

A central question is whether aneuploid phenotypes are the consequence of copy number changes of a few especially harmful genes that may be present on the extra chromosome or are caused by copy number alterations of many genes that confer no observable phenotype when varied individually. Bonney et al. used the proliferation defect exhibited by budding yeast strains carrying single additional chromosomes (disomes) to show that subtle changes in gene dosage across a chromosome can have significant phenotypic consequences.
Keywords: aneuploidy, dosage-sensitive genes, Down syndrome, proliferation
Abstract
Aneuploidy—the gain or loss of one or more whole chromosome—typically has an adverse impact on organismal fitness, manifest in conditions such as Down syndrome. A central question is whether aneuploid phenotypes are the consequence of copy number changes of a few especially harmful genes that may be present on the extra chromosome or are caused by copy number alterations of many genes that confer no observable phenotype when varied individually. We used the proliferation defect exhibited by budding yeast strains carrying single additional chromosomes (disomes) to distinguish between the “few critical genes” hypothesis and the “mass action of genes” hypothesis. Our results indicate that subtle changes in gene dosage across a chromosome can have significant phenotypic consequences. We conclude that phenotypic thresholds can be crossed by mass action of copy number changes that, on their own, are benign.
The impact of whole-chromosome gains and losses on human health is severe. Aneuploidy is the leading cause of miscarriages and mental retardation (Hassold and Jacobs 1984). It is also a hallmark of solid tumors: Seventy-five percent to 90% of tumors harbor abnormal karyotypes (Weaver and Cleveland 2006). Despite aneuploidy's dramatic impact on health, we are only beginning to understand how it affects cells and organisms.
Identifying the source of phenotypes observed when chromosome copy number is changed is central to understanding how aneuploidy affects cellular and organismal fitness. Changes in copy number (and thus expression level) of a relatively small subset of genes—known as dosage-sensitive genes (DSGs)—could be responsible for these phenotypes. Alternatively, aneuploid phenotypes may be the consequence of copy number changes of numerous genes, which have little phenotypic impact when misexpressed on their own. For instance, in Down syndrome (DS), caused by trisomy for human chromosome 21, the contribution of DSGs to phenotypes is debated. The “few critical genes” hypothesis predicts that a small number of triplicated genes causes the growth, developmental, and cognitive abnormalities characteristic of the condition. Indeed, previous studies mapped DS phenotypes to a genomic region known as the DS critical region (DSCR) (Rahmani et al. 1990; for review, see Rachidi and Lopes 2007). DSCR1, located in this region, was shown to protect individuals with DS from developing solid tumors (Baek et al. 2009). Triplication of APP (amyloid precursor protein) drives early-onset dementia in individuals with DS (Rumble et al. 1989; Rovelet-Lecrux et al. 2006; Theuns et al. 2006).
While some characteristics of DS could be attributed to changes in dosage of specific genes, other characteristics of the condition could not (Olson et al. 2004a,b, 2007; Korbel et al. 2009). In particular, phenotypes such as decreased proliferative capacity, a characteristic of not only human trisomy 21 cells but all constitutive aneuploidies (for review, see Torres et al. 2008), may be caused by cumulative effects of genes for which individual copy number alterations have minimal effects on fitness. A series of budding yeast strains harboring single additional chromosomes (disomes) is an ideal system to investigate which aspect of the aneuploid condition—individual DSGs or cumulative effects of many genes—is responsible for the decreased proliferative capacity in aneuploid cells across species. Here we show that changes in copy number of the most DSGs on a chromosome are insufficient to drive aneuploid proliferation defects. We conclude that the proliferation defect of disomes is largely caused by simultaneous copy number changes that are benign independently.
