

Abstract
A series of azidoaryl- and azidoalkyl(diphenyl)oxazole scaffolds were warranted for biofilm inhibition studies. Cyclization of azidoaryl- or azidoalkyl esters of benzoin with ammonium acetate in acetic acid gives 2-azidoaryl- or 2-azidoalkyl-4,5-diphenyloxazoles. The azidoaryl esters are prepared from the corresponding azidocarboxylic acids/acid chlorides while the azidoalkyl esters are prepared from the corresponding haloalkyl esters.
Keywords: Azides, Click chemistry, Oxazoles, Biofilms, Peptidomimetics
Introduction
Organic azides are utilized as precursors to amines, nitro compounds, and many types of nitrogen heterocycles such as aziridines, azetidines, pyrrolidones, oxazoles, indoles, and triazoles.1 In the area of biological probes, they are employed as readily-prepared reactive intermediates for labeling and bioconjugation.2 In terms of a functional group found in biologically active molecules, compounds such as AZT3a or azido-thalidomide3b are excellent examples of pharmacophores which exhibit the azide moiety as a necessary component of an active structure. More recently organic azides are commonly employed as reacting partners with acetylenes in the 1,3-dipolar cycloaddition reaction popularly known as ‘click chemistry’.4 While the dipolar cycloaddition of azides with terminal acetylenes was established through the pioneering work of Huisgen, the introduction of so called ‘click chemistry’ by Sharpless has served to exemplify its use in biomedical applications such as drug discovery and structural biology.5 Click chemistry may be used in conjunction with many types of scaffolds which bear either the acetylene or azide components. Scaffolds composed of heterocycles6, carbohydrates7, polypeptides8, terpenes,9 or lipids10 are but a few of the many possible structural families that may be functionalized for click chemistry. The 1, 2, 3-triazole ‘linker’ units that result from the dipolar cycloaddition come with a unique set of properties all their own.11 These units may hydrogen bond thereby increasing aqueous solubility or affinity for molecular targets. They are also poorly basic and, despite having a contiguous array of nitrogen atoms, are not protonated at physiological pH.
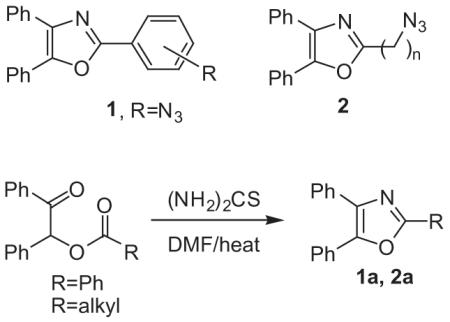
We are currently interested in the inhibition of biofilm formation using the techniques of click chemistry. Porphyromonas gingivalis is a major pathogen associated with periodontal disease and this organism colonizes the dental biofilm by interacting with oral streptococci. We previously showed that this interaction is inhibited by a peptide comprised of two distinct structural motifs which block the interaction of minor fimbrial antigen (Mfa) of P. gingivalis with the antigen I/II (AgI/II) of oral streptococci.12 Consequently, the design and study of small-molecule, non-peptide based inhibitors of the Mfa/AgI/II interaction can encompass the employment of two synthetic scaffolds joined together via a ‘click’ reaction. Within the expansive area of nitrogen/oxygen heterocycles, we have identified the substituted oxazole framework as a starting point for peptidomimetic inhibitors of Mfa/AgI/II interaction. Oxazoles and their semi-saturated congeners, the oxazolines have been proposed as backbone replacements for peptide bonds in peptidomimetic chemistry and protein-derived frameworks.13 Moreover, the oxazole framework is also a viable starting point as a basic scaffold for the construction of chemically diverse pharmacophores. The relative ease of installing an azido group on most molecules together with the diversity of oxazole substitution has led us to choose