

Abstract
Amide hydrogen/deuterium exchange (ssHDX-MS) and side-chain photolytic labeling (ssPL-MS) followed by mass spectrometric analysis can be valuable for characterizing lyophilized formulations of protein therapeutics. Labeling followed by suitable proteolytic digestion allows the protein structure and interactions to be mapped with peptide-level resolution. Since the protein structural elements are stabilized by a network of chemical bonds from the main-chains and side-chains of amino acids, specific labeling of atoms in the amino acid residues provides insight into the structure and conformation of the protein. In contrast to routine methods used to study proteins in lyophilized solids (e.g., FTIR), ssHDX-MS and ssPL-MS provide quantitative and site-specific information. The extent of deuterium incorporation and kinetic parameters can be related to rapidly and slowly exchanging amide pools (Nfast, Nslow) and directly reflects the degree of protein folding and structure in lyophilized formulations. Stable photolytic labeling does not undergo back-exchange, an advantage over ssHDX-MS. Here, we provide detailed protocols for both ssHDX-MS and ssPL-MS, using myoglobin (Mb) as a model protein in lyophilized formulations containing either trehalose or sorbitol.
Keywords: Chemistry, Issue 98, Amide hydrogen/deuterium exchange, photolytic labeling, mass spectrometry, lyophilized formulations, photo-leucine, solid-state, protein structure, protein conformation, protein dynamics, secondary structure, protein stability, excipients
Introduction
Protein drugs are the fastest growing sector of the biopharmaceutical industry and offer promising new treatments for previously intractable diseases, including hormonal disorders, cancers and autoimmune diseases1. In 2012, the global biotherapeutics market reached $138 billion and is expected to reach $179 billion by the year 20182. Proteins are larger and more fragile than conventional small molecule drugs and so are more susceptible to many types of degradation3. To ensure adequate shelf-life and stability, protein drugs are often formulated as lyophilized (i.e., freeze-dried) solid powders. However, a protein may still undergo degradation in the solid state, particularly if its native structure is not preserved during the lyophilization process4,5. Assuring that structure has been retained is feasible only if there are analytical methods that can probe protein conformation in the solid-state with sufficient resolution.
NMR spectroscopy6 and X-ray crystallography7 are the commonly used high resolution methods to assess protein structure in solution and crystalline solids8. Because of the nature of excipients and the processing methods used, lyophilized protein formulations are usually amorphous rather than crystalline9. The lack of homogeneity and microscopic order makes the above mentioned techniques impractical for proteins in amorphous solids. Fourier transform infrared spectroscopy (FTIR)10, Raman spectroscopy11 and near infrared spectroscopy (NIR)12 have been regularly used by the biopharmaceutical industry to compare protein secondary structure in lyophilized powders to that of the native solution-state structure. However, these methods are low resolution and can only provide information on global changes in secondary structure. Solid-state structural characterization using FTIR has shown either weak13,14 or poor15 correlation with long-term storage stability. These limitations highlight the need for suitable high resolution methods to identify protein structural perturbations in the solid-state.
Chemical labeling coupled with proteolysis and mass spectrometric analysis has emerged as a powerful approach to monitoring protein structure and molecular interactions in aqueous solution. In pharmaceutical development, HDX-MS has been used for epitope mapping in antigen-antibody interactions16,17, to map receptor-drug interactions18, to monitor the effects of post-translational modifications on the conformation of protein drugs19, and to compare batch-to-batch variation in developing biosimilars20. Similarly, photoactivatable ligands have been used to identify drug targets and to determine binding affinity and specificity of drug-receptor interactions21,22. To extend the application of these methods to lyophilized formulations, our group has developed solid-state hydrogen deuterium exchange mass spectrometry (ssHDX-MS) and solid-state photolytic labeling mass spectrometry (ssPL-MS) to study protein conformations and excipient interactions in lyophilized samples with high resolution.
In both ssHDX-MS and ssPL-MS, the protein is labeled under ideal reaction conditions in lyophilized solids, and the samples are then reconstituted and analyzed by mass spectrometry with or without proteolytic digestion. ssHDX-MS provides information on main chain exposure to deuterium vapor, while ssPL-MS provides information on the environment of side chains (Figure 1). The two methods thus can provide complementary information about protein conformation in the solid-state. Here, we provide a general protocol for studying proteins in lyophilized solids using ssHDX-MS and ssPL-MS (Figure 2), using Mb as a model protein. We show the ability of the two methods to distinguish differences in formulations with two different excipients.
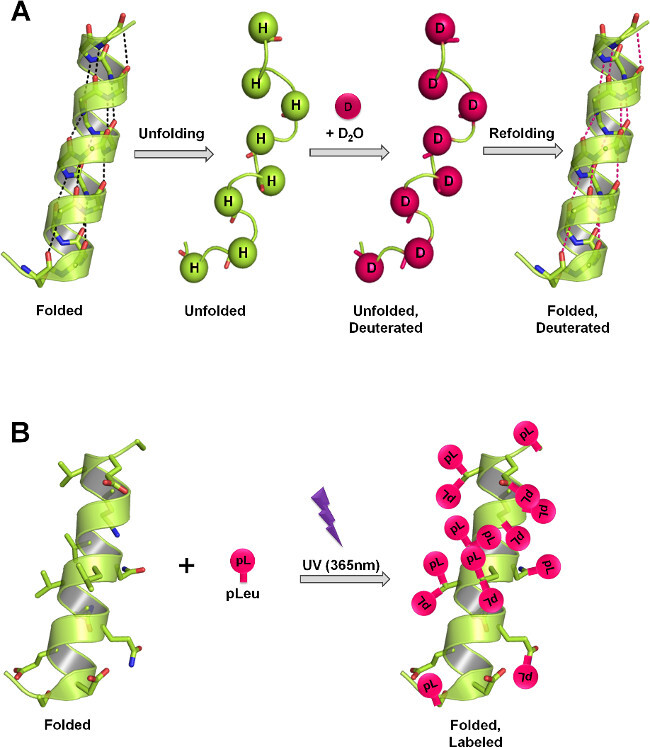 Figure 1: ssHDX and ssPL measure protein structure in lyophilized solids through different labeling mechanisms. (A) In HDX, the backbone amide hydrogens exchange with deuterium as a function of protein structure and D2O accessibility. In the solid-state, the rate and extent of deuterium exchange depend on the level of D2O sorption, protein mobility (unfolding and refolding events) and the nature of the excipients present in the solid matrix. (B) In PL, UV irradiation at 365 nm initiates the formation of a reactive carbene intermediate from the diazirine functional group of pLeu and is inserted non-specifically into any X-H bond (X = any atom), or added across a C=C bond in its immediate vicinity. In the solid-state, the rate and extent of labeling depend on the local concentration of the labeling agent, irradiation time, protein structure and the nature of excipients present in the solid matrix. Panels A and B show the maximum theoretical labeling that can occur on backbone and side-chains respectively in protein.