Results and Discussion
A method to determine the contribution of DSGs to the proliferation defect of disomic yeast
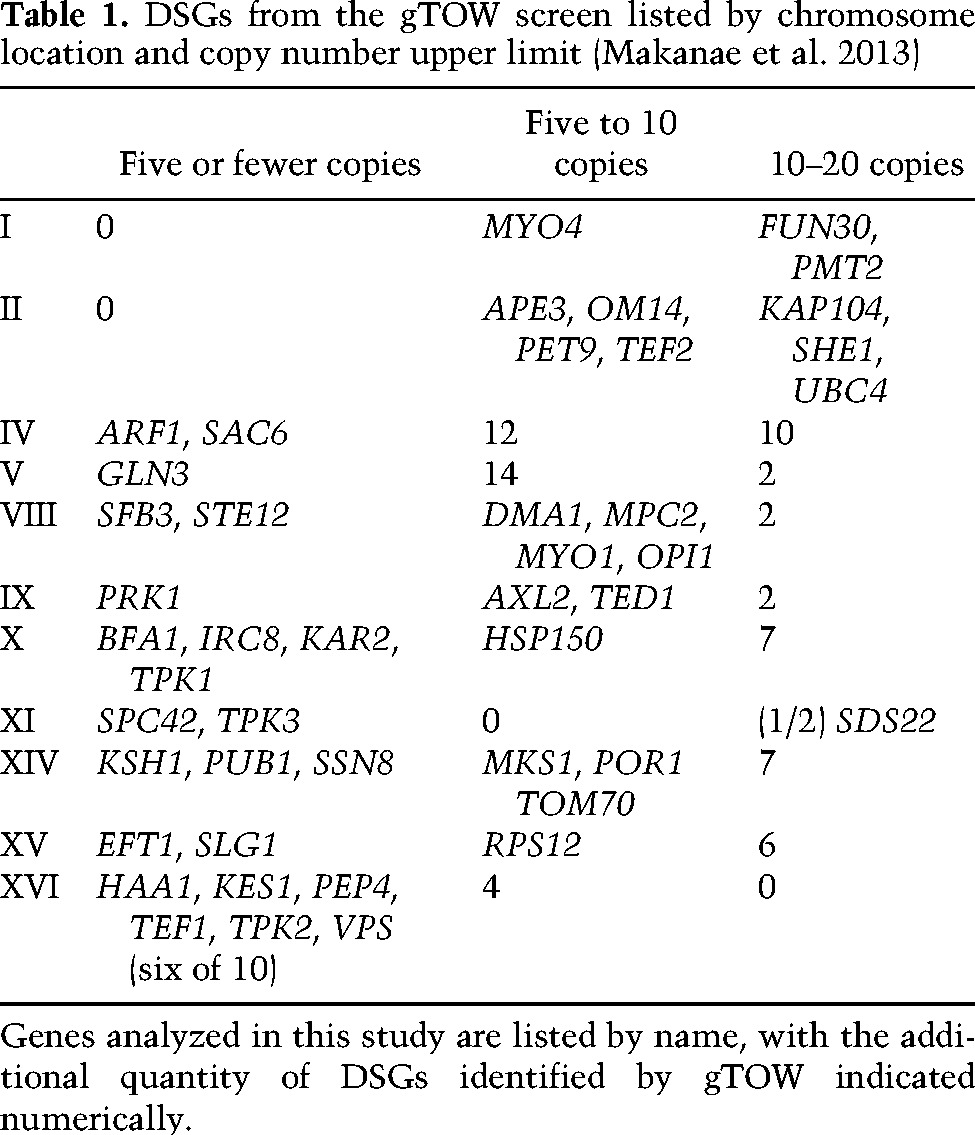
A previous genome-wide analysis shed light on which yeast genes cause proliferation defects when present in additional copies. The “genetic tug of war” (gTOW) method was used to determine the copy number at which a gene inhibits cell proliferation (Makanae et al. 2013). For gTOW, the gene of interest and its regulatory elements are present on a plasmid that can reach a high copy number to promote cell growth in medium lacking leucine due to the presence of a hypomorphic LEU2 allele (leu2-89) (Fig. 1A; Moriya et al. 2012). However, toxicity of the gene of interest prevents attaining high copy number even in growth conditions that select for the presence of the plasmid. Thus, plasmid copy number is a direct readout of maximal gene copy number tolerated by the cell (Moriya et al. 2012). The majority of yeast genes exhibit upper limits well over 75 copies (Makanae et al. 2013). However, 115 genes have an upper limit of ≤10 copies, and 55 genes are not tolerated at more than five copies per haploid genome (Makanae et al. 2013). Other approaches independently demonstrated that 76% of these genes were toxic upon overexpression (Boyer et al. 2004; Gelperin et al. 2005; Sopko et al. 2006; Douglas et al. 2012; Makanae et al. 2013), indicating that the genome-wide gTOW screen successfully identified DSGs in the yeast genome.
Figure 1.
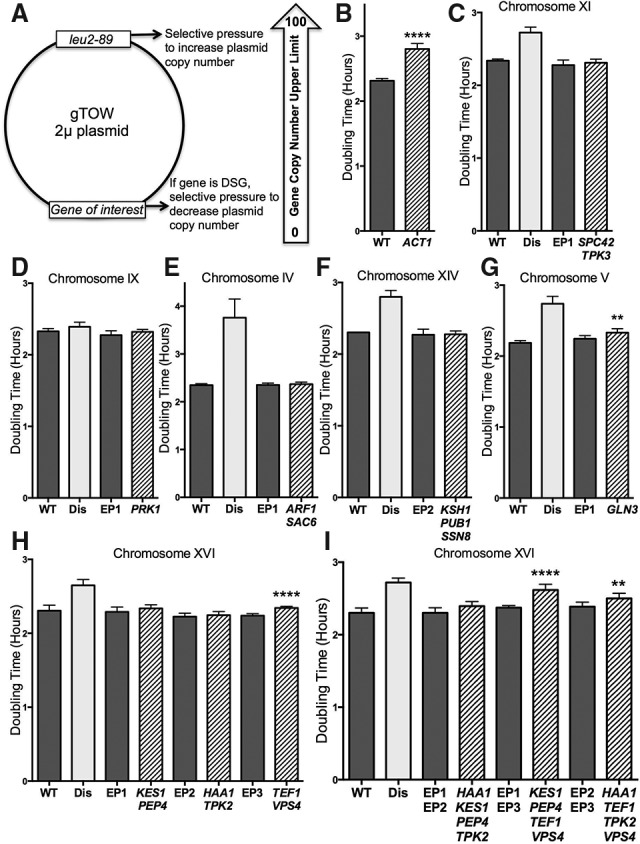
Effects of genes with copy number limits of five or fewer (DSGs[5]) on cell proliferation. (A) The gTOW plasmid contains a gene under the control of its endogenous regulatory elements, a multicopy 2-µm origin, and the hypomorphic leu2-89 allele (a LEU2 gene with a truncated promoter) (Moriya et al. 2012). (B–I) DSGs[5] on centromeric (CEN) plasmids were introduced into haploid wild-type (WT) cells. Doubling times of cells grown at 25°C in YEPD were determined by measuring OD600 every 15 min for 24 h. Genes were grouped into graphs according to their chromosomal location. Dark-gray bars represent control strains carrying empty plasmids, light-gray bars represent the corresponding disomic strain (Dis), and striped bars represent DSG plasmid-containing strains, with gene names listed directly below. Empty vector CEN plasmid controls were EP1-TRP1, EP2-HIS3, and EP3-LEU2. SD is shown. (B) (****) P < 0.0001. (G) (**) P = 0.0014. (H) (****) P < 0.0001; (I) (****) P < 0.0001, (**) P = 0.008; Student's t-test. The strains used, in order, were A2587 and A36983 (B); A22361, A28266, A36950, and A36937 (C); A22361, A13975, A36950, and A36931 (D); A22361, A24367, A36950, and A36926 (E); A22361, A28344, A36951, and A36941 (F); A22361, A28265, A36950, and A36927 (G); A22361, A34149, A36950, A36944, A36951, A36945, A36952, and A36946 (H); and A22361, A34149, A36953, A36947, A36954, A36948, A36955, and A36949 (I).
Previously, we generated 13 of 16 possible budding yeast disomies and found that all 13 disomies cause a decrease in fitness (Torres et al. 2007). We chose nine of the 13 disomic yeast strains (IV, V, VIII, IX, X, XI, XIV, XV, and XVI) to test whether the genes with copy number limits of five or fewer (henceforth DSGs[5]) (Table 1) could be responsible for the proliferation defects exhibited by these disomic yeast strains. We reasoned that genes that inhibit cell proliferation at five copies could slow proliferation when present at two copies, as occurs when haploid cells harbor an extra copy of a chromosome. We created centromeric (CEN) plasmids containing DSGs along with endogenous regulatory elements to mimic the single extra gene copy in disomes (Supplemental Table 1) and then examined the effects on cell proliferation in otherwise wild-type haploid strains.
