

Abstract
Background
Congenital heart diseases (CHDs) usually refer to abnormalities in the structure and/or function of the heart that arise before birth. GATA4 plays an important role in embryonic heart development, hence the aim of this study was to find the association of GATA4 mutations with CHD among the south Indian CHD patients.
Method
GATA4 gene was sequenced in 100 CHD patients (ASD, VSD, TOF and SV) and 200 controls. Functional significance of the observed GATA4 mutations was analyzed using PolyPhen, SIFT, PMut, Plink, Haploview, ESE finder 3.0 and CONSITE.
Results
We observed a total of 19 mutations, of which, one was in 5′ UTR, 10 in intronic regions, 3 in coding regions and 5 in 3′ UTR. Of the above mutations, one was associated with Atrial Septal Defect (ASD), two were found to be associated with Tetralogy of Fallot (TOF) and three (rs804280, rs4841587 and rs4841588) were strongly associated with Ventricular Septal Defect (VSD). Interestingly, one promoter mutation (−490 to 100 bp) i.e., 620 C>T (rs61277615, p-value = 0.008514), one splice junction mutation (G>A rs73203482; p-value = 9.6e-3, OR = 6.508) and one intronic mutation rs4841587 (p-value = 4.6e-3, OR = 4.758) were the most significant findings of this study. In silico analysis also proves that some of the mutations reported above are pathogenic.
Conclusion
The present study found that GATA4 genetic variations are associated with ASD, TOF and VSD in South Indian patients. In silico analysis provides further evidence that some of the observed mutations are pathogenic.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-015-0152-7) contains supplementary material, which is available to authorized users.
Keywords: Congenital heart disease, GATA4, Mutation, South Indian patients, ASD, TOF, VSD
Background
Congenital heart disease (CHD) usually refers to abnormalities in the structure and/or function of the heart that arise before birth. It has been estimated that about 1% and 6-8% of the newborns are affected with CHD in United States and worldwide, respectively [1]. Most of the studies conducted earlier reported that CHD has multi-factorial etiology, but recent studies indicate monogenic etiologies for a subset of CHDs [2-4]. Mutations in genes encoding transcription factor were found to cause nonsyndromic, human congenital heart disease. The knockout and transgenic mouse studies demonstrated that transcription factors play an important role during heart development. Approximately, more than 1,700 genes have been reported to be involved in the murine heart development [5]. GATA binding protein 4 (GATA4), NK2 homeobox 5 (NKX2.5), T-box 5 (TBX5), Zic family member 3 (ZIC3), myosin heavy chain 6 (MYH6) and NOTCH are some of the transcription factors, which play crucial role in heart development [6].
Several familial studies reported that mutations in GATA4 may cause different kind of CHD i.e., atrial septal defect (ASD), ventricular septal defect (VSD), tetralogy of fallot (TOF) and pulmonary stenosis (PS) [7-9]. Among all transcription factors, GATA4 is the most studied gene in different populations [9-13]. GATA4 has been mapped on 8p23.1 and has seven exons, which code for 442 amino acids and act as zinc finger transcription factor. GATA4 plays an important role in embryonic heart development such as proliferation of cardiomyocytes, endocardial cushion formation, development of right ventricle and septation of the outflow tract. GATA4 binds to the consensus sequence of the ANF promoter and interacts with other transcriptional factors such as; NKX 2.5 and TBX5 [14]. Subsequent studies have explained that TBX5, NKX2-5 and GATA4 interact during embryonic development and are involved in the regulation of heart developmental processes [6,15,16]. GATA4 knockout mice produce phenotypes like ventral morphogenesis and heart tube formation [5].
ASD and VSD are the commonest types of CHD and account for 50% of all cases of CHD. If proper precautionary measures are not taken, these defects result in ventricular dilation and heart failure, and ultimately cause a decrease in life expectancy [17,18]. VSD and ASD are defects in the ventricular and atrial septum, the wall dividing the left and right ventricles, and atrium of the heart, respectively. TOF is a congenital heart defect, which is classically understood to involve in four anatomical abnormalities of the heart i.e. right ventricular hypertrophy, ventricular septal defects, pulmonic stenosis (PS) and overriding aorta of the heart. ASD, VSD and TOF are common form of congenital heart disease (CHD) in India and other countries. Compared to other familial CHDs, the prevalence of ASD and VSD is significantly higher [19]. GATA4 has been identified as causative factors of familial ASD and VSD, and found to play a predominant role in the pathogenesis of both atrial and ventricular septal defects [7,20,21]. However, segregation of VSD and TOF with the GATA4 mutation needs to be explored.