Figure 1: ssHDX and ssPL measure protein structure in lyophilized solids through different labeling mechanisms. (A) In HDX, the backbone amide hydrogens exchange with deuterium as a function of protein structure and D2O accessibility. In the solid-state, the rate and extent of deuterium exchange depend on the level of D2O sorption, protein mobility (unfolding and refolding events) and the nature of the excipients present in the solid matrix. (B) In PL, UV irradiation at 365 nm initiates the formation of a reactive carbene intermediate from the diazirine functional group of pLeu and is inserted non-specifically into any X-H bond (X = any atom), or added across a C=C bond in its immediate vicinity. In the solid-state, the rate and extent of labeling depend on the local concentration of the labeling agent, irradiation time, protein structure and the nature of excipients present in the solid matrix. Panels A and B show the maximum theoretical labeling that can occur on backbone and side-chains respectively in protein.
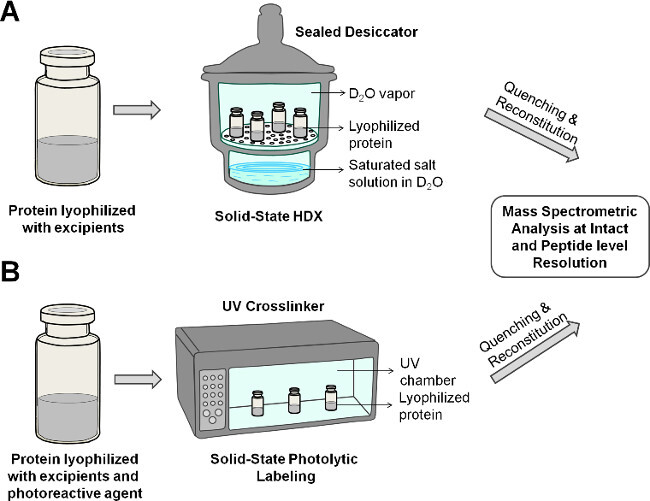 Figure 2: Schematic showing solid-state HDX-MS (A) and PL-MS (B) for protein in lyophilized formulation.
Figure 2: Schematic showing solid-state HDX-MS (A) and PL-MS (B) for protein in lyophilized formulation.
Protocol
1. Sample Preparation and Lyophilization
Dialyze the required volume of Mb stock solution against a suitable buffer and filter through a 0.22 µm sterile filter.
Prepare the required volume of excipients and photo-leucine (L-2-amino-4,4-azi-pentanoic acid; pLeu) stock solutions in suitable buffer. Filter the stock solutions through a 0.22 µm sterile filter.
Prepare formulations as shown in Table 1 using the stock solutions of protein, excipients, pLeu, and buffer.
Filter samples through a 0.22 µm sterile filter to remove any particles formed at step 1.3. Fill the samples separately as 0.2 ml into 2 ml glass vials. Use glass vials that are transparent to UV (365 nm) light in order to activate pLeu in the ssPL-MS studies.
- Load vials in a lyophilizer and initiate lyophilization by designing an appropriate lyophilization cycle.
- Here, freeze samples at -40 °C, followed by primary drying under vacuum (70 mTorr) at -35 °C for 12 hr and secondary drying at 25 °C for 12 hr. Other lyophilization cycles and drying methods (e.g., spray drying) may also be used.
Backfill the vials containing lyophilized samples with nitrogen before capping.
| Formulations | Composition (mg/ml) prior to lyophilization | ||||
| Mb | Trehalose | Sorbitol | pLeu c | Potassium, Phosphate, pH 7.4 | |
| MbT a | 1.7 | 3.4 | - | - | 0.4 |
| MbS a | 1.7 | - | 3.4 | - | 0.4 |
| MbT + pLeu b | 1.7 | 3.4 | - | 14.3 x 10-3 to 1.43 | 0.4 |
| MbS + pLeu b | 1.7 | - | 3.4 | 14.3 x 10-3 to 1.43 | 0.4 |
Table 1: Composition of lyophilized Mb formulations. a Formulations used for the ssHDX-MS study. b Formulations used for the ssPL-MS study. c L-2-amino-4,4-azipentanoic acid or photo-leucine (pLeu). pLeu at five different concentrations (14.3 x 10-3 to 1.43 mg/ml) corresponding to 1x, 10x, 20x, 50x and 100x molar excess relative to Mb were co-lyophilized with MbT and MbS formulations.
2. ssHDX-MS for Intact Protein
Add a saturating amount (~440 g) of K2CO3 to 200 ml of D2O previously placed in the lower compartment of a desiccator. Seal the desiccator air-tight and allow it to equilibrate at 5 °C until a stable relative humidity (RH) of ~43% is reached. Other RH values of interest can be obtained by selecting different saturated salt solutions23,24.
Initiate ssHDX reactions by placing uncapped vials containing the lyophilized protein in the upper compartment of the desiccator. Seal the desiccator air-tight and incubate at 5 °C to allow HDX to occur (Figure 2A).
Collect ssHDX samples at various times in triplicate. For Mb formulations, collect samples at nine time points 1, 2, 4, 8, 16, 32, 56, 92 and 144 hr.
Cap the vials immediately after withdrawing from the desiccator and quench the reactions by flash freezing the vials in liquid nitrogen. Store vials at -80 °C until mass spectrometric analysis.
Analyze samples using a suitable high resolution liquid chromatography-mass spectrometry (LC-MS) method. Design or purchase a suitable refrigerated LC system to minimize back-exchange during sample analysis. Use the setup of the column refrigeration unit and LC-MS method previously reported25. NOTE: Since the rate of amide proton exchange depends on pH and temperature, deuterons incorporated in the protein can exchange with hydrogen present in the mobile phase (“back exchange”), causing a loss of information. Although an acidic pH (pH 2.5) of quench buffer and HPLC solvents can minimize back-exchange to a large extent, reducing the temperature (≤0 °C) by means of a suitable column refrigeration system can further protect the protein from back-exchange.
Connect the sample loop and protein trap to the valve that automatically controls the desalting and elution process. Calibrate the mass spectrometer by injecting a TOF low concentration tuning mix into the mass spectrometer in the m/z range of 200-3,200. The immobilized pepsin column and analytical column are not required for intact protein analysis.
Set the temperature in the refrigerated system to ≤0 °C and wait for the system to reach a stable operating temperature of ~0 °C.
Quickly transfer the samples from -80 °C into liquid nitrogen for mass spectrometric analysis. Using forceps, carefully withdraw each vial from liquid nitrogen and reconstitute the sample by adding a specific volume of ice cold quench buffer containing 0.2% formic acid (FA) (pH 2.5) and 5% methanol in water to the vial.
