

In this study, Reizel et al. demonstrate that gender-specific demethylation occurs via a postnatal pathway found exclusively in the male liver. The secretion of testosterone at the time of sexual maturity regulates this process, providing new insights into how the environment impacts epigenetic regulation of genes in fully differentiated tissue.
Keywords: DNA methylation, liver, gender-specific genes, gene expression, epigenetic memory
Abstract
DNA methylation patterns are set up in a relatively fixed programmed manner during normal embryonic development and are then stably maintained. Using genome-wide analysis, we discovered a postnatal pathway involving gender-specific demethylation that occurs exclusively in the male liver. This demodification is programmed to take place at tissue-specific enhancer sequences, and our data show that the methylation state at these loci is associated with and appears to play a role in the transcriptional regulation of nearby genes. This process is mediated by the secretion of testosterone at the time of sexual maturity, but the resulting methylation profile is stable and therefore can serve as an epigenetic memory even in the absence of this inducer. These findings add a new dimension to our understanding of the role of DNA methylation in vivo and provide the foundations for deciphering how environment can impact on the epigenetic regulation of genes in general.
Although males and females are derived from genomes that differ only in their X and Y chromosomes, there appear to be many gender-specific functions that actually use autosomal genes, and very little is known about the mechanisms involved in this process. It has been shown, for example, that genes are differentially expressed in the male and female liver as a result of postnatal hormone secretion, and this is accompanied by changes in chromatin structure at nearby regulatory sequences (Waxman and Holloway 2009; Ling et al. 2010). These alterations not only bring about gender-specific changes in metabolic function but also may explain the predisposition to hepatocellular carcinoma seen specifically in the male. Other tissues of the body are also characterized by select gender-derived expression patterns (Yang et al. 2006).
DNA methylation is a key epigenetic factor that is known to play a role in gene regulation during development (Cedar and Bergman 2012), but aside from a few observations on individual genes (Weaver et al. 2004; Takasugi et al. 2013), little is known about how this modification may take part in the control of postnatal dynamics, as in the case of gender-specific gene expression. The basic DNA methylation pattern of animals is initially erased in the early embryo and then re-established at the time of implantation through a wave of global de novo methylation, with CpG islands being protected on the basis of underlying sequence motifs. Although the ability to de novo methylate the genome is only transient and subsequently undergoes down-regulation, once the original pattern is set up in the implanted embryo, it is then stably maintained through every cell division. Further adjustments then occur exclusively in a stage- or tissue-specific manner during embryonic development or adult stem cell differentiation (Sheaffer et al. 2014), with the changes being driven in a programmed manner by trans-acting factors that recognize the target sequences (Cedar and Bergman 2012).
One of the first events to occur after implantation is the inactivation of pluripotency genes such as Oct-3/4. Interestingly, this takes place in a three-step process by which the transcription of these genes is first turned off by repressor factors, and this is followed by G9a-mediated heterochromatinization. De novo methylation is a later event. Even though this epigenetic modification is not directly involved in repression, it does provide long-term stability, effectively preventing any reactivation (Feldman et al. 2006; Epsztejn-Litman et al. 2008). In a similar manner, tissue-specific genes undergo activation through interactions between trans-acting factors and cis-acting sequences, and this is accompanied by DNA demethylation, which may not only boost transcription but also contribute to stable gene accessibility (Cedar and Bergman 2012). While a considerable number of tissue-specific gene promoters have been shown to undergo preferential demethylation in their cell type of expression during development, recent high-throughput studies indicate that many other regulatory sequences are also differentially unmethylated in a tissue-specific manner (Hon et al. 2013; Ziller et al. 2013), but it is not yet known whether these changes take place as a part of normal embryonic development or occur postnatally.
Sexual dimorphism has an enormous effect on metabolism and physiology that has been documented in an extensive manner over the past 50 years (Waxman and Holloway 2009), with emphasis on the molecular biology of gene expression. In this study, we used high-throughput analysis to systematically characterize tissue differences in DNA methylation between males and females. We found that these variations come about as a result of programmed changes in methylation that take place at regulatory sequences and are driven by secretion of the male hormone testosterone. These gender-specific methylation patterns are stable and appear to influence gene expression in vivo.
Results
Gender-specific DNA methylation
In order to obtain a comprehensive picture of possible methylation differences between males and females, we isolated adult (20 wk) liver DNA and carried out reduced representation bisulfite sequencing (RRBS), which can scan a relatively large number of small loci with a high degree of molecular depth (Boyle et al. 2012). Under the assumption that there may be variations between individual animals, we assayed a large number of mice in each category. Using a strict statistical cutoff, we were able to identify 160 individual 100-base-pair (bp) tiles that were at least 25% less methylated in male as opposed to female animals, and these differences were all specific to the liver (Fig. 1; Supplemental Fig. S1A). Strikingly, we did not detect any equivalent sites that are significantly less methylated in the female. We also validated (Supplemental Fig. S1B,C) these results by carrying out a sample experiment using whole-genome bisulfite sequencing (WGBS), which demonstrated that, on average, these undermethylated regions span ∼400 bp (Fig. 1C). Furthermore, WGBS revealed that in the whole genome, there may be >1000 400-bp tiles that are specifically differentially undermethylated (>35%) in the male (Supplemental Fig. S1D,E), suggesting that this is indeed a widespread phenomenon.
Figure 1.