Table 1.
DSGs from the gTOW screen listed by chromosome location and copy number upper limit (Makanae et al. 2013)
To ensure that the CEN plasmids recapitulate copy number changes as they occur in cells harboring an extra chromosome, we conducted a series of control experiments. We first determined whether CEN plasmids themselves interfere with cell proliferation. Introducing three CEN plasmids with no coding sequence other than genes encoding selectable markers did not significantly alter cell proliferation, as judged by doubling time measurements (Supplemental Fig. 1). We then tested the consequences of including coding sequences on these plasmids. Cells bearing one or two CEN plasmids carrying a total of three to six genes that should not significantly slow doubling time did not interfere with proliferation. However, cells bearing three CEN plasmids—each containing multiple yeast genes, for a total of six to eight genes not among the DSGs[5]—did slow proliferation in haploid strains (Supplemental Fig. 1). The fact that introduction of multiple CEN plasmids slows cell proliferation (Futcher and Carbon 1986) could contribute to this phenotype. Based on these control experiments, we restricted our analysis to no more than two CEN plasmids.
Next, we verified whether it was possible to detect proliferation defects caused by a single additional copy of a DSG. Consistent with previous studies (Liu et al. 1992), we found that introduction of a CEN plasmid carrying the actin-encoding gene ACT1 impaired cell proliferation (Fig. 1B). We conclude that the addition of CEN plasmids to haploid wild-type strains can be used to mimic gene copy number changes as they occur in disomic strains.
Assessing the contribution of low-copy-limit DSGs to the proliferation defects of aneuploid cells
All disomic yeast strains exhibit proliferation defects (Torres et al. 2007), but only one disomy, disomy VI, is lethal, as this chromosome harbors the β-tubulin-encoding gene TUB2 (Neff et al. 1983; Katz et al. 1990; Liu et al. 1992). The reasons why disomes proliferate more slowly than euploid cells under standard growth condition are unknown. To determine whether the slow proliferation of disomes is caused by two copies of DSGs[5], we compared the doubling times of yeast strains harboring a specific disomy with wild-type strains containing CEN plasmids carrying DSGs[5] encoded by the disomic chromosome. For instance, chromosome XI encodes two DSGs[5]: SPC42 and TPK3 (Table 1). Introduction of a CEN plasmid carrying these two genes into wild-type cells did not significantly lengthen cell cycle time (Fig. 1C) even though expression of the two genes was comparable with that of disome XI cells (Supplemental Fig. 2N). Similar results were obtained for DSGs[5] encoded on chromosomes IX (PRK1) (Fig. 1D; Supplemental Fig. 2J), VIII (SFB3 and STE12) (Fig. 2A; Supplemental Fig. 2H), X (BFA1, KAR2, IRC8, and TPK1) (Fig. 2B; Supplemental Fig. 2L,M), and XV (EFT1 and SLG1) (Fig. 2C; Supplemental Fig. 2R). Introducing DSGs[5] encoded on chromosomes IV (ARF1 and SAC6) (Fig. 1E; Supplemental Fig. 2F) and XIV (KSH1, PUB1, and SSN8) (Fig. 1F; Supplemental Fig. 2P) into wild-type cells did not slow proliferation either; however, in these two cases, expression of one of the DSGs[5] (SAC6 or PUB1) (Supplemental Fig. 2F,P) from the CEN plasmid did not reach expression levels observed in the disomes.