Program a suitable HPLC and mass spectrometry method using the control software. For Mb formulations, desalt sample containing 20 pmol Mb in the protein trap for 1.7 min with 5% acetonitrile, 95% water and 0.1% formic acid (FA), and elute using a gradient increased to 80% acetonitrile, 20% water and 0.1% FA in 3.3 min. Collect mass spectra over the m/z range 200-3,200.
To determine the mass of intact protein, acquire data for an undeuterated protein sample (i.e., protein not subjected to ssHDX) in aqueous solution using the method of step 2.9.
Obtain the masses of undeuterated and deuterated samples by deconvoluting the raw spectra using data analysis software. Here, set the mass range to 15,000-18,000 Da, the mass resolution to 1.0 Da, and the peak height to 90% for calculating the mass of Mb.
Calculate the number of deuterons incorporated in the intact protein (here, Mb) by subtracting the mass of undeuterated protein from the mass of deuterated protein at each exchange time point.
Calculate the percent deuterium uptake relative to the theoretical maximum using the following equation (Equation 1)
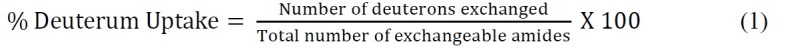 where total number of exchangeable amides = total number of amino acids – number of proline residues – 2 (“2” accounts for the N-terminal amino group and amide hydrogen that undergo rapid back-exchange).
where total number of exchangeable amides = total number of amino acids – number of proline residues – 2 (“2” accounts for the N-terminal amino group and amide hydrogen that undergo rapid back-exchange).Fit the ssHDX kinetic data using a suitable exponential equation. A biexponential equation (Equation 2) is usually the simplest that provides a reasonable fit to the ssHDX data. In this study, for MbT and MbS, fit the data to a biexponential model which assigns deuterons to “fast” and “slow” exchanging pools.
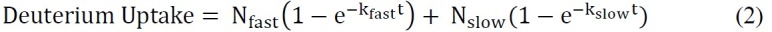 where Nfast and Nslow are the number of exchangeable amides in the “fast” and “slow” exchanging pools, respectively, and kfast and kslow are the first-order rate constants associated with the two pools.
where Nfast and Nslow are the number of exchangeable amides in the “fast” and “slow” exchanging pools, respectively, and kfast and kslow are the first-order rate constants associated with the two pools.
3. ssHDX-MS for Protein at Peptide Level
Perform ssHDX by following steps 2.1 to 2.8, with the following modifications in step 2.6. Connect the immobilized pepsin column and analytical column to the valve as reported previously25 and replace the protein trap connected to the valve with a peptide trap. Calibrate the mass spectrometer by setting the mass to charge ratio ranging from 100 to 1,700. NOTE: During the reconstitution step (step 2.8), a reducing agent and denaturing agent can be included in the quench buffer to facilitate pepsin digestion of proteins with disulfide bonds (e.g., monoclonal antibodies).
Program an appropriate HPLC and mass spectrometry method using the control software. For Mb formulations, digest samples containing 20 pmol Mb online with 0.1% FA, trap and desalt peptides for 1.7 min with 10% acetonitrile, 90% water and 0.1% FA in a peptide trap. Elute the fragments onto the analytical column with a gradient increase to 60% acetonitrile, 40% water and 0.1% FA in 4.0 min. Acquire mass spectra over the m/z range 100−1,700.
Identify the peptic fragments by MS/MS analysis of an undeuterated protein sample. Use mass spectrometry software to compare experimental masses of peptide fragment ions to the predicted masses of peptide fragments in a custom database. Set a mass cut-off point (e.g. 10 ppm) to identify masses with low error. For peptides matched, prepare a list comprised of (i) peptide sequence, (ii) charge state, and (iii) retention time.
Use the list generated in step 3.3 to map and determine the average number of deuterons incorporated for each pepsin digest fragment. This can be achieved by employing suitable HDX-MS data analysis software24.
To calculate the percent deuterium uptake and to fit the ssHDX kinetic data for each of the peptic fragments, follow steps 2.13 and 2.14. In this study, HDX kinetic data for each of six non-redundant pepsin digest fragments from MbT and MbS formulations were fitted to a biexponential association model (Equation 2).
4. ssPL-MS for Intact Protein
To begin the photolytic labeling reaction, first switch on the UV crosslinker and allow the lamps to warm up for 5 min. Make sure the UV source is equipped with lamps of wavelength 365 nm to activate the diazirine group of pLeu. CAUTION: Never open the door of the UV crosslinker when the lamps are on. Protect eyes and skin from exposure to UV light if the source is not enclosed by a UV-protective glass door.
Switch off the UV crosslinker before opening the door. Once the lamps are turned off, uncap the vials containing the lyophilized formulation and place them inside the UV crosslinker chamber as shown in Figure 2B. Irradiate the samples with UV light for 40 min.
Perform control experiments by following step 4.1 to 4.3 for (i) samples lyophilized without pLeu and (ii) samples lyophilized with pLeu reconstituted in water.
Cap and store the vials at -20 °C until mass spectrometric analysis.
Reconstitute the solid samples by adding a suitable volume of mass spectrometry grade distilled water to bring the concentration to 2 µM.
To begin sample analysis, follow steps 2.6 and 2.9. NOTE: Since back-exchange is not an issue with covalent labeling, ssPL-MS does not require a special refrigerated LC system.
To determine the native mass of the protein, acquire data for a protein sample that has not been subjected to ssPL by following step 2.9. Obtain the masses of unlabeled and labeled samples by deconvoluting the raw spectra as explained in step 2.11.
Calculate the number of pLeu incorporated using the following formula:
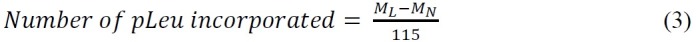 where ML is the mass of labeled protein, MN is the mass of native protein and 115 is the average mass (Da) added to native protein following single pLeu incorporation. Note that the labeling reaction occurs with the loss of N2 (28 Da). The monoisotopic mass of pLeu is 143.07.
where ML is the mass of labeled protein, MN is the mass of native protein and 115 is the average mass (Da) added to native protein following single pLeu incorporation. Note that the labeling reaction occurs with the loss of N2 (28 Da). The monoisotopic mass of pLeu is 143.07.Calculate the percentages of protein populations with different numbers of labels using peak heights from the extracted ion chromatograms.
 where “i” denotes the number of labels, PHi denotes the peak height for labeled protein Li and PHu denotes the peak height of the unlabeled protein as observed by mass spectrometry.
where “i” denotes the number of labels, PHi denotes the peak height for labeled protein Li and PHu denotes the peak height of the unlabeled protein as observed by mass spectrometry.
5. ssPL-MS for Protein at the Peptide Level
Perform ssPL by following steps 4.1 to 4.4.
For peptide level analysis, reconstitute solid samples in ammonium bicarbonate buffer (100 mM, pH 8.0).
After reconstitution, mix the labeled protein solution with trypsin at 10:1 molar ratio of protein to trypsin and incubate at 60 °C for 16 hr.