Tissue-specific demethylation in the male liver. (A) Heat map and histogram (average methylation for each sample) comparison (P < 10−4; see the Materials and Methods) of 160 tiles for male and female liver samples (n = 4–10) as a function of age as noted. Each column represents a single tissue sample. (B) Average methylation levels (±SD) for these same 160 tiles in different tissues in male (blue) and female (red) samples (n ≥ 3) from individual animals. A heat map of these samples is presented in Supplemental Figure S1. (C) Whole-genome bisulfite sequencing (WGBS) analysis shows the average differential methylation levels for all 160 tiles in the male liver as a function of distance from the original 100-bp tile detected by RRBS.
Since all of the male-specific sites were found to be highly methylated in other adult tissues as well as in the early embryo (embryonic day 6.5 [E6.5]–E7.5) (Fig. 1B; Supplemental Fig. S1A), it seemed likely that these sequences are initially de novo methylated at the time of implantation and maintained in this manner in all cell types, suggesting that the profile observed in the male liver was probably generated through a process of demethylation. Indeed, by following the modification patterns of gender-specific loci from the time of birth, we were able to confirm that, like the rest of the genome, these select sequences are, without exception, all methylated equivalently in both sexes as the mouse emerges from its embryonic development stage but then undergo a clear-cut process of demethylation specifically in the male (Fig. 1A,B), and this was confirmed by WGBS analysis as well (Supplemental Fig. S1). We also measured DNA methylation in purified neonatal versus adult hepatocytes (Materials and Methods), where the differences were found to be even more dramatic (Supplemental Fig. S1F), suggesting that this represents an inherent cell-autonomous process and cannot be the result of changes in whole liver cell composition during postnatal development.
Testosterone directs male-specific demethylation
We next asked whether this effect may be caused by hormonal differences that develop between male and female animals. To this end, we examined DNA methylation patterns in male mice castrated at the age of 20 d. Strikingly, these mice did not undergo any appreciable demethylation during postnatal development, suggesting that it is the male hormones that are responsible for this process (Fig. 2; Supplemental Fig. S2A). Furthermore, administration of testosterone following castration almost completely restored the demethylation pattern. These data strongly suggest that this hormone is both necessary and sufficient to drive male-specific demethylation in the liver, probably by bringing about the pulsatile secretion of GH from the pituitary gland (Waxman and O'Connor 2006). In contrast, ovariectomy of female mice was found to have no effect on these (Fig. 2) or any other DNA methylation sites. Since testosterone is first secreted a few weeks after birth (Selmanoff et al. 1977), the presence of this hormone seems to act as an “internal environmental factor” that can influence DNA methylation at specific sites. Indeed, these identical changes can even be induced in female mice by long-term administration of testosterone (Fig. 2). All of these findings were also confirmed for individual sites using single-molecule bisulfite analysis (Supplemental Fig. S2B).
Figure 2.
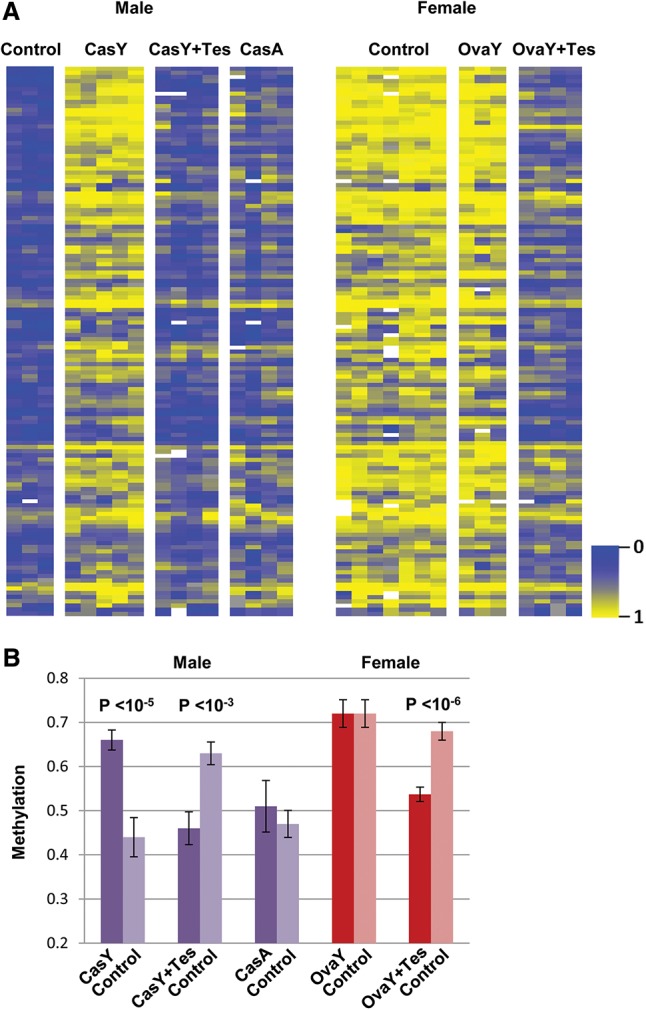
Demethylation is mediated by testosterone. Heat map (A) and average methylation levels (±SD) (B) of individual mice (each column) castrated (CasY) or oviarectomized (OvaY) by removal of the gonads or sham-operated (control) at 3 wk of age; liver DNA was assayed at 20 wk (n = 3–10). These same mice were treated with testosterone (Tes) or saline (control) starting at 8 wk for a period of 3 mo by means of an implanted pump; male mice were also castrated (CasA) or sham-operated (control) at 20 wk and assayed for liver DNA methylation after an additional 8 wk. A full heat map of the control samples is presented in Supplemental Figure S2.