the 4,5-disubstituted oxazole system with a spacer containing the azido group at the 2-position. The ‘spacer’ can take the form of either an aromatic ring with 1,3- or 1,4-substitution, or one or more methylene units which link the azido group to the heterocycle. Hence, the azidooxazole coupling partners described herein can be placed in two structural groups, 2-azidoaryl-4,5-disubstituted oxazoles 1 and 2-azidoalkyl-4,5-disubstituted oxazoles 2. Our initial experiments in the 2-azidoaryl-4,5-disubstitutedoxazole series entailed the preparation of benzoic or acetic esters of benzoin with the objective of thiourea-mediated cyclization to the corresponding 2-phenyl- or 2-methyl-4,5-diphenyloxazoles 1a, 2a (Eq. 1).14 One could also envision these intermediates as occurring en route to later-stage azido substitution. In experiments whereby the optimal temperatures required for heterocycle formation were explored, treatment of the known benzoyl ester of benzoin 6a
 |
(1) |
with thiourea in hot N,N-dimethylformamide (DMF) gave 2,4,5-triphenyloxazole 1a (69%) (Scheme 1).15 Next, the 4-azidobenzoyl ester 6b was prepared by diazotizing 4-aminobenzoic acid 3b (NaNO2/HCl/NaN3) in the presence of sodium azide.16 Following azidation of 3b, the resultant 4-azidobenzoic acid 4b was treated with thionyl chloride. The direct addition of the acid chloride 5b to benzoin then afforded the ester 6b.17 However, treatment of azidobenzoyl ester 6b with thiourea/DMF similar to the cyclization of 6a gave only products of decomposition and none of the expected 2-azidophenyl-4,5-diphenyloxazole 1b. Presumably, the highly reducing nature of thiourea at higher temperatures proved to be incompatible with the azide functionality of 6b or possibly the product 1b. Alternatively, the 4-azidobenzoyl ester 6b was treated with ammonium acetate in acetic acid (118 °C) which did provide 1b in 61% yield after purification by flash-column chromatography.18 Using a scheme similar to that which gave 1b, 2-(3-azidophenyl)-4,5-diphenyloxazole 1c was prepared accordingly. Starting with 3-aminobenzoic acid 3c, 3-azido-benzoic acid 4c was prepared using the NaNO2/HCl/NaN3 method followed by direct conversion to the acid chloride 5c. Reaction of 5c with benzoin afforded 3-azidobenzoyl ester 6c which was cyclized to 1c (80%) using the ammonium acetate/acetic acid protocol.
Scheme 1.

Synthesis of 2-azidoaryl-4,5-diphenyloxazoles. Reagents/conditions: (a) NaNO2/HCl/NaN3; (b) SOCl2/76 °C; (c) benzoin/pyridine/DMAP/0 °C; d. NH4OAc/HOAc/118 °C, or thiourea/DMF/150 °C.
The initial experiments in the 2-azidoalkyl-4,5-disubstituted oxazole series 2b-d entailed the preparation of acetic esters of benzoin with the same objective of thiourea-mediated cyclization to the analogous 2,4,5-substituted oxazoles. 2-Methyloxazole 2a would then be submitted to benzylic-type halogenation followed by substitution with azide ion (Scheme 2). Cyclization of the known acetylbenzoin 8a19 with thiourea in N,N-dimethylformamide (150 °C) gave 2-methyl-4,5-diphenyloxazole 2a in 74% yield.20 Halogenation of 2a with N-bromosuccinimide under a variety of conditions failed to give the expected benzylic bromide intermediate, thereby precluding installation of azide by nucleophilic substitution. When it became evident that the azide displacement would not be the last step in obtaining 2b, the alternative route involving the preparation and cyclization of the azidoacetyl ester was explored (Scheme 2).
Scheme 2.

Synthesis of 2-azidoalkyl-4,5-diphenyloxazoles. Reagents/conditions: (a) benzoin, pyridine/DMAP, 0 °C; (b) NaN3/DMF/80 °C; (c) NH4OAc/HOAc/118 °C; d. thiourea/DMF/150 °C.