Quench the reaction by adding 0.1% FA in water to the sample to yield final concentration to 2 µM protein.
Connect the sample loop, peptide trap and analytical column to the valve linked to the HPLC system.
Program a suitable HPLC and mass spectrometry method using the control software. For Mb formulations, inject 20 pmol of the digested protein into the sample loop, and desalt the peptides in a peptide trap for 1.5 min with 5% acetonitrile, 95% water and 0.1% FA, followed by an elution in the analytical column with gradient increase to 55% acetonitrile, 45% water and 0.1% FA in 22 min. Collect mass spectra over the m/z range 100-1,700.
Prepare a theoretical mass list for peptide-pLeu adducts using an online tool such as ExPASy26 with numbers of pLeu previously calculated from the intact protein analysis. Include at least 4 missed cleavages. Note that the labeling reaction occurs with the loss of N2. Therefore, the mass of peptide-pLeu adduct = mass of unlabeled tryptic peptide + n (mass of pLeu) – n (mass of N2), where “n” is the number of pLeu incorporated. NOTE: If the mass analysis of intact protein showed up to three labeled populations of protein, consider up to three possible pLeu labels per peptide. For a peptide with mass of 1,000 Da, the theoretical mass of peptide-pLeu adduct with 1 pLeu incorporation would be 1,000 + 1(143) - 1(28) = 1,115 Da. Similarly, the theoretical masses of peptide-pLeu adducts with 2 pLeu, 3 pLeu, etc. would be 1,230 Da, 1,345 Da, etc., respectively.
Use the mass analysis software to match the theoretical mass list generated in step 5.7 with the masses observed experimentally. Set a mass cut-off point (e.g., 50 ppm) to identify masses with low error.
For peptides matched, determine the actual number of pLeu incorporated using the formula under step 4.8 (Equation 3), where ML and MN are the masses of labeled and native peptide, respectively.
Representative Results
Here, ssHDX-MS and ssPL-MS have been used to study the effect of excipients on the conformation and solid-state interactions of lyophilized Mb formations. The concentrations of protein and excipients used in this study are given in Table 1. Representative results from the ssHDX-MS and ssPL-MS analysis of lyophilized Mb obtained by following the above protocols are presented.
Deuterium uptake at intact protein level
ssHDX-MS is able to distinguish between Mb formulations at intact level. The deconvoluted mass spectra of intact Mb following 144 hr of ssHDX from formulation MbS showed greater deuterium uptake than formulation MbT (Figure 3A). On an average, MbS showed 46% greater deuterium uptake than MbT (Table 2).
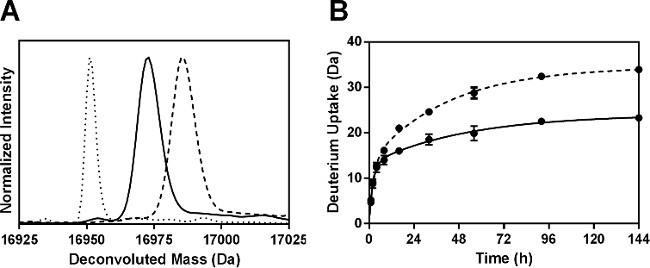 Figure 3: ssHDX-MS for intact Mb: (A) Deconvoluted mass spectra of deuterated intact Mb from formulations MbT (solid line) and MbS (dashed line) following 144 hr of ssHDX. The deconvoluted mass spectrum of undeuterated intact Mb is also shown (dotted line). (B) ssHDX kinetics for intact Mb in formulations MbT (solid line) and MbS (dashed line). The time course of ssHDX was fitted to an equation for two phase exponential association using Graph Pad Prism software version 5 (n = 3, ± SD).
Figure 3: ssHDX-MS for intact Mb: (A) Deconvoluted mass spectra of deuterated intact Mb from formulations MbT (solid line) and MbS (dashed line) following 144 hr of ssHDX. The deconvoluted mass spectrum of undeuterated intact Mb is also shown (dotted line). (B) ssHDX kinetics for intact Mb in formulations MbT (solid line) and MbS (dashed line). The time course of ssHDX was fitted to an equation for two phase exponential association using Graph Pad Prism software version 5 (n = 3, ± SD).
The deuteration kinetics for intact MbS and MbT are similar at early time points (1-4 hr), but MbS showed increased deuterium exchange with increase in time (8-144 hr) (Figure 3B). This suggests the importance of selecting longer time points for ssHDX at lower RH and temperature conditions. Also, the D2O sorption and diffusion process may affect the rate of ssHDX at the early time points. Our previous studies have shown that moisture sorption in ssHDX is complete in a period of hours, and has minimal contribution to exchange kinetics beyond this time. The observed rate and extent of exchange therefore are not simply measures of D2O adsorbtion27,28. The small error bars in Figure 3B, indicating standard deviations from three independent ssHDX-MS samples, show that the experiment is highly reproducible.
| Deuterium Uptake (%) b | Nfast c | kfast c | Nslow c | kslow c | |
| MbT a | 15.9 ± 0.5 | 13.1 (0.8) | 0.43 (0.03) | 11.0 (0.9) | 0.019 (0.001) |
| MbS a | 23.2 ± 0.5 | 15.4 (0.7) | 0.49 (0.04) | 19.2 (0.6) | 0.024 (0.002) |
| % change d | 46% | 18% | 14% | 75% | 26% |
Table 2: Quantitative measures of deuterium uptake in ssHDX studies of Mb formulations. a See Table 1 for composition. b Percent deuterium uptake relative to theoretical maximum by intact Mb after 144 hr of HDX at 5 °C, 43% RH (n = 3, mean ± SD). c Parameters determined by nonlinear regression of ssHDX-MS kinetic data. Time course of deuterium exchange for intact Mb was fitted to a biexponential association model (Eqn. 2). Values in parentheses are standard errors of the regression parameters. d The percent change in measurements were calculated as 100 x [(value from MbS – value from MbT) / (value from MbT)].
The regression parameters (Nfast, Nslow, kfast and kslow) for deuterium uptake kinetics for MbT and MbS are given in Table 2. Though the Nfast and Nslow values are larger for MbS than MbT, differences in the Nslow values were greater than differences in the Nfast values. Specifically, the Nfast value is only 18% greater in MbS than in MbT, whereas the Nslow value is 75% greater in MbS than in MbT. This suggests that the smaller Nslow values in MbT may be due to the higher retention of Mb structure or protection of amide groups by excipients that are exposed to D2O in MbS. However, the detailed mechanisms are not clearly understood. The rate constants (kfast and kslow) for both formulations are very similar.