Characterization of differentially methylated regions (DMRs)
We next asked about the nature of the DNA segments that undergo specific demethylation in response to testosterone. Very few were identified as known promoters (Fig. 3A). In contrast, chromatin immunoprecipitation (ChIP) combined with deep sequencing (ChIP-seq) analysis adapted from the literature (Rosenbloom et al. 2013; Sugathan and Waxman 2013) indicated that they are highly enriched in histone H3K4me1 and H3K27Ac, specifically in the adult liver (Fig. 3B), suggesting that this sequence set may be composed of enhancer elements (Heintzman et al. 2007), and bioinformatics analysis shows that it indeed contains a number of different common transcription factor-binding motifs (Fig. 3C) that are unique to nonpromoter regulatory sites (Supplemental Fig. S3A), with some of these known to be associated with sexual dimorphism in the liver (Laz et al. 2009). Furthermore, by using previously published data (Zhang et al. 2012; Kosters et al. 2013), we were able to demonstrate that some of these factors actually bind physically to their target sequences but only in the male liver, (Fig. 3D), suggesting that demethylation itself may contribute to factor accessibility.
Figure 3.
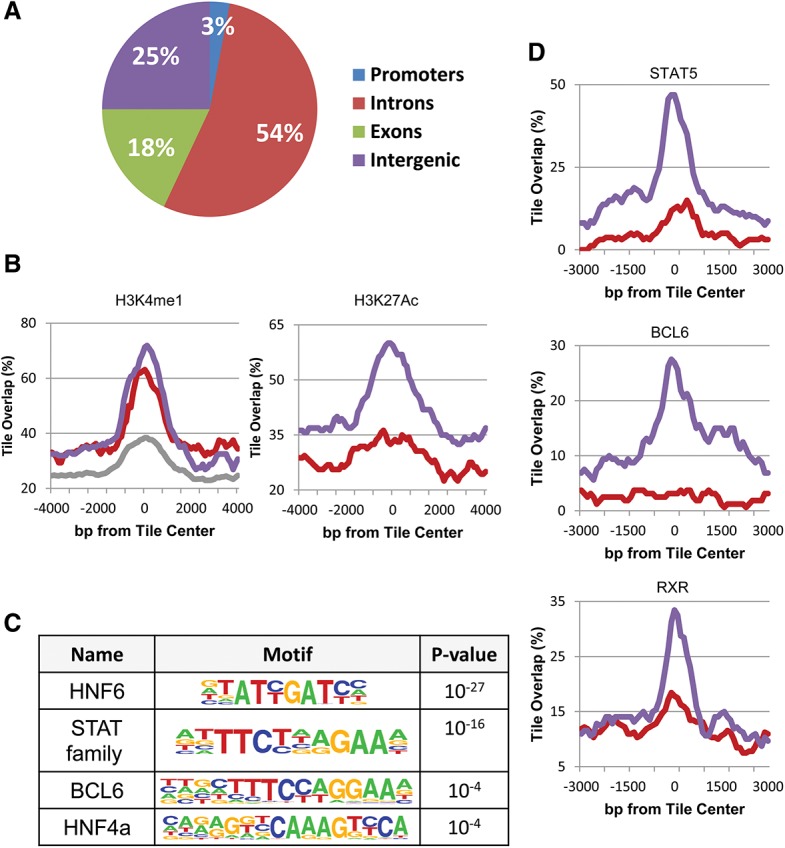
Enhancer regulation. (A) Genome distribution of demethylated regions (as determined by RRBS). (B) ChIP-seq (as adapted from Rosenbloom et al. 2013; Sugathan and Waxman 2013) of demethylated regions (RRBS) as a function of distance from their center tile for H3K4me1 and H3K27Ac in male (blue), female (red), and a mixture of other tissues (grey). (C) Motif analysis of demethylated regions as determined by WGBS. (D) ChIP-seq data of demethylated regions (RRBS) for transcription factors in male (blue) and female (red) liver DNA samples were adapted from the literature (Zhang et al. 2012; Kosters et al. 2013) and normalized as described in the Materials and Methods. Similar results for A, B, and D were also obtained using data from WGBS (data not shown).
It is well established that there are many hormone-responsive differences in expression between male and female livers (Wauthier and Waxman 2008; Waxman and Holloway 2009). Thus, on the assumption that the DMRs detected in our assay represent regulatory elements, we attempted to identify possible gene targets. To this end, we first selected (Fig. 4A) all of the demethylated tiles obtained by WGBS analysis that are located within or adjacent to (±50 kb) known gene sequences (n = 1034) and then characterized them for the presence of H3K4me1 or H3K27Ac as well as the binding of transcription factors (BCL6, STAT5, and RXR). Using RNA sequencing (RNA-seq) to assay the expression profiles in male and female livers, we found that a significant percentage of these (n = 171) are associated with genes (n = 88) that are differentially expressed (P < 0.05) in male mice, with the highest enrichment (∼30%) observed for those sites carrying histone and protein marks signifying active enhancers (Fig. 4B).
Figure 4.

Correlation with expression. (A) Heat map of methylation differences between male and female, histone marks (either H3K27Ac or H3K4me1), and transcription factor (TF) binding (STAT5, BCL6, or RXR) at 1034 demethylated regions (WGBS) that are located within or adjacent to (±50 kb) known gene sequences as a function of distance from their center tile. (B) Enhancement (Y-axis) of male-specific (blue) or female-specific (red) expression of all DMRs as compared with control regions that have none of these markers (χ2 test of proportions). Note that DMRs (asterisk) specifically labeled with histone marks and protein binding (n = 384) are highly enriched, with almost 30% of the tiles showing a correlation with male-specific expression as opposed to female-specific expression, and this is sixfold higher than a random sample of control regions. (C) Tissue expression level (arbitrary units; RNA-seq) heat map of 88 male-enhanced genes that harbor putative enhancer sequences ordered by unsupervised hierarchical clustering of normal young (Male Y) and adult (Male A and Female A) livers, gonadectamized (at 3 wk) livers (Cas Y and Ova Y) with or without the addition of testosterone (Tes), and other tissues. Enrichment of each cluster is statistically significant (P < 0.05, pvclust) (Suzuki and Shimodaira 2006). Each lane shows average data for at least two biological replicates. Testosterone levels and controls are presented in Supplemental Figure S5. (D) Gene ontology of male-specific demethylated regions (n = 1034).