Figure 2.
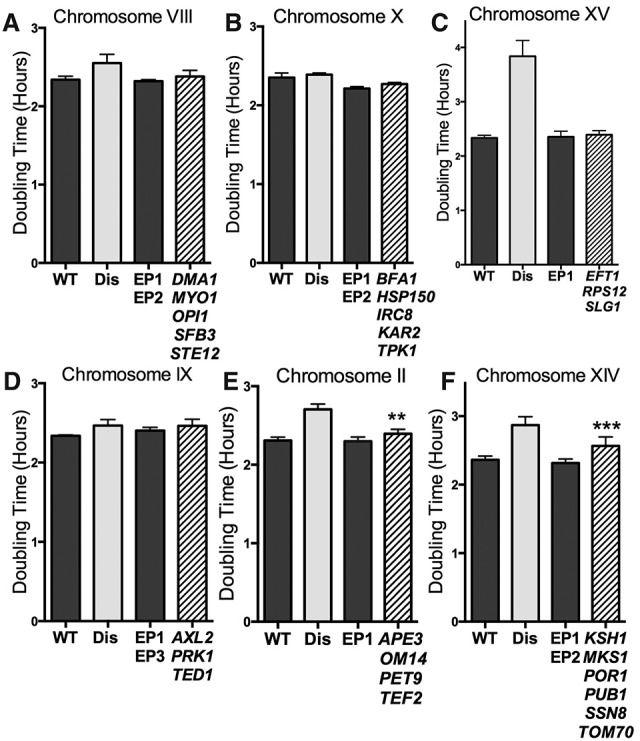
Consequences of an additional copy of genes with copy number limits of <10 (limit 0–10, henceforth DSGs[10]) on cell proliferation. (A–F) Cells were grown as described in Figure 1. Genes were grouped into graphs according to chromosomal location. Dark-gray bars represent control strains carrying empty plasmids, light-gray bars represent the corresponding disomic strain (Dis), and striped bars represent DSG plasmid-containing strains. Empty vector CEN plasmid controls were EP1-TRP1, EP2-HIS3, and EP3-LEU2. SD is shown. (E) (**) P = 0.001. (F) (***) P = 0.0002; Student's t-test. The strains, in order, were A22361, A27036, A35953, and A36930 (A); A22361, A27093, A36953, and A36936 (B); A22361, A29730, A36950, and A36943 (C); A22361, A13975, A36954, and A36933 (D); A22361, A12685, A36950, and A36923 (E); and A22361, A28344, A36953, and A36942 (F).
Only three DSGs[5] led to a significant proliferation defect when expressed from a CEN plasmid. GLN3, the sole DSG[5] encoded on chromosome V, slowed the doubling time of wild-type cells by 5.1 min (±1.3 min) (Fig. 1G; Supplemental Fig. 2G). For comparison, an additional copy of chromosome V increases doubling time by 33 min (±3.1 min). DSGs[5] encoded on chromosome XVI also slowed proliferation. Chromosome XVI encodes 10 DSGs[5] (Table 1). Cells disomic for chromosome XVI proliferate 25 min (±2.3 min) slower than wild type (Fig. 1H,I). We examined the effects of six chromosome XVI DSGs[5] individually and in combination. A plasmid containing TEF1 and VPS4 slowed doubling time by 6.3 min (±0.7 min) (Fig. 1H; Supplemental Fig. 2U). Additionally, when combined with a plasmid harboring the DSGs[5] KES1 and PEP4, time of doubling increased by 14.6 min (±1.8 min) compared with empty vector controls (Fig. 1I). The addition of HAA1 and TPK2 with TEF1 and VPS4 did not slow proliferation more than TEF1 and VPS4 alone (Fig. 1I).
To determine how an additional copy of KES1, PEP4, TEF1, and VPS4 interferes with cell proliferation, we analyzed progression through the cell cycle in cultures synchronized by centrifugal elutriation. Previous cell cycle studies of disome XVI cells revealed a substantial G1 delay, largely due to a growth defect (Thorburn et al. 2013) and a metaphase delay (Torres et al. 2007). Wild-type cells expressing an extra copy of KES1, PEP4, TEF1, and VPS4 exhibited a minor G1 delay, as judged by a subtle delay in bud formation (Supplemental Fig. 3A) and a 15-min-long metaphase delay (Supplemental Fig. 3B). TEF1 overexpression was previously proposed to interfere with bud formation, which could explain the slight delay in bud emergence (Munshi et al. 2001). We do not yet understand why expressing this gene combination causes a metaphase delay. Overall, however, we conclude that DSGs present in low copy number cannot account for the proliferation defect observed in disomic yeast strains.