Treatment of benzoin chloroacetate 8b21 with sodium azide in hot DMF (80 °C) gave the azido ester 9b in 22% yield after purification by flash-column chromatography. Cyclization of azidoacetyl ester 9b to 2-azidomethyloxazole 2b was accomplished in 46% yield by heating 9b with ammonium acetate (15 equiv) in acetic acid (118 °C). The analogous preparation of the benzoin haloesters 8c and 8d were accomplished from 4-bromobutyryl chloride 7c and 5-bromovaleryl chloride 7d.22 Preparation of haloesters 8c and 8d was followed by their conversion to the azidoesters 9c (85%) and 9d (56%) using sodium azide/DMF (80 °C).23 Through the employment of ammonium acetate under conditions similar to the preparation of 1b and 1c, the intermediate azidoesters 9c and 9d were cyclized to the 2-azidoalkyloxazoles 2c (64%) and 2d (81%).24 In summary, we have detailed a general method for the preparation of both 2-azidoaryl- and 2-azidoalkyloxazoles for employment in click chemistry. The scheme utilizes azido-substituted esters which are submitted to a relatively high-temperature aminative heterocyclization. A distinct improvement in the method would entail the introduction of a heterocyclization in which lower temperatures and neutral, non-reducing conditions are employed thereby allowing the presence of sensitive functionality and preserving the azide group. Ongoing synthetic studies currently involve the preparation of trisubstituted 2-azidooxazoles in which both (or one of) the 4,5-aryl groups described in the present work are interchanged with alkyl groups of varying substitution. The oxazole targets bearing more structural diversity together with their biological evaluation in biofilm inhibition will be reported by our laboratory in due course.
Acknowledgments
The measurement of high resolution mass spectra by the Texas A&M University Laboratory for Biological Mass Spectrometry and Dr. Vanessa Santiago are acknowledged. CML thanks the Department of Chemistry for financial support.
Footnotes
Presented by CML at the 242nd National Meeting of the American Chemical Society, Denver, Colorado, August 28, 2011; ORGN130.
References and notes
- 1.(a) Bräse S, Gil C, Knepper K, Zimmerman V. Angew. Chem., Int. Ed. 2005;44:5188–5240. doi: 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]; (b) Scriven EFV, Turnbull K. Chem. Rev. 1988;88:297–368. [Google Scholar]
- 2.Hein CD, Liu X-M, Wang D. Pharmaceutical Res. 2008;25:2216–2230. doi: 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Horowitz JP, Chua J, Noel M. J. Org. Chem. 1964;29:2076–2078. [Google Scholar]; (b) Capitosti SM, Hansen TP, Brown ML. Org. Lett. 2003;5:2865–2867. doi: 10.1021/ol034906w. [DOI] [PubMed] [Google Scholar]
- 4.(a) Huisgen R. Angew. Chem. 1963;75:604–637. [Google Scholar]; (b) Huisgen R. Angew. Chem., Int. Ed. Engl. 1963;2:565–598. [Google Scholar]; (c) Huisgen R. Angew. Chem. 1963;75:742–754. [Google Scholar]; (d) Huisgen R. Angew. Chem., Int. Ed. Engl. 1963;2:633–635. [Google Scholar]
- 5.Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 6.(a) Fiandanese V, Maurantonio S, Punzi A, Rafashieri GG. Org. Biomol. Chem. 2012;10:1186–1195. doi: 10.1039/c1ob06701j. [DOI] [PubMed] [Google Scholar]; (b) Wang M, Das MR, Li M, Boukerroub R, Szunerits S. J. Phys. Chem. C. 2009;113:17082–17086. [Google Scholar]; (c) Buck SB, Bradford J, Gee KR, Agnew BR, Clarke ST, Salic A. Biotechniques. 2008;44:927–929. doi: 10.2144/000112812. [DOI] [PubMed] [Google Scholar]; (d) Comstock LR, Rajski SR. J. Org. Chem. 2004;69:1425–1428. doi: 10.1021/jo035485z. [DOI] [PubMed] [Google Scholar]
- 7.(a) Stefani HA, Silva NCS, Manarin F, Lüdtke DS, Zukerman-Schpector J, Madureira LS, Tiekink ERT. Tetrahedron Lett. 2012;53:1742–1747. [Google Scholar]; (b) Pèrez-Castro I, Caamaño O, Fernández F, Garcia MD, López C, de Clercq E. ARKIVOK. 2010;(iii):152–168. [Google Scholar]