By using hierarchical clustering, we demonstrated that these 88 genes not only are expressed at higher levels in adult males than in females but are characterized by low activity in young animals as well as in castrated males, where these presumed enhancer elements are methylated. Furthermore, they show increased transcription in both females and castrated males treated with testosterone (Fig. 4C; Supplemental Fig. S4). Taken together, these data show that enhancer demethylation in the liver is highly correlated (P < 10−14) with gender-specific expression, and bioinformatics analysis indicates that it is preferentially associated with liver function (Fig. 4D), mainly lipid metabolism and drug detoxification, well-known features of sexual dimorphism.
The distribution of factor binding and histone H3K4me1 and H3K27Ac packaging (Fig. 4A) provides valuable information about the molecular events that are associated with demethylation. First, it should be noted that many tiles that have undergone demethylation do not carry either of these marks, suggesting that the demethylation may occur in a manner that is independent of these modifications. Furthermore, the fact that this epigenetic change is gender-specific raises the possibility that it may serve as the basis for selective binding of the transcription factors STAT5, BCL6, and RXR that are present ubiquitously in both male and female liver cells. Bioinformatics analysis shows that the lack of transcription factor binding to other demethylated sites is due to the absence of motifs for these factors. Interestingly, all of the demethylated sites are highly enriched for the HNF6 motif regardless of the presence or absence of other factors, perhaps suggesting that this family of liver-specific factors may actually be involved in initially recognizing these sequences and bringing about their demethylation.
Long-term effects of demethylation
It is well accepted that hormones can influence gene expression, but these effects are usually transient in the sense that transcription returns to normal levels after its removal. In order to test whether methylation changes induced by testosterone also respond in this manner, we castrated normal male mice at the age of 20 wk, at a time when gender-specific sites had already undergone maximum demethylation. We then followed the epigenetic fate of these loci over time (2 mo). Despite castration and the resulting removal of testosterone, the undermethylated DNA pattern in these regions remained relatively stable (Fig. 2; Supplemental Fig. S2A). This suggests that the methylation state serves as an epigenetic memory even though the original induction factors are no longer present.
We next tested whether the DNA methylation pattern at the enhancer sequences could also serve as a mechanism for regulating the expression patterns of target genes. To this end, we used RNA-seq to examine the expression patterns of all of the sexually dimorphic genes associated with enhancer demethylation (n = 88) in castrated males (with methylated enhancers) given an artificial pulse of testosterone at 20 wk of age. Expression of the target genes remained low as compared with control animals harboring undermethylated enhancers (Fig. 5A; Supplemental Fig. S5C). Since the level of testosterone is similarly high in both cases (Supplemental Fig. S5A), this experiment clearly indicates that the original state of enhancer methylation serves as a molecular memory that can predetermine the effect of transcription factors on gene expression. In a similar manner, we analyzed males castrated at 3 wk as compared with males castrated at 20 wk (Fig. 5B). Despite the fact that testosterone levels are very low in both of these cases (Supplemental Fig. S5A), a sample of genes (n = 24) associated with an undermethylated enhancer (old castrated males) is expressed at a higher level, and this was further confirmed by high-resolution RT–PCR analysis on selected hallmark genes (Supplemental Fig. S5B). This suggests that for some genes, the low methylation state initially induced by exposure to testosterone in the young male represents a stable memory marker for gene activation even after this hormone is no longer present.
Figure 5.
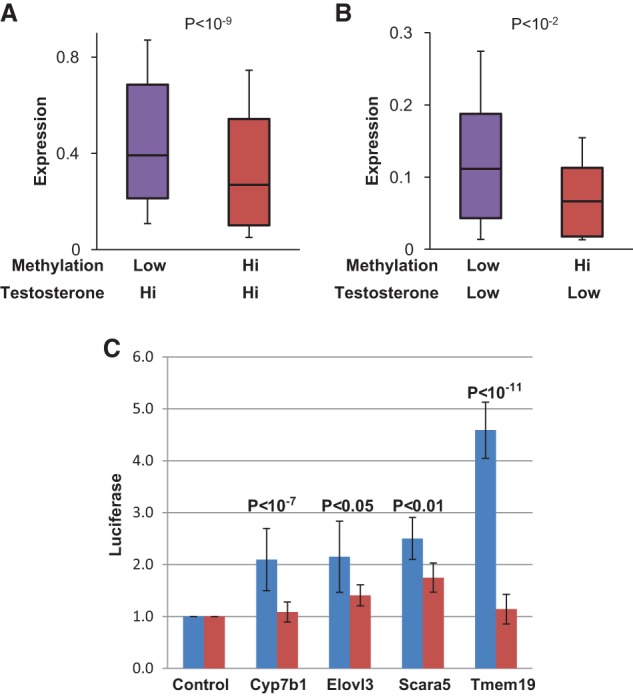
Effect of methylation on expression. (A) Box plot of expression levels (RNA-seq) of the same genes (n = 88) from 20-wk normal animals (undermethylated enhancer) or castrated (at 3 wk) animals treated with testosterone for 2 d starting at 20 wk of age (methylated enhancer). Error bars are from the ninth to the 91st percentile. (B) Box plot of expression levels (RNA-seq) in the absence of testosterone for genes in male livers (22 wk) from animals castrated at 3 wk (methylated enhancers) or at 20 wk (undermethylated enhancers). In order to improve RNA-level resolution, we carried out this analysis on a subset of genes (n = 24) that showed the largest differences (greater than twofold) between normal and castrated animals. (C) Putative enhancer sequences were cloned into a CpG-free reporter vector that was either left unmethylated (blue) or methylated (red) in vitro and transfected into a hepatocyte cell line, and the medium was then assayed for Luciferase activity (arbitrary units) after 48 h. For all cloned sequences, enhancement over control was significant (P < 10−4, n ≥ 10, t-test).
