groves AK, Anderson DJ. Cell. 1996;85:331–343. doi: 10.1016/s0092-8674(00)81112-5. [DOI] [PubMed] [Google Scholar]
- 10.Blank RS, Thompson MM, Owens GK. J Cell Biol. 1988;107:299–306. doi: 10.1083/jcb.107.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hautmann MB, Madsen CS, Owens GK. J Biol Chem. 1997;272:948–956. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- 12.Carroll SL, Bergsma DJ, Schwartz RJ. Mol Cell Biol. 1988;8:241–250. doi: 10.1128/mcb.8.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Owens GK. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 14.Kovacs AM, Zimmer WE. Gene Expr. 1998;7:115–129. [PMC free article] [PubMed] [Google Scholar]
- 15.Browning CL, Culberson DE, Aragon IV, Fillmore RA, Croissant JD, Schwartz RJ, Zimmer WE. Dev Biol. 1998;194:18–37. doi: 10.1006/dbio.1997.8808. [DOI] [PubMed] [Google Scholar]
- 16.McHugh KM, Lessard J. Mol Cell Biol. 1988;8:5224–5231. doi: 10.1128/mcb.8.12.5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McHugh KM. J Pediatr Gastroenterol Nutr. 1996;25:379–394. doi: 10.1097/00005176-199611000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Miwa T, Manabe Y, Kurokawa K, Kamada S, Kandu N, Burns G, Ueyama H, Kakunaga T. Mol Cell Biol. 1991;11:3296–3306. doi: 10.1128/mcb.11.6.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babij P, Kelly C, Periasamy M. Proc Natl Acad Sci U S A. 1991;88:10676–10680. doi: 10.1073/pnas.88.23.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miano J, Cserjesi P, Ligon K, Perissamy M, Olson EN. Circ Res. 1994;75:803–813. doi: 10.1161/01.res.75.5.803. [DOI] [PubMed] [Google Scholar]
- 21.Kallmeier RC, Somasundaram C, Babij P. J Biol Chem. 1995;220:30949–30957. doi: 10.1074/jbc.270.52.30949. [DOI] [PubMed] [Google Scholar]
- 22.Samaha FF, Ip HS, Morrisey EE, Seltzer J, Tang ZH, Solway J, Parmacek MS. J Biol Chem. 1996;271:395–403. doi: 10.1074/jbc.271.1.395. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Liu ZC, Mercer B, Overbeek P, Olson EW. Dev Biol. 1997;187:311–321. doi: 10.1006/dbio.1997.8621. [DOI] [PubMed] [Google Scholar]
- 24.Mossler H, Mericskag M, Li Z, Nagl S, Paulin D, Small JV. Development (Camb) 1996;122:2415–2425. doi: 10.1242/dev.122.8.2415. [DOI] [PubMed] [Google Scholar]
- 25.Solway J, Seltzer J, Samaha FF, Kim S, Alger LE, Nui Q, Morrisey EE, Ip HS, Parmacek MS. J Biol Chem. 1995;22:13460–13469. doi: 10.1074/jbc.270.22.13460. [DOI] [PubMed] [Google Scholar]
- 26.Collinge M, Matrisian PE, Zimmer WE, Shattuck RL, Lukas TJ, Van Eldik LJ, Watterson PM. Mol Cell Biol. 1992;12:2359–2371. doi: 10.1128/mcb.12.5.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallagher PJ, Herring BP. J Biol Chem. 1991;266:23945–23952. [PMC free article] [PubMed] [Google Scholar]
- 28.Mosse PR, Campbell GR, Campbell JH. Atherosclerosis. 1986;6:664–669. doi: 10.1161/01.atv.6.6.664. [DOI] [PubMed] [Google Scholar]
- 29.Ross R. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 30.MacDermott RP. Inflammatory Bowel Disease: Current Status and Future Approaches. Elsevier Scientific Publishers; New York: 1988. [Google Scholar]
- 31.Neurath MF, Meyer zum Buschenfelde KH. J Investig Med. 1996;44:516–520. [PubMed] [Google Scholar]