- 8.(a) Baccile JA, Morrell MA, Falotico RM, Milliken BT, Drew DL, Rossi FM. Tetrahedron Lett. 2012;53:1933–1935. [Google Scholar]; (b) Franke R, Doll C, Eichler J. Tetrahedron Lett. 2005;46:4479–4482. [Google Scholar]
- 9.Sall C, Dombrowsky L, Bottzeck O, Praud-Tabaries A, Blache Y. Bioorg. Med. Chem. Lett. 2011;21:1493–1497. doi: 10.1016/j.bmcl.2010.12.134. [DOI] [PubMed] [Google Scholar]
- 10.Bori ID, Hung H-Y, Qian K, Chen C-H, Morris-Natschke SL, Lee K-L. Tetrahedron Lett. 2012;53:1987–1989. doi: 10.1016/j.tetlet.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agalave SG, Maujan SR, Pore VS. Chem. Asian J. 2011;6:2696–2718. doi: 10.1002/asia.201100432. [DOI] [PubMed] [Google Scholar]
- 12.(a) Daep C, James D, Lamont R, Demuth D. Infection and Immunity. 2006;74:5756–5762. doi: 10.1128/IAI.00813-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Filoche S, Wong L, Sissons CH. J. Dent. Res. 2010;89:8–18. doi: 10.1177/0022034509351812. [DOI] [PubMed] [Google Scholar]; (c) Daep C, Lamont R, Demuth D. Infection and Immunity. 2008;76:3273–3280. doi: 10.1128/IAI.00366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Daep C, Novak E, Lamont R, Demuth D. Infection and Immunity. 2011;79:67–74. doi: 10.1128/IAI.00361-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Walsh CT, Malcolmson SJ, Young TS. ACS Chem. Biol. 2012;7:429–442. doi: 10.1021/cb200518n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davis MR, Sing EK, Wahyudi H, Alexander LD, Kunicki JB, Nazarova LA, Fairweather KA, Giltrap AM, Jolliffe KA, McAlpine SR. Tetrahedron. 2012;68:1029–1051. doi: 10.1016/j.tet.2011.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalko M, Dumats J. U.S. Patent 6333,414 B1 2001 Chem. Abs. 134:326522. See also: Turner CD, Liang SH. Current Org. Chem. 2011;15:2846–2870.
- 15.2,4,5-Triphenyloxazole (1a): Benzoyl ester 6a25 (200 mg, 0.63 mmol) was added to DMF (7.0 mL) followed by thiourea (96.5 mg, 1.26 mmol). The mixture was heated under a nitrogen atmosphere while stirring (150 °C, oil bath) under an atmosphere of nitrogen. Heating was continued for 16 h whereupon thin-layer chromatographic analysis indicated complete consumption of starting material and formation of product as evidenced by a more mobile spot. The DMF was removed under high vacuum and the solid residue was flash-chromatographed on silica gel (hexanes/ethyl acetate, 9:1) which provided 1a (130 mg, 69%) as a white crystalline solid: mp. 116–118 °C (Lit.26 116–117 °C).
- 16.General procedure for preparation of 4- and 3-azidobenzoic acid (4b, 4c): (Adapted from a procedure by Molina P, Diaz I, Tarraga A. Tetrahedron. 1995;51:5617–5630. 3-amino- or 4-aminobenzoic acid (3b or 3c, 1 equiv) was dissolved in aqueous HCl solution (10%) and cooled to 0 °C (ice bath). Aqueous sodium nitrite (20%, 1.2 equiv) was then added and the reaction mixture was allowed to stir at room temperature (15 min). A 20% aqueous solution of sodium azide (1.2 equiv) then was added at room temperature which resulted in a vigorous reaction and creating a foaming precipitate (Caution!) which filled the headspace of the reaction flask. The foam precipitate, which was the azidobenzoic acid product, was collected by vacuum filtration while washing the filter cake with water. Excess solvent was removed by slow rotary evaporation prior to vacuum filtration. The slightly yellow-white solids were stored damp in the refrigerator and were of sufficient purity to use in the next step (4b: mp 189-190 °C, Lit.27 188.5-190 °C; 4c: mp 176-178 °C, Lit.28176-177 °C). Prior to the treatment with thionyl chloride to produce 5b/5c, the azidobenzoic acids were dried at room temperature under high vacuum.