DMRs contain methyl-sensitive enhancers
Interestingly, all of the testosterone-responsive genes detected in our study have promoters that are equally modified in male and female DNA (Supplemental Fig. S3B). Thus, epigenetically, their differential expression pattern is best correlated with enhancer demethylation. In order to test whether these differentially methylated sequences do indeed represent enhancers, we cloned a sample of these loci into reporter plasmids containing a minimal promoter driving the luciferase gene (Hoivik et al. 2011) and transfected them into a hepatocyte cell line (Rienhoff and Groudine 1988). This experiment revealed that four out of five of these sequences indeed have the ability to enhance expression, but this effect was attenuated when the plasmids were artificially methylated in vitro prior to transfection (Fig. 5C). Since the entire test vector used in this experiment was designed to be lacking any CpG residues (Hoivik et al. 2011), the effect on expression can only be attributed to DNA methylation specifically on the putative enhancer sequences. These experiments thus provide strong evidence that testosterone-mediated demethylation itself may directly serve to functionally activate gene enhancers.
Gender-specific methylation in human livers
We next asked whether the epigenetic dimorphism seen in mouse livers is also a feature of human tissues. To this end, we carried out RRBS analysis on liver DNA from multiple adult individuals of both sexes. Strikingly, we were able to detect >450 tiles that are significantly undermethylated in the males (Fig. 6A), and, judging by their level of methylation in other tissues, it appears that this comes about through a process of demethylation (Fig. 6B). This phenomenon is very similar to what happens in mice, since these tiles are in nonpromoter regions that are highly enriched for H3K4me1, a strong indicator of enhancer activity. Furthermore, these presumed regulatory sequences carry transcription factor-binding motifs similar to those identified in mice (data not shown) and are highly associated with sexually dimorphic gene expression in human livers (Fig. 6C–E). Taken together, these data suggest that methylation could play a general role in regulating male/female characteristics independent of sex determination itself.
Figure 6.

DMRs in human livers. (A) Heat map of 460 tiles significantly (P < 10−4; see the Materials and Methods) more methylated in female versus male human livers. Each column represents a single tissue sample (n = 4–5) from adult tissue (average, 40 yr old). (B) Average methylation levels (±SD) for these same 460 tiles in a collection of different tissues (n > 8) in male (blue) and female (red) individual samples. (C) ChIP-seq adapted from the literature (Bernstein et al. 2010) of demethylated regions for H3K4me1 in the male (blue) and female (red). (D) Enhancement of male-specific (blue) or female-specific (red) expression for regions undermethylated in the male as compared with random tiles (P < 10−17, χ2 test of proportions) using published RNA-seq data (GSM916093 and GSM1067795). (E) Heat map of promoter methylation for genes (n = 73) that correlate with enhancer demethylation.
Discussion
We demonstrated that, while there are no apparent differences in DNA methylation in male and female livers at birth, gender-specific patterns can be generated postnatally through the influence of testosterone in the male. This demethylation takes place mostly on putative enhancer sequences, with a large percentage of them located within genes expressed differentially in the male liver, and experiments in tissue culture and in vivo strongly suggest that undermethylation itself may be a key factor in modulating the activity of these genes (Fig. 5). We envision that secretion of testosterone triggers a molecular pathway involving site- and tissue-specific factors that are able to recruit the demethylation machinery, thereby causing a slow progressive decrease in local methylation levels, which further “opens” these enhancers to increased activity, as reflected in their histone acetylation (Rosenbloom et al. 2013; Sugathan and Waxman 2013) and factor-binding patterns (Fig. 3).
These studies have revealed a new class of liver-specific genes that are expressed at low levels in both the male and female livers at birth as well as in other tissues (Fig. 4C), in keeping with their being driven by constitutively unmethylated promoters (Supplemental Fig. S3B). They then appear to undergo male-specific induction as a result of nearby enhancer demethylation. ChIP-seq analysis has shown that many of these gender-specific enhancers are packaged with nucleosomes carrying H3K4me1 over a very broad region of DNA (Fig. 4A) in both the male and female, and this marking most likely takes place after birth but prior to (Y Reizel, O Sabag, and H Cedar, unpubl.) and independent of (Fig. 4A) the much more localized demethylation event. Overall, this process appears to be involved in the fine-tuning of gene expression as an adaptive response to testosterone secretion in the male, perhaps serving to stably tailor liver function, including lipid metabolism and drug detoxification, in a gender-specific manner.
Once formed, the DNA methylation state at these sites remains stable over a long period of time and does not change even when the original trigger, testosterone, is no longer present. This provides a new perspective on the nature of postnatal gender-specific expression. Rather than being exclusively regulated by transcription factor interactions in a plastic manner, they actually appear to be programmed epigenetically and thus become a permanent feature in the adult. The most blatant sign of this cis-acting mechanism is the observation that several key transcription factors are bound to enhancer sequences exclusively in the male (Fig. 3D) even though these proteins are present ubiquitously and at similar levels in both the male and female livers (Zhang et al. 2012). In a sense, this process constitutes a continuation of the epigenetic programming that occurs during embryogenesis, whereby factor-induced alterations in DNA methylation serve as secondary effectors for stably maintaining developmental decisions over time (Kirillov et al. 1996; Feldman et al. 2006; Epsztejn-Litman et al. 2008; Stadler et al. 2011).