Deuterium uptake at the peptide level
Following pepsin digestion, a total of 52 peptides were identified. Six non-redundant fragments corresponding to 100% of the Mb sequence were used for the analysis reported here. Additional information can be obtained by using overlapping fragments, as reported by our group previously24. The percent deuterium uptake for each peptide was calculated and the results from 144 hr samples plotted (Figure 4A). HDX kinetics for the six peptic fragments showed biexponential behavior (Figure 4B), consistent with subpopulations of amide hydrogens undergoing “fast” and “slow” exchange.
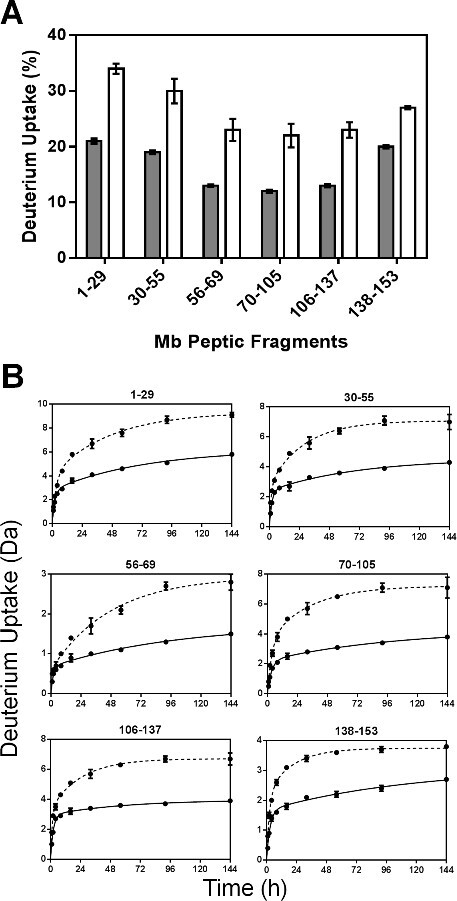 Figure 4: ssHDX-MS for Mb at the peptide level: (A) Percent deuterium uptake for 6 non-redundant peptic fragments from Mb in formulations MbT (gray) and MbS (white) following 144 hr of HDX. (B) ssHDX kinetics for six non-redundant peptic fragments from Mb in formulations MbT (solid line) and MbS (dashed line). The time course of ssHDX was fitted to an equation for two phase exponential association using Graph Pad Prism software version 5 (n = 3, ± SD).
Figure 4: ssHDX-MS for Mb at the peptide level: (A) Percent deuterium uptake for 6 non-redundant peptic fragments from Mb in formulations MbT (gray) and MbS (white) following 144 hr of HDX. (B) ssHDX kinetics for six non-redundant peptic fragments from Mb in formulations MbT (solid line) and MbS (dashed line). The time course of ssHDX was fitted to an equation for two phase exponential association using Graph Pad Prism software version 5 (n = 3, ± SD).
Regression parameters for the nonredundant peptides are presented in Figure 5. As the fitted rate constants for peptide fragments are not the average rate constant for individual amides, the observed rate constants for the peptic fragments cannot be linearly related to those for the intact protein. The Nfast values for most of the peptic fragments (except fragment 56-69) in formulations MbS were slightly greater than those in MbT (Figure 5A). Similarly, the kfast values generally showed little difference between formulations and in different regions of the Mb molecule (Figure 5B). However, the Nslow and kslow values for MbS are significantly greater in all fragments than for MbT (Figure 5C and 5D). The considerable increase in Nslow and kslow for MbS may reflect greater mobility of amide groups in the “slow” exchanging pools.
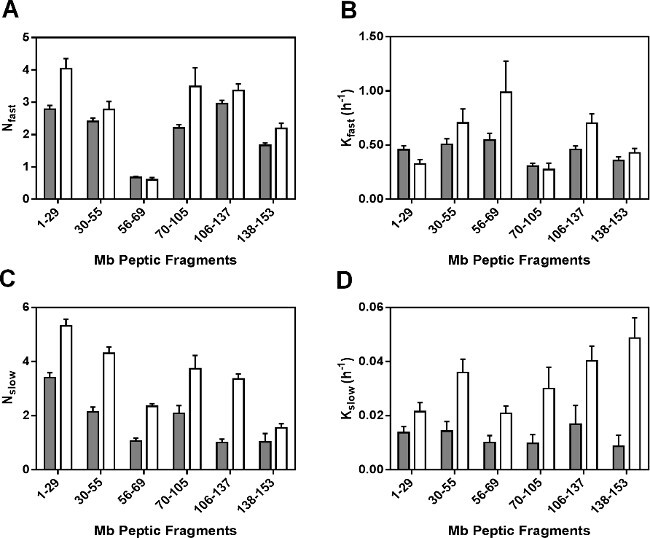 Figure 5: ssHDX kinetic parameters for Mb peptic peptides: Nfast (A), kfast (B), Nslow (C) and kslow (D) values obtained from nonlinear regression of ssHDX-MS kinetic data for six non-redundant peptic peptides from Mb in formulations MbT (gray) and MbS (white) (n = 3, ± SE).
Figure 5: ssHDX kinetic parameters for Mb peptic peptides: Nfast (A), kfast (B), Nslow (C) and kslow (D) values obtained from nonlinear regression of ssHDX-MS kinetic data for six non-redundant peptic peptides from Mb in formulations MbT (gray) and MbS (white) (n = 3, ± SE).
Photolytic labeling at intact protein level
Mb irradiated in the presence of 20x excess pLeu formed multiple Mb-pLeu adducts, as detected by LC-MS (Figure 6A). The deconvoluted spectra for MbT irradiated for 40 min with 20x pLeu showed up to 3 labels with the addition of +115, +230 and +345 Da to the mass of unlabeled Mb. MbS irradiated similarly with 20x pLeu showed less pLeu uptake at the intact level, with up to 2 labeled populations detected by LC-MS.
 Figure 6: ssPL-MS for intact Mb: (A) Deconvoluted mass spectra for MbT (solid line) and MbS (dashed line) labeled with 20x excess (5% w/w) pLeu. Deconvoluted mass spectrum of native Mb (Mb lyophilized and irradiated in the absence of pLeu) is shown as the dotted line. U denotes a population of protein that remains unlabeled after irradiation. Populations of protein carrying 1, 2 and 3 pLeu labels are represented as 1L, 2L and 3L respectively. (B) ssPL-MS kinetics for intact Mb in formulations MbT (closed circles) and MbS (open circles) as a function of pLeu concentration. All samples were irradiated for 40 min. Error bars are within the symbols. (C) ssPL-MS kinetics for intact Mb in formulations MbT (closed circles) and MbS (open circles) lyophilized and irradiated in the presence of 100x excess pLeu (20.7 % w/w) as a function of irradiation time. Error bars are within the symbols.