- 32.Majesky MW, Lindner V, Twurdzik DR, Schwartz SM, Reidy MA. J Clin Investig. 1991;88:904–910. doi: 10.1172/JCI115393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf VG, Rasmussen LM, Ruoslahti E. J Clin Investig. 1994;93:1172–1178. doi: 10.1172/JCI117070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powrie FT. Immunity. 1995;3:171–174. doi: 10.1016/1074-7613(95)90086-1. [DOI] [PubMed] [Google Scholar]
- 35.Jester JV, Barry-Lane PA, Cavanaugh HD, Petroll WM. Cornea. 1996;15:505–516. [PubMed] [Google Scholar]
- 36.Landerholm TE, Dong XR, Lu J, Belaguli NS, Schwartz RJ, Majesky MW. Development (Camb) 1999;126:2053–2062. doi: 10.1242/dev.126.10.2053. [DOI] [PubMed] [Google Scholar]
- 37.Kim E, Walters SH, Hule LE, Hect NB. Mol Cell Biol. 1989;9:1875–1881. doi: 10.1128/mcb.9.5.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blank RS, McQuinn TC, Yin KC, Thompson MM, Takegasa K, Schwartz RJ, Owens GK. J Biol Chem. 1992;267:984–989. [PubMed] [Google Scholar]
- 39.Kim S, Ip HS, Lu M, Clendenin C, Parmacek MS. Mol Cell Biol. 1997;17:2266–2278. doi: 10.1128/mcb.17.4.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miano JM, Carlson MJ, Spencer JA, Misra RP. J Biol Chem. 2000;275:9814–9822. doi: 10.1074/jbc.275.13.9814. [DOI] [PubMed] [Google Scholar]
- 41.Smith AF, Bigsby M, Word RA, Herring BP. Am J Physiol. 1998;274:C1188–C1195. doi: 10.1152/ajpcell.1998.274.5.C1188. [DOI] [PubMed] [Google Scholar]
- 42.Belaguli NS, Schildmeyer L, Schwartz RJ. J Biol Chem. 1997;272:18222–18231. doi: 10.1074/jbc.272.29.18222. [DOI] [PubMed] [Google Scholar]
- 43.Croissant JD, Kim JH, Eichele G, Geering C, Lough J, Prywes R, Schwartz RJ. Dev Biol. 1996;177:250–264. doi: 10.1006/dbio.1996.0160. [DOI] [PubMed] [Google Scholar]
- 44.Hirschi KK, Rohovsky SA, D'Amore PA. J Cell Biol. 1998;141:805–814. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruch RJ, Bonney WJ, Sigler K, Guan X, Matesic D, Schafer LD, Dupont E, Trosko JE. Carcinogenesis. 1994;15:301–306. doi: 10.1093/carcin/15.2.301. [DOI] [PubMed] [Google Scholar]
- 46.Nurrish S, Treisman R. Mol Cell Biol. 1995;15:4076–4085. doi: 10.1128/mcb.15.8.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmer WE, Browning CL, Kovacs AM. Dev Biol. 1996;175:399. [Google Scholar]
- 48.Hirschi KK, Rohovsky SA, Smith SR, Beck LH, D'Amore PA. Circ Res. 1999;84:298–305. doi: 10.1161/01.res.84.3.298. [DOI] [PubMed] [Google Scholar]
- 49.Qian J, Kumas A, Szucsik JC, Lessard JL. Dev Dyn. 1996;207:135–144. doi: 10.1002/(SICI)1097-0177(199610)207:2<135::AID-AJA2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 50.Grueneberg DA, Nateson S, Alexandre L, Gilmun M. Science. 1992;257:1089–1095. doi: 10.1126/science.257.5073.1089. [DOI] [PubMed] [Google Scholar]
- 51.Chen CY, Croissant S, Majesky MW, Topouzis S, McQuinn T, Frankovsky M, Schwartz RJ. Dev Genet. 1996;19:119–130. doi: 10.1002/(SICI)1520-6408(1996)19:2<119::AID-DVG3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 52.Carson JA, Fillmore RA, Schwartz RJ, Zimmer WE. J Biol Chem. 2000;275:39061–39072. doi: 10.1074/jbc.M006532200. [DOI] [PubMed] [Google Scholar]
- 53.Hungerford JE, Owens GK, Argraves WS, Little CD. Dev Biol. 1996;178:375–392. doi: 10.1006/dbio.1996.0225. [DOI] [PubMed] [Google Scholar]