This study provides the first identification of a factor that is capable of altering DNA methylation patterns in differentiated cells postnatally. As proof of this concept, we show that treatment of female animals with testosterone can also bring about enhancer-specific demethylation even though females are normally exposed to only very low concentrations of this hormone. Many cases of polycystic ovary syndrome, a disease associated with abnormally high levels of testosterone in the female, develop complications of metabolism (Franks 1995), and it is compelling to think that alterations in DNA methylation may be involved. In contrast, adult males who have undergone feminization with its consequent loss of testosterone could theoretically still carry male-specific demethylation marks, such as those seen in the liver, and thus may retain some memory of male-type expression.
Pioneering experiments by Meany and colleagues (Weaver et al. 2004) have shown that maternal behavior can induce a gene-specific change in DNA methylation in pups during the first week after birth that is then maintained in the adult, and our data on postnatal demethylation in the mouse liver suggest that this may be part of a more general phenomenon. In support of this concept, we demonstrated that there are extensive male-specific undermethylated sites in the human liver as well, and, here too, they seem to be associated with gender-specific gene expression (Fig. 6). Interestingly, previous studies have shown that every tissue of the body contains thousands of cell type-specific, undermethylated sites that are enriched for enhancer sequences (Hon et al. 2013; Ziller et al. 2013). While it is usually thought that these are established during prenatal development, there is already evidence that some sites actually undergo demethylation postnatally (Gilsbach et al. 2014). Furthermore, it has recently been shown that macrophages all over the body may adapt to local tissue environment in the adult by activating specific enhancers (Gosselin et al. 2014; Lavin et al. 2014), and it is very likely that this is accompanied by changes in methylation as well. Taken together, these observations suggest that there may be a wide range of non-stem cell-related epigenetic changes that take place postnatally.
Even though many methylation changes in the genome are programmed to occur as part of embryonic development, it has been postulated that environmental factors may be able to further impact on DNA methylation, perhaps in a stochastic manner (Jaenisch and Bird 2003), in either the embryo (Kaminen-Ahola et al. 2010) or the adult (Meaney and Szyf 2005). Our studies provide a new model for how this may occur by showing that the “internal environment” of an animal (e.g., hormones) can actually act in a programmed manner to bring about extensive postnatal changes in DNA methylation by using specific protein factor DNA interactions at enhancer sequences. In a similar manner, “external environmental” factors may operate by impacting on a variety of signaling molecules or metabolites that may be capable of inducing specific changes in DNA methylation.
Materials and methods
Mice
C57/bl mice were sacrificed at different ages, and their liver, spleen, cerebellum, and muscle were isolated. Hepatocytes were prepared as described (Erhardt et al. 2011). Mice were anesthetized and either sham-operated or castrated by removal of the gonads at the age of 3 or 20 wk. Testosterone (5 mg/mL) or vehicle alone was dissolved as described (Shao et al. 2006) and then injected into an osmotic pump (Alzet, model 2004) that was incubated in saline for 48 h at 37°C and then implanted subcutaneously into castrated males or females. For long-term testosterone administration, the pumps were replaced every 28 d. All animal experiments were performed in accordance with the guidelines of the Institutional Committee for the Use of Animals for Research.
RRBS
DNA was isolated from snap-frozen mouse tissues or frozen human samples (purchased from Cancer Center Tissue and Tumor Bank of the University of Massachusetts Medical School) and incubated in lysis buffer (25 mM Tris-HCl at pH 8, 2 mM EDTA, 0.2% SDS, 200 mM NaCl) supplemented with 300 μg/mL proteinase K (Roche) followed by phenol:chloroform extraction and ethanol precipitation. RRBS libraries were prepared as described (Boyle et al. 2012) and were found to be highly robust and reproducible. Samples were run on HiSeq 2500 (Illumina).
Bisulfite methylation analysis
Bisulfite conversion of genomic DNA was carried out and analyzed using tissue samples from individual mice using the Zymo bisulfite kit according to the manufacturer's instructions. Specific primers of chosen tiles were designed with Methyl Primer Express software version 1.0 (Applied Biosystems). After PCR amplification, the DNA was extracted from the gel with the Qiagen MiniElute gel extraction kit and sequenced by MiSeq (Illumina). Reads were trimmed using trim galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore), and methylation levels were called using Bismark (Krueger and Andrews 2011). The average coverage of the sequenced regions was >1000.
WGBS
Genomic DNA was isolated and pooled from eight individual animals per sample. Unmethylated λ phage DNA was spiked in for a final concentration of 0.5%. One microgram of gDNA was sonicated using the S2 Sonicator (Covaris, Inc.) to the desired range of ∼125-bp median size, with the majority of inserts falling within the range of 100–200 bp as determined by Agilent Bioanalyzer examination and quantified using the Kapa Biosystems kit. End repair, A-tailing, and adapter ligation were carried out using the standard Illumina TruSeq protocol. We used the methylated bar-coded adapters included in the Illumina TruSeq DNA sample preparation kit. Sodium bisulfite conversion of all DNA samples following adapter ligation was performed using the EZ DNA methylation gold kit or the EZ DNA methylation lightning kit (Zymo Research). Four to eight rounds of PCR were performed using the KAPA HiFi +uracil DNA polymerase (Kapa Biosystems). AMPure XP purification (Agencourt) was used to retain fragments with insert sizes between 100 and 200 bp, and this range was verified after sequencing. Cluster generation and 75-bp paired-end sequencing were performed on the HiSeq 2000 instrument.