Figure 6: ssPL-MS for intact Mb: (A) Deconvoluted mass spectra for MbT (solid line) and MbS (dashed line) labeled with 20x excess (5% w/w) pLeu. Deconvoluted mass spectrum of native Mb (Mb lyophilized and irradiated in the absence of pLeu) is shown as the dotted line. U denotes a population of protein that remains unlabeled after irradiation. Populations of protein carrying 1, 2 and 3 pLeu labels are represented as 1L, 2L and 3L respectively. (B) ssPL-MS kinetics for intact Mb in formulations MbT (closed circles) and MbS (open circles) as a function of pLeu concentration. All samples were irradiated for 40 min. Error bars are within the symbols. (C) ssPL-MS kinetics for intact Mb in formulations MbT (closed circles) and MbS (open circles) lyophilized and irradiated in the presence of 100x excess pLeu (20.7 % w/w) as a function of irradiation time. Error bars are within the symbols.
In kinetic studies, the percent of labeled protein increased exponentially for both MbT and MbS with increasing irradiation time (Figure 6B). MbS showed less pLeu uptake than MbT at every irradiation time. Both formulations appeared to reach a plateau at 40 min. Thus, a kinetic study can be useful to determine the duration of irradiation needed to obtain complete pLeu activation. Labeling kinetics were also studied as a function of pLeu concentration (Figure 6C). The percent of labeled protein increased with pLeu concentration for both MbT and MbS. However, at 20.7% w/w pLeu, MbT showed a decrease in pLeu uptake. This may be due to exclusion of pLeu from the surface of the protein at high pLeu concentration. Hence, a study with varying concentration of pLeu should be performed to select the appropriate pLeu concentration that allows for sufficient labeling across the protein surface without surface exclusion. In this study, 20x excess pLeu was selected for further peptide-level studies.
The overall decreased labeling observed for MbS suggests poor side-chain accessibility to the matrix containing pLeu. This is consistent with a conformational change in the presence of sorbitol that results in reduced labeling.
Photolytic labeling at the peptide level
Based on the intact protein labeling studies, 20x excess pLeu was selected to compare MbT and MbS at the peptide level. Labeled samples were digested with trypsin and analyzed by LC-MS. A total of 40 peptides corresponding to 100% of the Mb sequence were detected for MbT and MbS samples. In some cases, tryptic digestion may provide limited protein sequence coverage if Lys and/or Arg residues are heavily labeled. To improve sequence coverage, a mixture of trypsin and chymotrypsin can be used to digest the labeled protein.
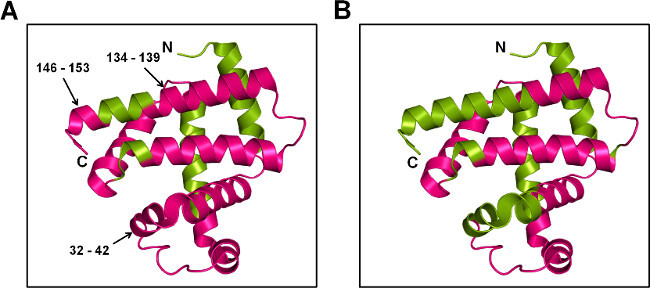 Figure 7: ssPL-MS for Mb at peptide level: Cartoon representation of Mb labeled with 20x excess pLeu (5% w/w) in the presence of trehalose (A) and sorbitol (B). The labeled protein was digested with trypsin and labeled peptides were mapped on to the crystal structure of Mb (PDB ID 1WLA). The labeled and unlabeled regions are colored magenta and green, respectively.
Figure 7: ssPL-MS for Mb at peptide level: Cartoon representation of Mb labeled with 20x excess pLeu (5% w/w) in the presence of trehalose (A) and sorbitol (B). The labeled protein was digested with trypsin and labeled peptides were mapped on to the crystal structure of Mb (PDB ID 1WLA). The labeled and unlabeled regions are colored magenta and green, respectively.
ssPL-MS with trypsin digestion provides qualitative information about the peptides being labeled. Given the different labeled populations at the intact level, the promiscuous mechanism of pLeu labeling and differences in ionization efficiencies of labeled and unlabeled peptides, it is difficult to obtain quantitative metrics for ssPL-MS after digestion. However, the qualitative information can still provide insight into protein conformational changes at the peptide level. In this study, both MbT and MbS formulations showed pLeu uptake across most of the protein surface. When compared to MbS, peptide fragments 32-42, 134-139 and 146-153 from MbT showed pLeu labeling (Figure 7). This suggests that the side-chains of these amino acids are exposed to pLeu, as the helices in these regions are intact in the MbT matrix. In contrast, protection from pLeu labeling in the MbS matrix is consistent with structural perturbations in these regions.
Overall, the results from ssHDX-MS and ssPL-MS suggest that the methods can provide complementary high-resolution peptide-level information about backbone (ssHDX-MS) and side-chain (ssPL-MS) exposure and excipient effects in lyophilized protein formulations.
Discussion
Several studies have suggested that the local environment in lyophilized samples affects protein degradation5,29,30. However, establishing a direct relationship between protein structure and stability in the solid state has not been feasible due to the lack of high-resolution analytical methods. The application of existing high resolution methods such as HDX and PL to lyophilized powders requires modification of solution protocols and careful data interpretation. HDX-MS and PL-MS have been successively adopted to monitor protein conformations in the solid-state. The results presented here and elsewhere27,28,31-33 have demonstrated the ability of these methods to monitor protein conformation with high resolution in the solid environment. Though the critical steps in data analysis do not vary from labeling in solution34-36, important considerations during experimental setup and data interpretation are required for solid-state chemical labeling.
Selection of the labeling reagent must be based on size and mechanism of labeling. The small size of deuterium allows the peptide backbone to be probed easily, whereas the relatively larger size of pLeu limits labeling to the side-chains. Both ssHDX and ssPL show no preference for any amino acid, so that labeling depends only on backbone and side-chain exposure to the matrix. To effectively probe protein conformations in solid-state, the external factors affecting the labeling process must be carefully controlled. The total amount and the spatial distribution of labeling agent in lyophilized solids is different from aqueous solutions.
In ssHDX, the amount of D2O in the solid matrix may affect the rate of protein unfolding (or partial unfolding), refolding, and deuterium exchange. This is not the case with solution HDX, in which the protein sample is normally diluted with an ample volume of D2O. Careful screening of the effects of hydration on the ssHDX rate can inform the selection of ideal RH conditions. To control the rate of moisture sorption and avoid collapse of the powder in formulations containing hygroscopic excipients (e.g. sucrose and trehalose), ssHDX may be carried out under refrigerated conditions (2-8 °C). Our previous study on hydration effects showed increased rate and extent of exchange with increase in moisture content, as expected. In much of our work, an intermediate RH of 43% at 5 °C has proven to be ideal to distinguish formulations in a reasonable time24. The reaction is usually carried out until a plateau is reached. This ensures that moisture sorption and diffusion into the solid do not control the HDX rate. The use of small solid sample sizes with pre-lyophilization volume of ≤2 ml also helps to ensure that D2O vapor sorption is essentially complete early in the exchange period. Though ssHDX-MS provides quantitative information on the conformation of protein in solid-state, there are certain conditions where interpretation of data cannot be entirely based on the ssHDX study alone. It is possible that the decreased deuterium uptake observed in a sample (when compared to control) may be due to the higher retention of protein structure or the significant amount of protein aggregates present in the sample. In such case, interpretation of ssHDX data requires results from other complementary methods. Peak broadening in deuterated mass spectra was observed for several Mb formulations27,28. This could be due to various factors like the presence of partially unfolded protein population, spatial heterogeneity in the sample, or the spatial gradients in the D2O concentration. However, these factors were not distinguished in ssHDX-MS and needs further investigation.