Ten-copy-limit DSGs cause a minor cell proliferation defect
Next, we investigated the possibility that synergistic effects of genes with slightly higher copy number limits account for the proliferation defects of disomes. We examined the consequences of expressing genes with copy number limits of <10 (limit 0–10, henceforth DSGs[10]) from CEN plasmids. Chromosome VIII encodes six DSGs[10] (Table 1). Expression of five of these genes (DMA1, MYO1, OPI1, SFB3, and STE12) together did not interfere with cell proliferation (Fig. 2A; Supplemental Fig. 2H,I). Similar results were obtained for DSGs[10] encoded on chromosomes X (BFA1, KAR2, HSP150, IRC8, and TPK1) (Fig. 2B; Supplemental Fig. 2L,M), XV (EFT1, RPS12, and SLG1) (Fig. 2C; Supplemental Fig. 2R), and IX (AXL2, PRK1, and TED1) (Fig. 2D; Supplemental Fig. 2J,K). Coexpression of DSGs[10] from either chromosome II or XIV conferred a measurable proliferation defect. APE3, OM14, PET9, and TEF2 expressed from a CEN plasmid lengthened doubling time by 5.8 min (±1.4 min) compared with control cells (Fig. 2E; Supplemental Fig. 2D). Cells with plasmids containing the six genes with limits from 0 to 10 on chromosome XIV exhibited doubling times 15 min (±3.2 min) slower than control cells (KSH1, MKS1, POR1, PUB1, SSN8, and TOM70) (Fig. 2F; Supplemental Fig. 2P,Q). For comparison, the doubling time of disome XIV cells is 32 min (±5.2 min) slower than that of wild-type cells (Fig. 2F).
Furthermore, for chromosomes I, II, and XI, we determined the effects of expressing all genes with upper limits of ≤20 copies (DSGs[20]; for chromosome XI, we were only able to analyze three of four DSGs[20]). Proliferation was not significantly slowed when DSGs[20] encoded on chromosome I or XI were expressed from CEN plasmids (Fig. 3A,B; Supplemental Fig. 2B,C,N,O). Coexpression of all DSGs[20] located on chromosome II led to an increase in doubling time by 6.6 min (±1.4 min) (Fig. 3C; Supplemental Fig. 2D,E) compared with cells containing empty vectors, which is a slightly slower rate of doubling than seen in cells harboring a plasmid containing chromosome II DSGs[10] alone (Fig. 2E).
Figure 3.
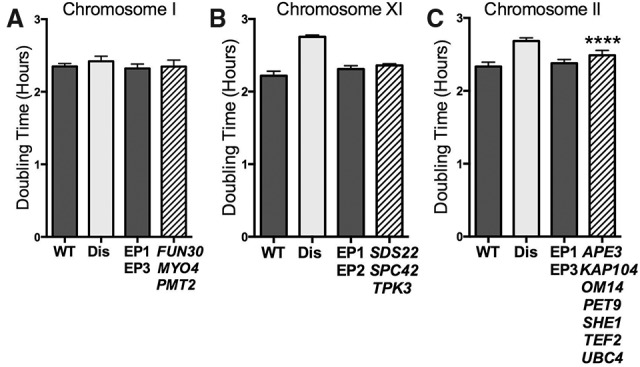
Effects of DSGs[20] on cell proliferation. (A–C) Cells were grown as described in Figure 1. Genes were grouped into graphs according to their chromosomal location. Dark-gray bars represent control strains carrying empty plasmids, light-gray bars represent the corresponding disomic strain (Dis), and striped bars represent DSG plasmid-containing strains. Empty vector CEN plasmid controls were EP1-TRP1, EP2-HIS3, and EP3-LEU2. SD is shown. (****) P < 0.0001; Student's t-test. The strains used, in order, were A22361, A12683, A36954, and A36922 (A); A22361, A28266, A36953, and A36939 (B); and A22361, A12685, A35954, and A36925 (C).
Additionally, we note that an extra copy of DSGs did not cause other phenotypes characteristic of disomic yeast strains such as sensitivity to high temperature, cycloheximide, or phleomycin (Supplemental Fig. 4). This was not unexpected given that gTOW DSGs were identified based on their proliferation defect, not sensitivity to adverse growth conditions.
Our results indicate that a doubling in gene dosage of DSGs[5] is partly responsible for the proliferation defect of only two out of nine disomic yeast strains analyzed (disomes V and XVI). A portion of the proliferation defect of an additional two disomies (disomes II and XIV) can be explained by a doubling in gene dosage of DSGs[10]. Nevertheless, it is important to state that no plasmids containing DSGs slowed doubling times to rates seen in cells expressing an extra copy of the corresponding whole chromosome. These results support the conclusion that the proliferation defect observed in disomic yeast strains is caused by increased expression of genes that, altered individually, have little or no observable effect rather than a few proliferation antagonists at altered copy number. We suspect that this concept applies to other phenotypes widespread among aneuploid cells. For instance, in other work, we were not able to identify a single gene responsible for the hydroxyurea sensitivity of disome VIII strains (Blank et al. 2015). Additionally, Chen et al. (2015) were unable to ascribe the hygromycin sensitivity of disome XV cells to a specific gene combination.