Gene expression
RNA was isolated from tissues using the RNeasy kit according to the manufacturer's instructions (Qiagen). One microgram of RNA was taken from different samples and reversed to cDNA using the verso kit. Specific primers for RT–PCR to different genes were designed with Primer3 software, and the expression levels of these genes were tested and normalized to three different housekeeping genes. For RNA-seq experiments, RNA was isolated using miRNeasy (Qiagen), and TruSeq RNA sample preparation kit version 2 (Illumina) was used for library preparation.
Enhancer assay
DNA was amplified by PCR using presumed enhancer tile primers containing AvrtII/BamHI restriction sites and cloned into a CpG-free vector containing a minimal promoter and reporter gene (Invivogene). Cloned tiles were transfected into a transformed hepatocyte cell line (BNL) grown in DMEM medium supplemented with 10% FCS, 2 mM glutamine, 1000 U/mL penicillin, and 100 μg/mL streptomycin. After 48 h, medium was collected, and reporter expression levels were measured using the Renilla kit (Promega). To test the effect of methylation, templates were in vitro methylated with SssI methylase (New England Biolabs) prior to transfection.
Data analysis
All data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE60012. One-hundred-base-pair paired-end sequencing reads from RRBS were obtained using the HiSeq 2500. Adapter trimming and quality filtering were performed with the trim galore software using default parameters for RRBS analysis. BSMAP (Xi and Li 2009) was used for read alignment (genome builds mm9 and hg19) and extraction of single-base resolution methylation levels. One-hundred-base-pair tiles and DMRs were calculated with the MethylKit package (Akalin et al. 2012) using a minimum coverage of 10 per tile, a methylation difference of 25%, and a Q-value ≤0.01. In order to increase sensitivity, DMRs were further filtered for multiple alignments in the genome using blat (Kent 2002) and excluding tiles that were mapped more than once. The statistical significance of the DMR set was calculated by repeating the above steps on random permutations of male and female samples. For each permutation, the number of DMRs was calculated, and a P-value was obtained by measuring the quantile of the original DMRs. For tissues with at least eight samples (liver and muscle), we included the condition that the difference between the second highest and second lowest values would not exceed 30%.
DMR tiles were subject to extensive bioinformatics analysis. Transcription factor-binding sites and their promoter association were analyzed using Genomatix (http://www.genomatix.de). Gene ontology analysis was performed using GREAT (McLean et al. 2010). RNA-seq 50-bp single-end reads were obtained from the HiSeq 2500 and analyzed by TopHat2 (Kim et al. 2013). Differential expression between male and female mice was analyzed by CuffDiff (Q-value ≤ 0.01) (Trapnell et al. 2010). Promoter methylation levels were calculated by summing the methylated calls of all single CpGs in the promoter and dividing by the sum of their coverage. Regions with a minimal total coverage of 10 were reported. Motif analysis was carried out by HOMER (Heinz et al. 2010). ChIP-seq data were obtained from the publicly available GEO database: BCL6 and STAT5 (GSE31578), RXR (GSE21696), histone modification of the male and female (GSE44571) (see the Supplemental Material from Sugathan and Waxman 2013), H3K4me1 and H3K27Ac (ENCODE annotation data) (Rosenbloom et al. 2013), and human tissues (GSE17312). Genomic distribution was analyzed using HOMER (annotatePeaks.pl).
WGBS data analysis
Quality score recalibration, local realignment, and methylation calling were performed using the Bis-SNP version 0.82.1 pipeline (Liu et al. 2012). Average bisulfite conversion was determined to be >99.65% for all libraries. After removing PCR duplicates and improperly paired read pairs, genomic coverage was determined to be 14.2× (20 d male), 14.7× (20 wk female), and 14.9× (20 wk male). One-hundred-base-pair tiles and methylation levels were calculated as was done for the RRBS data, and the methylation difference (20-wk male minus 20-wk female) profile (DMRs) was determined. In order to reduce noise, we analyzed 400-bp tiles and required a minimum coverage of 20 and a threshold of 35%. We marked a DMR as valid only if the 20-d methylation level was at least 25% higher than the adult methylation level. This yielded 1630 DMRs hypomethylated in the 20-wk male sample and only 600 DMRs hypomethylated in the 20-wk female sample. In order to validate the significance of these DMRs, we performed ChIP-seq analysis for H3K4me1, H3K27Ac, BCL6, STAT5, and RXR. As seen in Figure 4, A and B, many of the 1630 male hypomethylated regions exhibit a pattern of “enhancer-like” behavior, similar to the RRBS DMRs. In contrast, the 600 female hypomethylated regions did not show significant enrichment for any of these markers (data not shown). In addition, the correlation to expression for the male demethylated DMRs was also found to be much more significant than the female demethylated DMRs. Since no significant female demethylated sequences were detected in the RRBS data set, we assume that the large majority of female demethylated tiles from the WGBS (n = 600) probably represents noise resulting from low sample density. Considering that there may be an equivalent number of nonspecific male demethylated DMRs, we estimate that there are ∼1000 authentic male demethylated regions in the liver.
Supplementary Material
Acknowledgments
We thank A. Meissner for his extensive and patient help in setting up the RRBS system, E. Pikarsky for helpful discussions, and N. Mayorek for assistance with the mice. We thank the University of Southern California Epigenome Center Sequencing Core for WGBS. This research was supported by grants from the European Research Council (ERC; 268614), the Israel Cancer Research Fund (ICRF; 210919), Rosetrees Foundation, the Israel Science Foundation (ISF; 419/10), the Israel Centers of Excellence Program (I-CORE), Lew Sanders, and the Lady Davis Fellowship Program (Y.R.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.259309.115.