- 54.Massaque J, Chen VG. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- 55.Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich N. Cell. 1998;94:585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- 56.Labbe E, Silvestri C, Hoodless PA, Wiana JL, Attisano L. Mol Cell. 1998;2:109–120. doi: 10.1016/s1097-2765(00)80119-7. [DOI] [PubMed] [Google Scholar]
- 57.Hata A, Sevane J, Lugna G, Montalvo E, Hemmati-Brivanlou A, Massague J. Cell. 2000;100:229–240. doi: 10.1016/s0092-8674(00)81561-5. [DOI] [PubMed] [Google Scholar]
- 58.Germain S, Howell M, Esslemont GM, Hill CS. Genes Dev. 2000;14:435–451. [PMC free article] [PubMed] [Google Scholar]
- 59.Wong C, Rougier-Chapman EM, Frederick JP, Datto MR, Liberati NT, Li JM, Wang XF. Mol Cell Biol. 1999;19:1821–1830. doi: 10.1128/mcb.19.3.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hua X, Miller ZA, Wu G, Shi V, Lodish HF. Proc Natl Acad Sci U S A. 1999;96:13130–13135. doi: 10.1073/pnas.96.23.13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spencer JA, Misra RP. J Biol Chem. 1996;271:16535–16543. doi: 10.1074/jbc.271.28.16535. [DOI] [PubMed] [Google Scholar]
- 62.Rydzeil S, Varghese S, Canalis E. J Cell Physiol. 1997;170:145–152. doi: 10.1002/(SICI)1097-4652(199702)170:2<145::AID-JCP6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 63.Yamamoto K, Yamamoto M, Akazawa K, Tajima S, Wakimoto H, Aoyagi M. Cell Biol Int. 1997;21:605–611. doi: 10.1006/cbir.1997.0192. [DOI] [PubMed] [Google Scholar]
- 64.Parks WC. Am J Respir Cell Mol Biol. 1997;17:1–2. doi: 10.1165/ajrcmb.17.1.f135. [DOI] [PubMed] [Google Scholar]
- 65.Yamaguchi M, Endo H, Maeda T, Tahara M, Ono M, Miki M, Miyake A. Biochem Biophys Res Commun. 1994;204:1206–1211. doi: 10.1006/bbrc.1994.2591. [DOI] [PubMed] [Google Scholar]
- 66.Amara FM, Cheun FY, Wright JA. Nucleic Acids Res. 1995;23:1461–1467. doi: 10.1093/nar/23.9.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amara FM, Entwistle T, Kuschack TI, Turley EA, Wright JA. J Biol Chem. 1996;271:15279–15284. doi: 10.1074/jbc.271.25.15279. [DOI] [PubMed] [Google Scholar]
- 68.van der Velden AW, Thomas AA. Int J Biochem Cell Biol. 1999;31:87–106. doi: 10.1016/s1357-2725(98)00134-4. [DOI] [PubMed] [Google Scholar]
- 69.Willis AE. Int J Biochem Cell Biol. 1999;31:73–86. doi: 10.1016/s1357-2725(98)00133-2. [DOI] [PubMed] [Google Scholar]
- 70.Roberts DJ, Johnson RL, Burke AL, Nelson CE, Morgan BA, Taziz C. Development (Camb) 1995;121:3163–3174. doi: 10.1242/dev.121.10.3163. [DOI] [PubMed] [Google Scholar]
- 71.Tsai SY, Tsai M. Endocr Rev. 1997;18:229–240. doi: 10.1210/edrv.18.2.0294. [DOI] [PubMed] [Google Scholar]
- 72.Blank RS, Swartz EA, Thompson MM, Olson EN, Owens GK. Circ Res. 1995;76:742–749. doi: 10.1161/01.res.76.5.742. [DOI] [PubMed] [Google Scholar]
- 73.Gazit D, Ebner R, Kahn AJ, Derynck R. Mol Endocrinol. 1993;7:189–198. doi: 10.1210/mend.7.2.8385738. [DOI] [PubMed] [Google Scholar]
- 74.Okuno M, Moriwaki H, Imal S, Muto Y, Kawada N, Suzuki Y, Kojima S. Hepatology. 1997;26:913–921. doi: 10.1053/jhep.1997.v26.pm0009328313. [DOI] [PubMed] [Google Scholar]
- 75.Phiel CJ, Gabbeta V, Parsons LM, Rothblat D, Harvey RP, McHugh KM. J Biol Chem. 2001;276:34637–34650. doi: 10.1074/jbc.M105826200. [DOI] [PubMed] [Google Scholar]