Deletion of DSGs from disomic strains does not improve cellular fitness
Our results reveal that an additional copy of some DSGs is sufficient to explain a small portion of the proliferation delay in disomic cells, but are DSGs necessary for the proliferation defects of disomic yeast strains? To address this question, we investigated the consequences of deleting one copy of GLN3 in cells disomic for chromosome V. Introduction of an extra copy of GLN3 into wild-type cells subtly impaired proliferation (Fig. 1G). However, disome V cells lacking one copy of GLN3 did not proliferate faster than cells disomic for a wild-type copy of chromosome V (Fig. 4A). Similar results were obtained for TEF1 and VPS4. Expression of these DSGs slows proliferation (Fig. 1H), but deleting one copy in disome XVI cells did not improve proliferation (Fig. 4B). Deletion of one copy of other chromosome XVI DSGs[5] (TPK2 or KES1) did not improve proliferation rate either (Fig. 4B).
Figure 4.
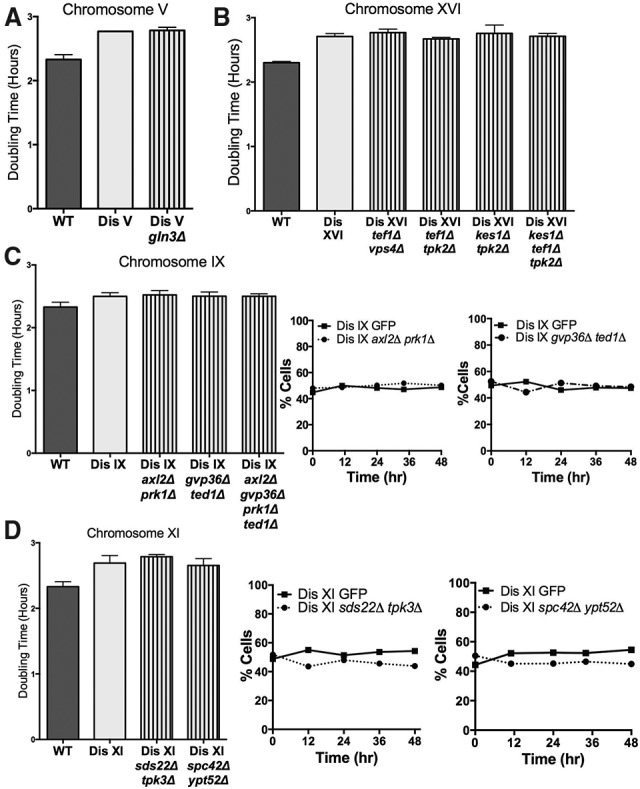
Deletion of one copy of DSGs from disomic yeast strains does not improve cellular fitness. (A) Cells harboring an additional copy of chromosome V (A28265) or an additional copy of chromosome V lacking GLN3 (A36470) were grown as described in Figure 1 to measure their doubling time. (B) Cells harboring an additional copy of chromosome XVI (A34149) or an additional copy of chromosome XVI lacking the indicated genes (A36585, A36650, A36689, and A36859) were grown as described in Figure 1 to measure their doubling time. (C, left graph) Cells with an additional copy of chromosome IX (A13975) or an additional copy of chromosome IX lacking the indicated genes (A36233, A36215, and A36069) were grown as described in Figure 1 to measure their doubling time. The middle and right graphs show competition experiments in which equal numbers of disome IX cells harboring GFP (A36219) and disome IX cells lacking the indicated genes (A36233 and A36215) were coinoculated in –His G418 medium. The percentages of GFP-positive and GFP-negative cells were monitored at the indicated times. (D, left graph) Cells with an additional copy of chromosome XI (A28266) or an additional copy of chromosome XI lacking the indicated genes (A36919 and A36336) were grown as described in Figure 1 to measure their doubling time. The middle and right graphs show competition experiments in which equal numbers of disome XI cells expressing GFP (A36222) and GFP− disome XI cells lacking the indicated genes (A36919 and A36336) were coinoculated in −His G418 medium. The percentages of GFP-positive and GFP-negative cells were monitored at the indicated times.
Similarly, we asked whether low-copy-limit genes might be necessary for disomic proliferative defects even if they were not sufficient. We deleted one copy of the DSG[5] PRK1, the DSGs[10] TED1 and AXL2, and the DSG[20] GVP36 in disome IX cells. Doubling time measurements did not reveal either a decrease in cell cycle duration or a fitness advantage when competed against the parental disome IX strain (Fig. 4C). The parental and DSG deletion disomic strains maintained an approximately equal share of the population following serial dilutions for 48 h. Similar results were obtained for DSGs encoded by chromosome XI (Fig. 4D). These data demonstrate that individual genes whose alteration in copy number is most toxic are not solely responsible for the proliferation defect of cells harboring an additional copy of the entire chromosome.