References
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. 2012. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13: R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol 28: 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle P, Clement K, Gu H, Smith ZD, Ziller M, Fostel JL, Holmes L, Meldrim J, Kelley F, Gnirke A, et al. 2012. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol 13: R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. 2012. Programming of DNA methylation patterns. Annu Rev Biochem 81: 97–117. [DOI] [PubMed] [Google Scholar]
- Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H, et al. 2008. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol 15: 1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt A, Wegscheid C, Claass B, Carambia A, Herkel J, Mittrucker HW, Panzer U, Tiegs G. 2011. CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J Immunol 186: 5284–5293. [DOI] [PubMed] [Google Scholar]
- Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, Cedar H, Bergman Y. 2006. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol 8: 188–194. [DOI] [PubMed] [Google Scholar]
- Franks S. 1995. Polycystic ovary syndrome. N Engl J Med 333: 853–861. [DOI] [PubMed] [Google Scholar]
- Gilsbach R, Preissl S, Gruning BA, Schnick T, Burger L, Benes V, Wurch A, Bonisch U, Gunther S, Backofen R, et al. 2014. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun 5: 5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. 2014. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159: 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoivik EA, Bjanesoy TE, Mai O, Okamoto S, Minokoshi Y, Shima Y, Morohashi K, Boehm U, Bakke M. 2011. DNA methylation of intronic enhancers directs tissue-specific expression of steroidogenic factor 1/adrenal 4 binding protein (SF-1/Ad4BP). Endocrinology 152: 2100–2112. [DOI] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B. 2013. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet 45: 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33Suppl: 245–254. [DOI] [PubMed] [Google Scholar]
- Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, Whitelaw E, Chong S. 2010. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet 6: e1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T, Bergman Y. 1996. A role for nuclear NF-kB in B-cell-specific demethylation of the Igk locus. Nat Genet 13: 435–441. [DOI] [PubMed] [Google Scholar]
- Kosters A, Sun D, Wu H, Tian F, Felix JC, Li W, Karpen SJ. 2013. Sexually dimorphic genome-wide binding of retinoid X receptor α (RXRα) determines male-female differences in the expression of hepatic lipid processing genes in mice. PLoS One 8: e71538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27: 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. 2014. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159: 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laz EV, Sugathan A, Waxman DJ. 2009. Dynamic in vivo binding of STAT5 to growth hormone-regulated genes in intact rat liver. Sex-specific binding at low- but not high-affinity STAT5 sites. Mol Endocrinol 23: 1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling G, Sugathan A, Mazor T, Fraenkel E, Waxman DJ. 2010. Unbiased, genome-wide in vivo mapping of transcriptional regulatory elements reveals sex differences in chromatin structure associated with sex-specific liver gene expression. Mol Cell Biol 30: 5531–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Siegmund KD, Laird PW, Berman BP. 2012. Bis-SNP: combined DNA methylation and SNP calling for bisulfite-seq data. Genome Biol 13: R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. 2005. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci 28: 456–463. [DOI] [PubMed] [Google Scholar]
- Rienhoff HY Jr, Groudine M. 1988. Regulation of amyloid A gene expression in cultured cells. Mol Cell Biol 8: 3710–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. 2013. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41: D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmanoff MK, Goldman BD, Ginsburg BE. 1977. Developmental changes in serum luteinizing hormone, follicle stimulating hormone and androgen levels in males of two inbred mouse strains. Endocrinology 100: 122–127. [DOI] [PubMed] [Google Scholar]
- Shao TC, Li HL, Kasper S, Matusik R, Ittmann M, Cunningham GR. 2006. Comparison of the growth-promoting effects of testosterone and 7-α-methyl-19-nor-testosterone (MENT) on the prostate and levator ani muscle of LPB-tag transgenic mice. Prostate 66: 369–376. [DOI] [PubMed] [Google Scholar]
- Sheaffer KL, Kim R, Aoki R, Elliott EN, Schug J, Burger L, Schubeler D, Kaestner KH. 2014. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes Dev 28: 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, Wirbelauer C, Oakeley EJ, Gaidatzis D, Tiwari VK, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495. [DOI] [PubMed] [Google Scholar]
- Sugathan A, Waxman DJ. 2013. Genome-wide analysis of chromatin states reveals distinct mechanisms of sex-dependent gene regulation in male and female mouse liver. Mol Cell Biol 33: 3594–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H. 2006. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22: 1540–1542. [DOI] [PubMed] [Google Scholar]
- Takasugi M, Hayakawa K, Arai D, Shiota K. 2013. Age- and sex-dependent DNA hypomethylation controlled by growth hormone in mouse liver. Mech Ageing Dev 134: 331–337. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauthier V, Waxman DJ. 2008. Sex-specific early growth hormone response genes in rat liver. Mol Endocrinol 22: 1962–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman DJ, Holloway MG. 2009. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 76: 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman DJ, O'Connor C. 2006. Growth hormone regulation of sex-dependent liver gene expression. Mol Endocrinol 20: 2613–2629. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. 2004. Epigenetic programming by maternal behavior. Nat Neurosci 7: 847–854. [DOI] [PubMed] [Google Scholar]
- Xi Y, Li W. 2009. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA, Lusis AJ. 2006. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res 16: 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Laz EV, Waxman DJ. 2012. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol 32: 880–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. 2013. Charting a dynamic DNA methylation landscape of the human genome. Nature 500: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.