As ssPL-MS is relatively new when compared to other methods, continuous learning about its applications and limitations is required. In ssPL, the photo-cross-linker is lyophilized with the protein. The lack of moisture limits the mobility of components within the solid matrix, and the partial structural relaxation that may occur with moisture sorption in ssHDX is not a phenomenon in ssPL. This limits labeling in ssPL to the immediate vicinity of the photo-cross-linker. However, unlike HDX-MS, MS/MS analysis of the covalently labeled protein can provide residue-level structural information. Since ssPL labeling is covalent and irreversible, back exchange does not occur and samples can be prepared and analyzed without concern for loss of label. To facilitate diffusion of labeling agent and improve labeling efficiency in solid-matrix, ssPL may be performed with increasing % RH. pLeu uptake can also be improved by increasing the concentration of photoreactive agent. The molar ratio of protein to pLeu can be varied as desired. In general, a 100x molar excess of pLeu to protein will ensure adequate labeling. However, high pLeu concentration may result in loss of protein tertiary structure in the solid matrix. Hence, In addition to labeling kinetics and formulation composition, selection of pLeu concentration must also be based on maintaining protein structural integrity. As pLeu nonselectively labels X-H (where X = C, N, O) group, it is possible that excipients with similar labeling sites can greatly affect the level of protein labeling. The interference of excipients in the availability of pLeu for protein labeling is yet to be characterized. It is known that the carbene generated from diazirine activation is not residue-specific, however one study reports bias towards Asp and Glu36. While it is good to learn about residue-specific interactions, peptide-level information is also useful and can be used to design excipients to block regions with high matrix exposure in the solid state. ssPL-MS provides detailed qualitative information, however quantitative data needs to be obtained and robust metrics need to be developed to analyze formulation differences across a variety of lyophilized systems.
The use of a residue-specific label combined with MS/MS analysis can further enhance resolution to the amino-acid level. Labeling reagents such as 2,3-butanedione to label Arg, N-hydroxysuccinimide derivatives for Lys and N-alkylmaleimide derivatives for Cys can be used to precisely map molecular interactions in lyophilized powder. However these reagents are pH-dependent and the reactions may not be as well-controlled as photolytic labeling in solid-state. An alternate approach is to incorporate the photo-cross-linker into the protein sequence with the use of auxotrophic cell lines, site-directed mutagenesis or side chain derivatization.
Our previous ssHDX-MS and ssPL-MS studies have shown that labeling of protein depends on the nature and amount of excipients used24,27,28,31-33,37,38. ssHDX-MS of Mb co-lyophilized with guanidine hydrochloride (Gdn.HCl) showed greater deuterium uptake than Mb co-lyophilized with low-molecular-weight sugars32. In a separate ssPL-MS study, Mb co-lyophilized with Gdn.HCl showed greater protection from photolytic labeling than Mb with sucrose33. Further, quantitative measurements from ssHDX-MS have been highly correlated with the stability of protein during long-term storage28. These studies suggest that ssHDX or ssPL of protein reflects the extent of structural retention of the protein in lyophilized powder. We believe that the retention of secondary structure in lyophilized powders provides favorable environment for side chain labeling with pLeu and protection of amide hydrogen from deuterium exchange. However, detailed comparison of the informative content from these methods needs to be performed in the future. Though establishing the utility of ssHDX-MS and ssPL-MS as a formulation screening tool will ultimately require that it be applied to many proteins, results from our recent studies supports its wider adoption. With further development, these methods are expected to be widely useful for characterizing solid-state protein formulations in the biopharmaceutical industry.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors gratefully acknowledge financial support from NIH R01 GM085293 (PI: E. M. Topp) and from the College of Pharmacy at Purdue University.
References
- Lawrence S. Billion dollar babies--biotech drugs as blockbusters. Nat. Biotechnol. 2007;25(4):380–382. doi: 10.1038/nbt0407-380. [DOI] [PubMed] [Google Scholar]
- Global Markets and Manufacturing Technologies for Protein Drugs. BCC Research; 2013. p. BIO021D. [Google Scholar]
- Lai MC, Topp EM. Solid-state chemical stability of proteins and peptides. J. Pharm. Sci. 1999;88(5):489–500. doi: 10.1021/js980374e. [DOI] [PubMed] [Google Scholar]
- Carpenter JF, Pikal MJ, Chang BS, Randolph TW. Rational design of stable lyophilized protein formulations: some practical advice. Pharm. Res. 1997;14(8):969–975. doi: 10.1023/a:1012180707283. [DOI] [PubMed] [Google Scholar]
- Carpenter JF, Chang BS, Garzon-Rodriguez W, Randolph TW. Rational design of stable lyophilized protein formulations: theory and practice. Pharm. Biotechnol. 2002;13:109–133. doi: 10.1007/978-1-4615-0557-0_5. [DOI] [PubMed] [Google Scholar]
- Wüthrich K. Protein structure determination in solution by NMR spectroscopy. J. Biol. Chem. 1990;265(36):22059–22062. [PubMed] [Google Scholar]
- Ilari A, Savino C. Protein structure determination by x-ray crystallography. Methods. Mol. Biol. 2008;452:63–87. doi: 10.1007/978-1-60327-159-2_3. [DOI] [PubMed] [Google Scholar]
- Brunger AT. X-ray crystallography and NMR reveal complementary views of structure and dynamics. Nat. Struct. Biol. 1997;4:862–865. [PubMed] [Google Scholar]
- Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv Drug. Deliv. Rev. 2001;48(1):27–42. doi: 10.1016/s0169-409x(01)00098-9. [DOI] [PubMed] [Google Scholar]
- Manning MC. Use of infrared spectroscopy to monitor protein structure and stability. Expert. Rev. Proteomics. 2005;2(5):731–743. doi: 10.1586/14789450.2.5.731. [DOI] [PubMed] [Google Scholar]
- Grohganz H, Gildemyn D, Skibsted E, Flink JM, Rantanen J. Rapid solid-state analysis of freeze-dried protein formulations using NIR and Raman spectroscopies. J. Pharm. Sci. 2011;100(7):2871–2875. doi: 10.1002/jps.22490. [DOI] [PubMed] [Google Scholar]