- 17.General procedure for preparation of 2-oxo-1,2-diphenylethyl-4-azidobenzoate (6b) and 2-oxo-1, 2-diphenylethyl-3-azidobenzoate (6c) through acid chlorides 5b and 5c: The azidobenzoic acid 4b or 4c (1 e0071uiv) was dissolved in thionyl chloride (4.5 equiv). The mixture is heated to reflux (75 °C), and allowed to stir (5 h). The reaction mixture was then allowed to stir overnight at room temperature. Thionyl chloride was then removed by adding dichloromethane (10 mL) and concentrating with the rotary evaporator under aspirator vacuum. The addition of dichloromethane and vacuum rotary evaporation was repeated (3 ×) to give the acid chlorides 5b or 5c as oils. The azidobenzoyl chlorides (5b/5c) were used in the next esterification step without further purification. Benzoin (1 equiv), triethylamine (1 equiv), and 4-dimethylaminopyridine (0.1 equiv) are dissolved in dichloromethane (10 mL) at 0 °C (ice water bath). The azidobenzoyl chloride (5b or 5c, 1 equiv) dissolved in dry dichloromethane (5 mL) was gradually introduced dropwise into the reaction flask while stirring. The reaction mixture was allowed to stir (3 h) at room temperature while monitoring by TLC. Upon completion of the reaction, the reaction mixture was dissolved in diethyl ether (100 mL) and washed with 5% aqueous HCl solution (4 × 50 mL) followed by 5% aqueous sodium bicarbonate (2 × 100 mL). The organic layer was separated and dried over anhydrous sodium sulfate. Removal of the drying agent by filtration and rotary evaporation of the solvent gave a crude solid that was purified by flash chromatography on silica gel (hexane/EtOAc, 4:1) to obtain 6b (80%) and 6c (72%). 2-oxo-1,2-diphenylethyl-4-azidobenzoate (6b): Rf: 0.42 (hexane/ethyl acetate, 4:1); FTIR 2118.00, 1174.79, 1710.42, 1693.73, 1448.18, 1504.39 cm−1; 1H NMR: (400 MHz, CDCl3) δ: 6.96 (s, 1H); 6.99-7.33 (m, 14H); 7.90-7.92 (d, 2H, J = 7.99); 8.01-8.04 (d, 2H); 13C NMR (100 MHz, CDCl3) δ: 193.62, 165.20, 145.29, 118.81-145.20, 78.06; HRMS calcd for C21H15N3O3 (M+H)+ 358.1192, Found: 358.1195. 2-oxo-1,2-diphenylethyl-3-azidobenzoate (6c): Rf: 0.53 (hexane/ethyl acetate, 4:1); FTIR 2124.85, 1299.51, 1711.90, 1695.45, 1482.43, 1448.61 cm−1; 1H NMR: (400 MHz, CDCl3) δ: 7.00 (s, 1H); 7.08–7.90 (m, 14H); 13C NMR (100 MHz, CDCl3) δ: 193.42, 165.20, 120.16-140.62, 78.33; HRMS calcd for C21H15N3O3 (M+Li)+ 364.1273, Found: 364.1266.
- 18.General procedure for preparation of 2-(4-azidophenyl)-4,5-diphenyloxazole (1b) and 2-(3-azidophenyl)-4,5-diphenyl-oxazole (1c): The azidobenzoic esters 6b or 6c (1 equiv) and ammonium acetate (15 equiv) are combined in glacial acetic acid (10 mL). The mixture is then heated (oil bath) at reflux (118 °C) for 2 hours under an atmosphere of nitrogen. The reaction mixture was monitored by TLC and when complete, the reaction mixture was dissolved in diethyl ether (110 mL) and washed with NaOH solution (3 × 100 mL). The diethyl ether layer is separated and dried over anhydrous sodium sulfate. Removal of the drying agent by filtration and rotary evaporation of the solvent gave a crude oil that was purified by flash chromatography on silica gel (hexane/ethyl acetate, 4:1) to obtain 1b (61%) and 1c (80%) as amorphous solids. 2-(4-azidophenyl)-4,5-diphenyloxazole (1b): Rf: 0.60 (hexane/ethyl acetate, 4:1); FTIR: 2088.97, 1278.93, 1608.72, 1493.77, 1087.95 cm−1; 1H NMR: (500 MHz, CDCl3) 7.21-8.13 (m, 14H, aromatic); 13C NMR (125 MHz, CDCl3) δ: 159.46, 145.61, 142.26, 136.47, 119.34-132.01; HRMS calcd for C21H14N4O (M+H)+ 339.1246, Found: 339.1243. 2-(3-azidophenyl)-4,5-diphenyloxazole (1c): Rf: 0.53 (hexane/ethyl acetate, 4:1); FTIR: 2146.37, 1276.93, 1590.12, 1590.35 cm−1; 1H NMR: (400 MHz, CDCl3) δ: 7.09-7.94 (m, 14H, aromatic); 13C NMR (100 MHz, CDCl3) δ: 159.09, 145.96, 140.87, 136.85, 116.84-132.25; HRMS calcd for C21H14N4O (M+H)+ 339.1246, Found: 339.1241.