CHD is the leading cause of infant morbidity and mortality in India. Congenital heart diseases are found in 8–10 of every 1,000 live births [22] and nearly 1,80,000 children are born with CHD each year in India [23]. Of these, nearly 60,000 to 90,000 suffer from critical CHD requiring early intervention [23]. Because of higher rate of consanguineous marriages, frequency of congenital heart disease is very high in Andhra Pradesh state of southern India, compared to rest of India [24]. In spite of higher number of CHD in India, there are very few studies to find a link between GATA4 mutation and congenital heart disease. Only two studies with CHD patients were conducted to find the association between few selected GATA4 SNP with CHD, however, their data was not significant [25,26]. Therefore, in the present study we have sequenced the complete coding region including exon-intron boundaries of GATA4 to elucidate the role of GATA4 in the etiology of CHD in South Indian patients.
Methods
Clinical evaluation of CHD
Babies (including pre-mature infants), who were attending Innova Children’s Heart Hospital, Hyderabad were screened for CHD according to a pre-designed protocol, that include 2D echocardiography, color doppler and ECG. Of these different phenotypes, four different types of CHDs were considered for the present investigation; they are: atrial septal defect (ASD), ventricular septal defect (VSD), tetralogy of fallot (TOF), and single ventricle (SV).
Patient and the control samples
A total of 100 patients, who fulfilled the criteria of CHD were selected for the study. Two hundred individuals, who had no CHD or family history of CHD or any heart disease, were also included in this study as control. The case samples (CHD) were drawn from Innova Children’s Heart Hospital, Hyderabad; while ethnically matched control samples were collected from Innova and other hospital in Hyderabad. Informed written consent was obtained from the parents of CHD patients and the control subjects prior to the collection of blood sample. In addition, we also took consent for ‘publication of individual patient data’ for all those individuals included in the manuscript (i.e.. all of those indicated in the family trees)”. All patient samples were collected from specialized hospital and they were belongs to same ethnic group (Dravidian). This study was conducted with prior approval of Institutional Ethical Committee of the Innova Children’s Heart Hospital, Hyderabad (IEC/IRB No. 001/2010) and IICT, Hyderabad.
DNA isolation and sequencing
We collected about 2.0 ml of peripheral blood samples from all the patients and controls in EDTA-coated vacutainer and genomic DNA was isolated according to the protocol of Sambrook et al. 1989 [27]. GATA4 sequence from the ENSEMBL (ID: ENSG00000136574; www.ensembl.org) was used to design primer employing primer 3 software (http://frodo.wi.mit.edu/) and was synthesized commercially (Eurofins, India). Detailed sequences of all primers used in this study have been summarized in Additional file 1: Table S1. The amplification reactions contained 40 ng of DNA, 10 mM dNTP mix, 10X PCR buffer, 2 U of Taq polymerase (Takara, catalogue no:R001B) and 5 pM of both the primers. PCRs were carried out using ABI GeneAmp PCR System 9700 (Forster city, USA). Amplification conditions used were as follows; an initial denaturation at 95°C for 5 min., followed by 35 cycles of denaturartion at 94°C for 1 min, annealing at 55-64°C for 30 sec (Additional file 1: Table S1) and at 72°C for 1 min. A final extension was carried out at 72°C for 10 min. PCR products were cleaned up using Exo-SAP-IT (USB, Affymetrix, USA) and 1.0 μl of the purified products were directly used as templates for sequencing using di-deoxy chain terminator cycle sequencing protocol (BigDye V3.1, Applied Biosystems, Forster city, USA) [28]. The extended products were purified by ethanol precipitation and run in an ABI 3730 Automated DNA Analyzer (Applied Biosystems, Forster city, USA). Sequencing was carried out using both forward and reverse primer independently.