Concluding remarks
In summary, our data indicate that DSGs with low copy number limits can interfere with cell proliferation when present in two copies in haploid strains, but their effects cannot explain the significant proliferation defect seen in cells harboring an additional copy of the entire chromosome on which they are located. We surveyed 11 chromosomes for effects of an extra copy of low-copy-limit DSGs on proliferation, and none revealed substantial growth alteration. Thus, our study conclusively excludes the “few critical genes” hypothesis as a possible explanation for the growth defect of disomic yeast strains and implies that this aneuploidy-associated phenotype is caused by an increase in dosage of a large number of genes not harmful when amplified individually. To our knowledge, this is the first systematic analysis to demonstrate that subtle changes in gene dosage have significant phenotypic consequences when they occur at a chromosome-wide scale. This conclusion may not only apply to the proliferation defect of aneuploid yeast strains. Previous studies of segmental aneuploidies in Drosophila melanogaster showed that larger deletions and duplications were more deleterious than smaller ones (Lindsley et al. 1972).
Our findings may also have implications for understanding the origins of phenotypes of chromosome gains and losses in humans. Phenotypes observed in monosomies are likely due to a combination of specific gene copy number alterations and mass effects of individually harmless genes, as 1.5%–5% of the human genome is predicted to be haploinsufficient (Dang et al. 2008; Huang et al. 2010). However, contribution of mass action of genes with little or no phenotypic effect when varied individually could significantly impact phenotypes associated with trisomy. For example, despite intense efforts, no specific gene or chromosomal region has been identified as responsible for the general growth retardation, developmental abnormalities, and cognitive deficiencies characteristic of DS. In developing a framework to think about treating diseases involving whole-chromosome or segmental aneuploidies, our data suggest that, rather than solely searching for therapies that target single genes, we also need to identify methods to increase the overall fitness of aneuploid cells.
Materials and methods
Yeast strains and plasmids
Plasmids used in this study are listed in Supplemental Table S1. Plasmids were created via cloning as described in Gibson et al. (2009) or were from the MoBY ORF collection (Ho et al. 2009). Representative plasmids were sequenced to ensure that no alterations in sequence arose during cloning. Strains used in this study are listed in Supplemental Table S2. All strains are derivatives of W303. Disomic strain generation is described in Torres et al. (2007). Karyotypes of all aneuploid strains were confirmed by comparative genomic hybridization (CGH) as performed in Torres et al. (2007) and analyzed with Java TreeView.
Doubling time analysis
Strains were grown overnight at room temperature in medium to select for maintenance of plasmids. The following morning strains were diluted to OD600 = 0.1 in yeast extract/peptone medium containing 2% glucose (YEPD). The growth rate was measured in triplicate using a Biotek plate reader at 25°C. OD600 measurements were taken every 15 min for 24 h. Only the period of exponential growth was used to determine doubling time. Two to four transformants per plasmid were tested on at least two different days and compared with a wild-type strain carrying the relevant empty vector plasmid. Data were accumulated using Gen5 software. Statistics were performed using GraphPad Prism software.
Competition experiments
Approximately equal amounts of cells with and without integrated PGK1-GFP were mixed in selective medium (−His +G418) at OD600 = 0.15 and maintained in exponential growth phase at room temperature. Samples were taken every 12 h for 48 h. Each sample was fixed for 5 min in 3.7% formaldehyde in KPi (pH = 6.4). Relative cell populations in the cultures were measured by quantifying the percentage of GFP-positive versus GFP-negative cells via flow cytometry.
Real-time RT–PCR
Analysis of gene expression was performed as previously described (Thorburn et al. 2013).
Elutriation
Elutriation performed as described in Thorburn et al. (2013) with the exception that elutriated cells were resuspended in YEPD at 25°C.
Indirect in situ immunofluorescence
Indirect in situ immunofluorescence of α-tubulin was performed as described previously (Kilmartin and Adams 1984). Cells were imaged with a Zeiss Axioplan 2 microscope (Zeiss).
Supplementary Material
Acknowledgments
We thank Frank Solomon and members of the Amon laboratory for suggestions and critical reading of this manuscript. This work was supported by the National Institutes of Health (GM056800 to A.A). A.A is also an investigator of the Howard Hughes Medical Institute.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.261743.115.