- 19.Butler RN, Hanahoe AB, King WB. J. Chem. Soc. 1978;8:881–884. [Google Scholar]
- 20.2-Methyl-4,5-diphenyloxazole (2a): 2-Oxo-1, 2-diphenylethyl acetate 8a (0.20 g, 0.79 mmol) was dissolved in DMF (10 mL). Thiourea was then added and the reaction was heated (150°, oil bath) under a nitrogen atmosphere. As the reaction progressed, the color changed from colorless to a light yellow-orange and had an odorous smell. The reaction was monitored by TLC and when complete, the reaction mixture was dissolved in dichloromethane (40 mL) and then washed with water (3 × 30 mL). The dichloromethane layer was separated and dried over anhydrous sodium sulfate. Removal of the drying agent by filtration and rotary evaporation of the solvent gave a crude oil that was purified by flash chromatography on silica gel (dichloromethane) to provide 2a (74%): Rf: 0.098 (hexane/ethyl acetate, 2:1); FTIR 2920.50; 1220.30; 1502.00, 1588.24 cm−1; 1H NMR (400 MHz, CDCl3)δ: 2.617 (s, 3H); 7.25-7.66 (m, 10H, aromatic); 13C NMR (100 MHz, CDCl3) δ 160.35, 145.41, 134.82, 126.46-132.14, 13.92; HRMS calcd for C16H13NO (M+H)+ 236.1075, Found: 236.1077.
- 21.1-Oxo-1, 2-diphenylethyl-2-chloroacetate (8b): Benzoin (0.50 g, 2.37 mmol) and 4-dimethylaminopyridine (0.29 g, 2.37 mmol) were dissolved in dichloromethane (20 mL) and the solution was allowed to stir at 0 °C. Commercially-available chloroacetyl chloride 7b (0.21 mL, 2.60 mmol) was added dropwise by syringe. The reaction mixture was then stirred under nitrogen at 0 °C (4 h) while monitoring by TLC. Upon completion of the reaction, the reaction mixture was dissolved in diethyl ether (100 mL) and washed with water (2 × 90 mL), 5% aqueous HCl (1 × 90 mL), and 5% aqueous sodium bicarbonate (1 × 90 mL). The diethyl ether layer was separated and dried over anhydrous sodium sulfate. After removal of the drying agent by filtration and removal of solvent by rotary evaporation, the product chloroacetyl ester 8b was obtained in 95% yield and found to be of reasonable purity as evidenced by 1H NMR and TLC. Rf: 0.51 (hexane/ethyl acetate, 4:1); FTIR 2960, 1734, 1694, 1597, 1495, 1224 cm−1; 1H NMR (500 MHz, CDCl3) δ: 7.25-7.91 (m, 10H); 6.94 (s, 1H); 3.96-4.11 (dd, 2H); 13C NMR (125 MHz, CDCl3) δ: 192.62, 167.91, 128.71-134.17, 78.75, 50.11.