- Bai S, Nayar R, Carpenter JF, Manning MC. Noninvasive determination of protein conformation in the solid state using near infrared (NIR) spectroscopy. J. Pharm. Sci. 2005;94(9):2030–2038. doi: 10.1002/jps.20416. [DOI] [PubMed] [Google Scholar]
- Pikal MJ, et al. Solid state chemistry of proteins: II. The correlation of storage stability of freeze-dried human growth hormone (hGH) with structure and dynamics in the glassy solid. J. Pharm. Sci. 2008;97(12):5106–5121. doi: 10.1002/jps.21374. [DOI] [PubMed] [Google Scholar]
- Wang B, Tchessalov S, Cicerone MT, Warne NW, Pikal MJ. Impact of sucrose level on storage stability of proteins in freeze-dried solids: II. Correlation of aggregation rate with protein structure and molecular mobility. J. Pharm. Sci. 2009;98(9):3145–3166. doi: 10.1002/jps.21622. [DOI] [PubMed] [Google Scholar]
- Schule S, Friess W, Bechtold-Peters K, Garidel P. Conformational analysis of protein secondary structure during spray-drying of antibody/mannitol formulations. Eur. J. Pharm. Biopharm. 2007;65(1):1–9. doi: 10.1016/j.ejpb.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Baerga-Ortiz A, Hughes CA, Mandell JG, Komives EA. Epitope mapping of a monoclonal antibody against human thrombin by H/D-exchange mass spectrometry reveals selection of a diverse sequence in a highly conserved protein. Protein. Sci. 2002;11(6):1300–1308. doi: 10.1110/ps.4670102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coales SJ, Tuske SJ, Tomasso JC, Hamuro Y. Epitope mapping by amide hydrogen/deuterium exchange coupled with immobilization of antibody, on-line proteolysis, liquid chromatography and mass spectrometry. Rapid. Commun. Mass. Spectrom. 2009;23(5):639–647. doi: 10.1002/rcm.3921. [DOI] [PubMed] [Google Scholar]
- Pacholarz KJ, Garlish RA, Taylor RJ, Barran PE. Mass spectrometry based tools to investigate protein-ligand interactions for drug discovery. Chem. Soc. Rev. 2012;41(11):4335–4355. doi: 10.1039/c2cs35035a. [DOI] [PubMed] [Google Scholar]
- Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol. Cell. Proteomics. 2010;9(8):1716–1728. doi: 10.1074/mcp.M900540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D, Berkowitz SA, Engen JR. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 2011;100(6):2071–2086. doi: 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman G, Prestwich GD. Using photolabile ligands in drug discovery and development. Trends. Biotechnol. 2000;18(2):64–77. doi: 10.1016/s0167-7799(99)01402-x. [DOI] [PubMed] [Google Scholar]
- Robinette D, Neamati N, Tomer KB, Borchers CH. Photoaffinity labeling combined with mass spectrometric approaches as a tool for structural proteomics. Expert. Rev. Proteomics. 2006;3(4):399–408. doi: 10.1586/14789450.3.4.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan L. Humidity fixed points of binary saturated aqueous solutions. Journal of Research of the National Bureau of Standards. 1977;81A(1):8. [Google Scholar]
- Sophocleous AM, Zhang J, Topp EM. Localized hydration in lyophilized myoglobin by hydrogen-deuterium exchange mass spectrometry. 1. Exchange mapping. Mol. Pharm. 2012;9(4):718–726. doi: 10.1021/mp3000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel TR, Jacques ME, Young RW, Ratzlaff KL, Weis DD. An efficient and inexpensive refrigerated LC system for H/D exchange mass spectrometry. J. Am. Soc. Mass. Spectrom. 2011;22(8):1472–1476. doi: 10.1007/s13361-011-0152-6. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, et al. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic. Acids. Res. 2003;31(13):3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sophocleous AM, Topp EM. Localized hydration in lyophilized myoglobin by hydrogen-deuterium exchange mass spectrometry. 2. Exchange kinetics. Mol. Pharm. 2012;9(4):727–733. doi: 10.1021/mp2004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy BS, Schultz SG, Kim SG, Topp EM. Predicting Protein Aggregation during Storage in Lyophilized Solids Using Solid State Amide Hydrogen/Deuterium Exchange with Mass Spectrometric Analysis (ssHDX-MS) Mol. Pharm. 2014;11(6):1869–1879. doi: 10.1021/mp500005v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000;203(1-2):1–60. doi: 10.1016/s0378-5173(00)00423-3. [DOI] [PubMed] [Google Scholar]
- Sarciaux JM, Mansour S, Hageman MJ, Nail SL. Effects of buffer composition and processing conditions on aggregation of bovine IgG during freeze-drying. J. Pharm. Sci. 1999;88(12):1354–1361. doi: 10.1021/js980383n. [DOI] [PubMed] [Google Scholar]
- Li Y, Williams TD, Schowen RL, Topp EM. Characterizing protein structure in amorphous solids using hydrogen/deuterium exchange with mass spectrometry. Anal. Biochem. 2007;366(1):18–28. doi: 10.1016/j.ab.2007.03.041. [DOI] [PubMed] [Google Scholar]
- Sinha S, Li Y, Williams TD, Topp EM. Protein conformation in amorphous solids by FTIR and by hydrogen/deuterium exchange with mass spectrometry. Biophys. J. 2008;95(12):5951–5961. doi: 10.1529/biophysj.108.139899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LK, Moorthy BS, Topp EM. Photolytic labeling to probe molecular interactions in lyophilized powders. Mol. Pharm. 2013;10(12):4629–4639. doi: 10.1021/mp4004332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze N, Mayer MP. Analyzing protein dynamics using hydrogen exchange mass spectrometry. J. Vis. Exp. 2013. p. e50839. [DOI] [PMC free article] [PubMed]
- Kaltashov IA, Bobst CE, Abzalimov RR. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: is there a need for a top-down approach. Anal. Chem. 2009;81(19):7892–7899. doi: 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper CC, Schriemer DC. Mass spectrometry of laser-initiated carbene reactions for protein topographic analysis. Anal. Chem. 2011;83(8):2913–2920. doi: 10.1021/ac102655f. [DOI] [PubMed] [Google Scholar]
- Li Y, Williams TD, Topp EM. Effects of excipients on protein conformation in lyophilized solids by hydrogen/deuterium exchange mass spectrometry. Pharm. Res. 2008;25(2):259–267. doi: 10.1007/s11095-007-9365-6. [DOI] [PubMed] [Google Scholar]
- Li Y, Williams TD, Schowen RL, Topp EM. Trehalose and calcium exert site-specific effects on calmodulin conformation in amorphous solids. Biotechnol. Bioeng. 2007;97(6):1650–1653. doi: 10.1002/bit.21362. [DOI] [PubMed] [Google Scholar]