References
- Baek K-H, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park I-H, Yoon SS, Minami T, et al. 2009. Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 459: 1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank HM, Sheltzer JM, Meehl CM, Amon A. 2015. Mitotic entry in the presence of DNA damage is a widespread property of aneuploidy in yeast. Mol Biol Cell 10.1091/mbc.E14-10-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J, Badis G, Fairhead C, Talla E, Hantraye F, Fabre E, Fischer G, Hennequin C, Koszul R, Lafontaine I, et al. 2004. Large-scale exploration of growth inhibition caused by overexpression of genomic fragments in Saccharomyces cerevisiae. Genome Biol 5: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Mulla WA, Kucharavy A, Tsai H-J, Rubinstein B, Conkright J, McCroskey S, Bradford WD, Weems L, Haug JS, et al. 2015. Targeting the adaptability of heterogeneous aneuploids. Cell 160: 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VT, Kassahn KS, Marcos AE, Ragan MA. 2008. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet 16: 1350–1357. [DOI] [PubMed] [Google Scholar]
- Douglas AC, Smith AM, Sharifpoor S, Yan Z, Durbic T, Heisler LE, Lee AY, Ryan O, Göttert H, Surendra A, et al. 2012. Functional analysis with a barcoder yeast gene overexpression system. G3 (Bethesda) 2: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futcher B, Carbon J. 1986. Toxic effects of excess cloned centromeres. Mol Cell Biol 6: 2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, et al. 2005. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev 19: 2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Hassold TJ, Jacobs PA. 1984. Trisomy in man. Annu Rev Genet 18: 69–97. [DOI] [PubMed] [Google Scholar]
- Ho CH, Magtanong L, Barker SL, Gresham D, Nishimura S, Natarajan P, Koh JLY, Porter J, Gray CA, Andersen RJ, et al. 2009. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat Biotechnol 27: 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME. 2010. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6: e1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz W, Weinstein B, Solomon F. 1990. Regulation of tubulin levels and microtubule assembly in Saccharomyces cerevisiae: consequences of altered tubulin gene copy number. Mol Cell Biol 10: 5286–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. 1984. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. J Cell Biol 98: 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Tirosh-Wagner T, Urban AE, Chen X-N, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, et al. 2009. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci 106: 12031–12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, Hall JC, Jacobs PA, Miklos GL, Davis BK, Gethmann RC, et al. 1972. Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics 71: 157–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Krizek J, Bretscher A. 1992. Construction of a GAL1-regulated yeast cDNA expression library and its application to the identification of genes whose overexpression causes lethality in yeast. Genetics 132: 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makanae K, Kintaka R, Makino T, Kitano H, Moriya H. 2013. Identification of dosage-sensitive genes in Saccharomyces cerevisiae using the genetic tug-of-war method. Genome Res 23: 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya H, Makanae K, Watanabe K, Chino A, Shimizu-Yoshida Y. 2012. Robustness analysis of cellular systems using the genetic tug-of-war method. Mol Biosyst 8: 2513–2522. [DOI] [PubMed] [Google Scholar]
- Munshi R, Kandl KA, Carr-Schmid A, Whitacre JL, Adams AE, Kinzy TG. 2001. Overexpression of translation elongation factor 1A affects the organization and function of the actin cytoskeleton in yeast. Genetics 157: 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff NF, Thomas JH, Grisafi P, Botstein D. 1983. Isolation of the β-tubulin gene from yeast and demonstration of its essential function in vivo. Cell 33: 211–219. [DOI] [PubMed] [Google Scholar]
- Olson LE, Richtsmeier JT, Leszl J, Reeves RH. 2004a. A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 306: 687–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson LE, Roper RJ, Baxter LL, Carlson EJ, Epstein CJ, Reeves RH. 2004b. Down syndrome mouse models Ts65Dn, Ts1Cje, and Ms1Cje/Ts65Dn exhibit variable severity of cerebellar phenotypes. Dev Dyn 230: 581–589. [DOI] [PubMed] [Google Scholar]
- Olson LE, Roper RJ, Sengstaken CL, Peterson EA, Aquino V, Galdzicki Z, Siarey R, Pletnikov M, Moran TH, Reeves RH. 2007. Trisomy for the Down syndrome ‘critical region’ is necessary but not sufficient for brain phenotypes of trisomic mice. Hum Mol Genet 16: 774–782. [DOI] [PubMed] [Google Scholar]
- Rachidi M, Lopes C. 2007. Mental retardation in Down syndrome: from gene dosage imbalance to molecular and cellular mechanisms. Neurosci Res 59: 349–369. [DOI] [PubMed] [Google Scholar]
- Rahmani Z, Blouin JL, Créau-Goldberg N, Watkins PC, Mattei JF, Poissonnier M, Prieur M, Chettouh Z, Nicole A, Aurias A. 1990. Down syndrome critical region around D21S55 on proximal 21q22.3. Am J Med Genet 7: 98–103. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, et al. 2006. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38: 24–26. [DOI] [PubMed] [Google Scholar]
- Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R, Hockey A, Montgomery P, Beyreuther K, Masters CL. 1989. Amyloid A4 protein and its precursor in Down's syndrome and Alzheimer's disease. N Engl J Med 320: 1446–1452. [DOI] [PubMed] [Google Scholar]
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, et al. 2006. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 21: 319–330. [DOI] [PubMed] [Google Scholar]
- Theuns J, Brouwers N, Engelborghs S, Sleegers K, Bogaerts V, Corsmit E, De Pooter T, van Duijn CM, De Deyn PP, Van Broeckhoven C. 2006. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet 78: 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn RR, Gonzalez C, Brar GA, Christen S, Carlile TM, Ingolia NT, Sauer U, Weissman JS, Amon A. 2013. Aneuploid yeast strains exhibit defects in cell growth and passage through START. Mol Biol Cell 24: 1274–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924. [DOI] [PubMed] [Google Scholar]
- Torres EM, Williams BR, Amon A. 2008. Aneuploidy: cells losing their balance. Genetics 179: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BAA, Cleveland DW. 2006. Does aneuploidy cause cancer? Curr Opin Cell Biol 18: 658–667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.