- 22.General procedure for preparation of bromoacyl esters (8c and 8d) through acid chlorides (7c, 7d: 5-bromovaleric acid (1 equiv) or 4-bromobutyric acid (1 equiv) was dissolved in thionyl chloride (4.5 equiv). The reaction mixture was then heated (75 °C, oil bath) under a nitrogen atmosphere overnight. The excess thionyl chloride was removed by adding dichloromethane (25 mL) followed by rotary evaporation under aspirator vacuum. The addition of the dichloromethane and rotary evaporation was repeated (3×) which yielded the crude acid chloride as an oil. The 5-bromobutyryl chloride 7c or the 4-bromovaleryl chloride 7d were used without further purification in the next step. Benzoin (1 equiv) was dissolved in pyridine (12 mL) followed by cooling the solution to 0 °C (ice water bath). The acid chloride 7c, 7d (1 equiv) was then added dropwise to the stirred solution while cooling and stirring. The reaction flask was capped, and after 30 min, the cooling bath was removed. The reaction mixture was then stirred (4 h) at room temperature while monitoring by TLC. After the starting materials were consumed, the reaction mixture was then dissolved in dichloromethane (300 mL) and washed with 5% aqueous HCl (5 × 120 mL). The organic layer was then separated and dried over anhydrous sodium sulfate. Flash chromatography on silica gel (hexane/ethyl acetate, 6:1) afforded esters 8c (70%) and 8d (23%) as oils. 2-oxo-1,2-diphenylethyl-4-bromobutanoate (8c): Rf: 0.49 (hexane/ethylacetate, 4:1); FTIR: 3063.60, 1708.60, 1692.03, 1588.70, 1531.70 cm−1; 1H NMR (400 MHz, CDCl3) δ: 2.21-2.23 (t,2H); 2.60-2.69 (m, 2H); 3.45-3.47 (t, 2H); 6.85 (s, 1H); 7.28-8.01 (m, 10H); 13C NMR (100 MHz, CDCl3) δ: 193.66, 172.08, 134.56, 133.57, 128.70-129.43, 77.85, 32.55, 32.34, 27.83; FTIR: 3063.60, 1708.60, 1692.03, 1588.70, 1531.70 cm−1. 2-oxo-1,2-diphenylethyl-5-bromopentanoate (8d): Rf: 0.47 (hexane/ethyl acetate, 4:1); FTIR: 1734.38, 1693.88, 1597.38, 1448.37 cm−1; 1H NMR (400 MHz, CDCl3) δ: 1.81-1.87 (m, 2H); 1.91-1.98 (m, 2H); 2.44-2.59 (m, 2H); 3.40-3.43 (t, 2H); 6.85 (s, 1H), 7.35-7.93 (m, 10H); 13C NMR (100 MHz, CDCl3) δ: 193.75, 172.63, 134.59, 133.49, 128.07-128.83, 60.25, 44.55, 33.09, 31.76, 23.38.
- 23.General procedure for conversion of the halogenated esters 8b, 8c or 8d to azido esters (9b, 9c or 9d): The halogenated ester 8b, 8c or 8d (1 equiv) was dissolved in DMF. Sodium azide (1.1 equiv) was then added and the reaction mixture was heated (80 °C, oil bath) under a nitrogen atmosphere (4 h). The reaction mixture was monitored by TLC and when complete, the DMF was removed under high vacuum. The crude oil was purified by flash chromatography on silica gel (hexane/ethyl acetate, 9:1) to obtain 9b (22%) 9c (85%) and 9d (56%). 2-Oxo-1,2-diphenylethyl-2-azidoacetate (9b): Rf: 0.49 (hexane/ethyl acetate, 4:1); FTIR 2104.56, 1173.01, 1747.90, 1692.58, 1597.22, 1448.76 cm−1; 1H NMR: (500 MHz, CDCl3) δ: 7.38-7.94 (m, 10H); 6.97 (s, 1H); 4.03-4.10 (dd, 2H); HRMS calcd for C16H13N3O3 (M+Li)+ 302.1117, Found 302.1121. 2-oxo-1,2-diphenylethyl-4-azidobutanoate (9c): Rf: 0.44 (hexane/ethylacetate, 4:1); FTIR: 2096.16, 1448.47, 1734.69, 1693.83, 1580.74, 1496.07 cm−1; 1H NMR (500 MHz, CDCl3) δ: 1.82-1.92 (t, 2H); 2.41-2.60 (m, 2H); 3.28-3.38 (t, 2H); 6.82 (s, 1H); 7.25-7.98 (m, 10H, aromatic); 13C NMR (125 MHz, CDCl3) δ: 193.65, 172.23, 134.55, 133.56, 128.69-129.19, 77.86, 50.54, 30.95, 24.33; HRMS calcd for C18H17N3O3 (M+H)+ 324.1348, Found: 324.1346. 2-oxo-1,2-diphenylethyl-5-azidopentanoate (9d): Rf: 0.22 (hexane/ethyl acetate, 4:1); FTIR 2092.58, 1224.81, 1734.84, 1694.21, 1597.46, 1448.81 cm−1; 1H NMR: (500 MHz, CDCl3) δ: 1.52-1.61 (m, 2H); 1.62-1.71 (m, 2H); 2.32-2.49 (m, 2H); 3.14-3.22 (t, 2H); 6.782 (s, 1H); 7.24-7.85 (m, 10H); 13C NMR (125 MHz, CDCl3) δ: 193.76, 172.64, 134.60, 133.55, 128.66-129.36, 77.66, 51.02, 33.33, 28.13, 22.04. HRMS calcd for C19H19N3O3 (M+H)+ 338.1505, Found: 338.1500.