Mutation analysis
The raw sequence data were analyzed and carefully edited using the Sequence Analysis Software. The edited sequences were assembled with reference sequence using DNA Star and Auto Assembler software (Applied Biosystems, USA). All the variant sites, compared to the reference sequence, were noted down. Genetic Association, Hardy–Weinberg equilibrium and Chi-square test were computed by using plink software [29]. Pathogenic potential of identified missense mutations from CHD patients was predicted by three different softwares. The prediction of PolyPhen software (www.tux.embl-heidelberg.de/ramensky/polyphen.cgi) was determined based on sequence comparison between homologous proteins. Profile scores position-specific independent counts (PSIC) were generated for the allelic variants. A PSIC score of the variant with more than 2 indicates damaging effect, scores between 1.5 and 2 indicate possibly damaging effects, score 1.5 indicates that the variant is probably damaging and score less than 1.5 indicates that the variant is benign. PMut, software for mutation prediction, is pathogenicity index ranging from 0 to 1. Indices greater than 0.5 indicate pathological mutation while less than 0.5 indicate neutral. SIFT is another sequence homology-based tool that identifies intolerant from tolerant amino acid substitutions. This software predicts whether an amino acid substitution at a particular position in a defined protein will have a phenotypic effect or not. For prediction of mutation effect on splicing of intron, we used ESE finder 3.0 [30]. This software finds the changes in the binding of splicing enhancers. ‘R’ packages were used for generation of plot. Promoter mutation’s function was predicted by CONSITE software, which finds the difference of transcription factor binding into wild type and mutant genomic sequences.
Effect of 3’ UTR region mutations on microRNA-target interactions
The interaction of 3′UTR with microRNA was determined by a bioinformatics tool as described by Kertesz et al. 2007 [31]. This bioinformatics tool finds microRNA-target interactions by an energy score, ΔΔG that is equal to the difference between the energy gained by binding of the microRNA to the target, and dGopen, the energy required to make the target region accessible for microRNA binding. dGduplex is the binding free energy of the microRNA-target duplex structure in which the microRNA and target are paired according to pairing constraints imposed by the seed. ΔΔG is an energetic score, the lower (more negative) the value, the stronger the binding of the microRNA to the given site is expected to be. As a rough rule of thumb, sites having ΔΔG values below −10 are likely to be functional in endogenous microRNA expression levels.
Statistical analysis
Statistical analysis was performed with Plink software [29]. For checking markers, whether they are in hardy Weinberg equilibrium, we used cut off p-value 0.01; and for association analysis we used same cut off p-value (0.01). Haploview was used to find blocks in data [32]. For finding the blocks, we considered r2 value and LOD score. Further, we used PHASE software for making haplotype for these blocks [33] and did association analysis with R basic packages (R version 3.0.2, 2013) [34].
Results
Clinical evaluation
In the present study, we have analyzed a total of 100 CHD patients. The percentages of CHD patients belonging to different categories were as follows; ASD: 33%, VSD: 32%, TOF: 32%, and SV: 3%. Age of all CHD patients ranged from 0.35 to 10.79 years. However, maximum number of CHD patients taking part in this study were of <5 years (Table 1).
Table 1.
Clinical classification of CHD patients studied
| S. no | Type of CHD | Patients (n = 100) | Age | ||||
|---|---|---|---|---|---|---|---|
| >1 year | 1-5 years | 5-10 years | 10-15 years | 15-20 years | |||
| 1 | ASD | 33(33%) | 2 | 18 | 8 | 3 | 2 |
| 2 | VSD | 32(32%) | 11 | 17 | 3 | 1 | - |
| 3 | TOF | 32(32%) | 12 | 16 | 3 | 1 | - |
| 4 | SV | 3(3%) | 2 | 1 | - | - | - |
| Total | 100 | 27 | 52 | 14 | 5 | 2 | |
Mutation analysis in GATA4 gene
We investigated the genomic DNA of CHD patients for variations in the entire coding regions, exon-intron boundaries, and untranslated regions (3′ UTR and 5′ UTR) of GATA4 gene. Our analysis revealed a total of 19 mutations, of which 1 each in promoter and splicing regions, 9 were in intronic regions, 3 were in exonic regions (2 missense and one synonymous) and 5 were in 3′ UTR (Tables 2 and 3). In silico analysis showed that one missense mutation was conserved and predicted to be pathogenic (Figure 1). Figure 2 shows sequence electropherogram of two mutations, which were evidenced in promoter and splicing regions. We evidenced the presence of homozygous mutation (rs61277615) at the promoter region in two ASD and one VSD patients, and (rs73203482) at the splice site in one TOF patients. We have collected parent’s samples for those patients who have either rs61277615 or rs73203482 variations in GATA4 gene. Analysis of parents’ samples revealed that parents of all the three patients had heterozygous mutation (Figure 2).
Table 2.
Detailed description of GATA4 mutations identified in this study
| S. no. | dbSNP | Exon/intron | Coordinate position | Nucleotide variation | Mutation type | A.A change | Frequency of mutation | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ASD | VSD | TOF | SV | Control | |||||||
| 33 | 32 | 32 | 3 | 200 | |||||||
| 1 | rs61277615 | Exon1 | 11561728 | C>T | 5′UTR | - | 0.06061 | 0.03125 | 0.03125 | 0 | 0.0125 |
| 2 | rs73203482 | Intron1 | 11561818 | G>A | Splice junction | - | 0.0303 | 0.03125 | 0.04688 | 0 | 0.0075 |
| 3 | CM051488 | Exon3 | 11606451 | G>A | Synonymous | G214G | 0.04545 | 0 | 0 | 0 | 0.0125 |
| 4 | rs804280 | Intron5 | 11612698 | A>C | Intronic | - | 0 | 0.07812 | 0.0625 | 0 | 0.0125 |
| 5 | rs2645457 | Intron5 | 11614112 | T>G | Intronic | 0.01515 | 0.01562 | 0.01562 | 0 | 0.005 | |
| 6 | rs4841587 | Intron5 | 11614175 | G>T | Intronic | - | 0.0303 | 0.07812 | 0.03125 | 0 | 0.0175 |
| 7 | rs4841588 | Intron5 | 11614225 | G>T | Intronic | - | 0.01515 | 0.1562 | 0.0625 | 0 | 0.02 |
| 8 | rs111272281 | Intron5 | 11614264 | A>del | Intronic | - | 0 | 0.0625 | 0.03125 | 0 | 0.005 |
| 9 | rs3729853 | Intron5 | 11614316 | C>T | Intronic | - | 0.0303 | 0.2188 | 0.07812 | 0 | 0.055 |
| 10 | rs3729854 | Intron5 | 11614329 | C>T | Intronic | - | 0.01515 | 0.04688 | 0.04688 | 0 | 0.01 |
| 11 | rs142395583 | Intron5 | 11614337 | T>A | Intronic | - | 0 | 0.01562 | 0.01562 | 0 | 0.0125 |
| 12 | rs745379 | Intron6 | 11615695 | A>G | Intronic | - | 0.04545 | 0.1094 | 0.1719 | 0.16 | 0.01 |
| 13 | rs200319078 | Exon7 | 11615835 | C>A | Missence | P394T | 0.01515 | 0 | 0 | 0 | 0.005 |
| 14 | rs56208331 | Exon7 | 11615928 | G>A | Missence | D425N | 0 | 0.01562 | 0 | 0 | 0.0025 |
| 15 | rs884662 | Exon7 | 11616501 | T>C | 3′UTR | - | 0 | 0.01562 | 0.01562 | 0 | 0.0125 |
| 16 | rs904018 | Exon7 | 11616516 | C>T | 3′UTR | - | 0.04545 | 0.01562 | 0.03125 | 0 | 0.0225 |
| 17 | rs12825 | Exon7 | 11616547 | C>G | 3′UTR | - | 0.01515 | 0.09375 | 0.1094 | 0 | 0.0825 |
| 18 | rs12458 | Exon7 | 11617240 | A>T | 3′UTR | - | 0 | 0 | 0 | 0 | 0.01 |
| 19 | rs3203358 | Exon7 | 11617505 | C>G | 3′UTR | - | 0.01515 | 0.01562 | 0.01562 | 0 | 0.0125 |
Table 3.