- 24.General Procedure for preparation of the azidoalkyl oxazoles (2b, 2c, 2d): The azido esters 9b, 9c or 9d (1 equiv) were dissolved in glacial acetic acid (10 mL). Ammonium acetate (15 equiv) was then added and the reaction mixture was heated (118 °C, oil bath) under a nitrogen atmosphere (2 h). The reaction mixture was monitored by TLC, and when complete as evidenced by the disappearance of the ester, the reaction mixture was dissolved in diethyl ether (100 mL) and washed with aqueous sodium hydroxide (3 × 100 mL). The organic layer was separated and dried over anhydrous sodium sulfate. Flash chromatography on silica gel (hexane/ethyl acetate, 8:1) afforded 2b (46%), 2c (64%) and 2d (81%). 2-(azidomethyl)-4,5-diphenyloxazole (2b): Rf: 0.58 (hexane/ethyl acetate, 4:1); FTIR 2098, 1444.19, 1569.48, 1604.78, 1251.13 cm−1; 1H NMR (400 MHz, CDCl3) δ: 4.44 (s, 2H); 7.18-7.59 (m, 10H); 13C NMR (125 MHz, CDCl3) δ: 157.23, 146.77, 135.52, 126.69-131.80, 46.75. HRMS calcd for C16H13N4O (M+H)+ 277.1089, Found: 277.1090. 2-(3-azidopropyl)-4,5-diphenyl-oxazole (2c): Rf: 0.30 (hexane/ethyl acetate, 8:1); FTIR 2094.21, 1218.91, 2933.26, 1570.34, 1501.98, 1218.91 cm−1; 1H NMR: (500 MHz, CDCl3) δ: 2.16-2.19 (m, 2H); 3.02-3.06 (t, 2H); 3.49-3.51 (t, 2H); 7.35-7.67 (m, 10H); 13C NMR (125 MHz, CDCl3) δ: 162.68, 145.69, 134.22, 126.54-131.34, 50.55, 26.31, 25.18; HRMS calcd for C18H16N4O (M+H)+ 305.1402, Found: 305.1407. 2-(4-azidobutyl)-4,5-diphenyl-oxazole (2d): Rf: 0.50 (hexane/ethyl acetate, 4:1); FTIR 2936.78, 2091.06, 1218.94, 1570.01, 1501.95, 1157.14 cm−1; 1H NMR: (500 MHz, CDCl3) δ: 1.77-1.80 (m, 2H); 1.96-2.0 (m, 2H); 2.92-2.95 (t, 2H); 3.36-3.39 (t, 2H); 7.336-7.668 (m, 10H); 13C NMR (125 MHz, CDCl3) δ: 162.99, 145.35, 132.23, 126.47-128.92, 51.04, 28.35, 27.65, 24.27; HRMS calcd for C19H18N4O (M+H)+ 319.1559, Found: 319.1564.
- 25.Cutulic SPY, Findlay NJ, Zhou SZ, Chrystal EJT, Murphy JA. J. Org. Chem. 2009;74:8713–8718. doi: 10.1021/jo901815t. [DOI] [PubMed] [Google Scholar]
- 26.Davidson D, Jelling M. J. Org. Chem. 1937;2:328–334. [Google Scholar]
- 27.Goddar-Borger ED, Stick RV. Org. Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 28.Boyer JH, Elizey S., Jr. J. Org. Chem. 1958;23:127–129. [Google Scholar]