The allelic distributions of GATA4 polymorphism among patients and controls
| dbSNP | Control allele (n = 400) | ASD allele (n = 66) | VSD allele (n = 64) | TOF allele (n = 64) | Control vs. ASD | Control vs. VSD | Control vs. TOF | |||
|---|---|---|---|---|---|---|---|---|---|---|
| p-value | OR (95% CI) | p-value | OR (95% CI) | p-value | OR (95% CI) | |||||
| rs61277615 | 5(0.0125) | 4(0.06061) | 2(0.03125) | 2(0.03125) | 0.008514 | 5.0968(1.3322 - 19.4998) | 0.2532 | 2.54849(0.4838 - 13.4239) | 0.2532 | 2.54849(0.4838 - 13.4239) |
| rs73203482 | 3(0.0075) | 2(0.0303) | 2(0.03125) | 3(0.04688) | 0.09573 | 4.1354(0.6777 - 25.2338) | 0.08752 | 4.2688(0.6992 - 26.0617) | 0.009632 | 6.5082(1.2843 - 32.9812) |
| rs804280 | 5(0.0125) | 0 | 5(0.07812) | 4(0.0625) | - | - | 0.0007886 | 6.6949(1.8812 - 23.8262) | 0.007083 | 5.2667(1.3755 - 20.1654) |
| rs4841587 | 7(0.0175) | 2(0.0303) | 5(0.07812) | 2(0.03125) | 0.4838 | 1.7545(0.3565 - 8.6342) | 0.004553 | 4.7579(1.4622 - 15.4814) | 0.459 | 1.8111(0.3678 - 8.9182) |
| rs4841588 | 8(0.02) | 1(0.01515) | 10(0.1562) | 4(0.0625) | 0.7909 | 0.7538(0.0927 - 6.1274) | 0.0000001597 | 9.0741(3.4321 - 23.9913) | 0.04671 | 3.2667(0.9542 - 11.1831) |
Figure 1.
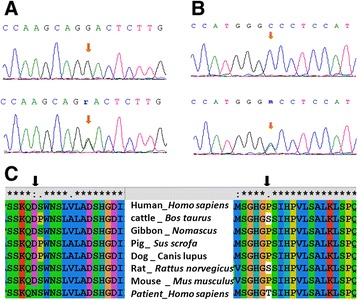
DNA sequencing electropherogram showing wild type (upper panel) and heterozygous (G → A) mutation (lower panel) in GATA4 coding region (arrows). A. Mutation Asp425Asn, B. Mutation Pro394Thr, C. Multiple amino acid sequence alignment of different species shows the conservation of the mutated amino acid residue (amino acid marked) across species.
Figure 2.

DNA sequencing electropherogram of promoter mutation (rs61277615) and a splice junction mutation (rs73203482) showing heterozygous condition in parents and homozygous condition and the patients.
Genetic studies
To find out association between genetic markers and CHD, we initially analyzed the effect of SNPs independently and then analyzed the haplotype level. We classified the samples on the basis of phenotype: ASD, VSD, TOF, and SV and removed 6 markers, which were not following Hardy-Weinberg equilibrium in ethnically matched control samples. These markers were rs12825 (HWE p-value = 1.296e-7), rs745379 (HWE p-value = 1.894e-5), rs3203358 (HWE p-value = 9.47e-5), rs3729853 (HWE p-value = 1.855e-15), rs111272281 (HWE p-value = 2.506e-3) and rs904018 (HWE p-value = 2.35e-3) (Additional file 1: Figure S1).
Further, we performed Chi-square analysis for finding the statistical significance and identified one 5′ UTR (promoter region −490 to 100 bp) mutation (rs61277615, p-value = 0.008514) in ASD. In TOF, rs73203482 (p-value = 9.6e-3, OR = 6.508) and rs804280 (p-value = 7.08e-3, OR = 5.267) were found to be associated with the disease. In case of VSD, 3 mutations were found to have strong association with the disease; they were rs804280 (p-value = 7.9e-4, OR = 6.695), rs4841587 (p-value = 4.6e-3, OR = 4.758) and rs4841588 (p-value = 1.6e-7, OR = 9.074). In case of SV, we did not find any variation associated with the CHD phenotype (Figure 3, Table 4).
Figure 3.

Chi-square P-value of variations found in GATA4 of all types of CHD. A. atrial septal defect (ASD), B. ventricular septal defect (VSD), C. tetralogy of fallot (TOF) and D. single ventricle (SV).
Table 4.
Statistical data of GATA4 mutations and their association with ASD, VSD, TOF and SV
| S. no. | dbSNP | Observed heterozygosity | Expected heterozygosity | Hardy Weinberg P-value | P-value | |||
|---|---|---|---|---|---|---|---|---|
| ASD | VSD | TOF | SV | |||||
| 1 | rs61277615 | 0.04 | 0.0392 | 1 | 0.008514 | 0.2532 | 0.2532 | 0.7829 |
| 2 | rs73203482 | 0.015 | 0.01489 | 1 | 0.09573 | 0.08752 | 0.009632 | 0.8314 |
| 3 | CM051488 | 0.01 | 0.00995 | 1 | 0.003122 | 0.5708 | 0.5708 | 0.8622 |
| 4 | rs804280 | 0.025 | 0.02469 | 1 | 0.3611 | 0.0007886 | 0.007083 | 0.7829 |
| 5 | rs2645457 | 0.01 | 0.00995 | 1 | 0.3394 | 0.3248 | 0.3248 | 0.8622 |
| 6 | rs4841587 | 0.025 | 0.03439 | 0.05197 | 0.4838 | 0.004553 | 0.459 | 0.7438 |
| 7 | rs4841588 | 0.03 | 0.0392 | 0.06886 | 0.7909 | 1.597e-07 | 0.04671 | 0.7264 |
| 8 | rs111272281 | 0 | 0.00995 | 0.002506 | - | - | - | - |
| 9 | rs3729853 | 0.01 | 0.104 | 1.855e-15 | - | - | - | - |
| 10 | rs3729854 | 0.01 | 0.0198 | 0.01502 | 0.7067 | 0.02464 | 0.02464 | 0.8056 |
| 11 | rs142395583 | 0.025 | 0.02469 | 1 | 0.3611 | 0.8372 | 0.8372 | 0.7829 |
| 12 | rs745379 | 0 | 0.0198 | 1.894e-05 | - | - | - | - |
| 13 | rs200319078 | 0.01 | 0.00995 | 1 | 0.3394 | 0.5708 | 0.5708 | 0.8622 |
| 14 | rs56208331 | 0.005 | 0.004987 | 1 | 0.6843 | 0.1367 | 0.6888 | 0.9024 |
| 15 | rs884662 | 0.015 | 0.02469 | 0.02497 | 0.3611 | 0.8372 | 0.8372 | 0.7829 |
| 6 | rs904018 | 0.025 | 0.04399 | 0.002346 | - | - | - | |
| 17 | rs12825 | 0.075 | 0.1514 | 1.296e-07 | - | - | - | |
| 18 | rs12458 | 0.01 | 0.0198 | 0.01502 | 0.4145 | 0.4217 | 0.4217 | 0.0606 |
| 19 | rs3203358 | 0.005 | 0.02469 | 9.47e-05 | - | - | - | |
Bold indicates: The SNP which is not in HW-equilibrium and P-value < 0.01.
In silico analysis
In coding region, we found a total of two missense mutations and one synonymous mutation in CHD patients that include Pro394Thr, Asp425Asn and Gly214Gly. Multiple alignments of GATA4 amino acid sequences from human, cattle, gibbon, pig, dog, rat and mouse found that asparagine at 425 position (Figure 1B) is highly conserved during evolution. But other missense mutation i.e., Pro394Thr is not conserved. To understand the functional significance of missense mutations, we performed bioinformatics analysis with all of identified two missense mutations using PolyPhen-2, PMut and SIFT softwares. Results of all the three analyses strongly indicate that Asp425Asn mutation was pathogenic (Table 5). We analyzed functional significance of splice junction mutation (rs73203482) by ESE finder 3.0 and found that this mutation is present near splice site of exon 1 (ENST00000335135) and intron 1. The data has shown the effect of binding of SFRS6, a splicing factor to the splicing region (Figure 4). This splicing factor has a role in site selection in alternative splicing. This also explains that the binding of splice enhancer will be affected in the presence of this splice mutation. We computed the functional significance of promoter mutation (promoter region −490 to 100 bp, rs61277615) and found the transcription factor binding in the wild-type and mutant promoter region by CONSITE software. Our data indicated that mutant sequence strongly binds with the transcription factor Myf1 with score of 7.360 in comparison to wild type sequence. The score with more than 7.0 represents the stronger binding of transcription factor with the DNA sequence. Except Myf1, binding of other transcription factors for both wild and mutant sequence is the same (Additional file 1: Table S2).
Table 5.
Functional significance of mutation prediction done by using PolyPhen-2, PMut and SIFT
| P394T | D425N | |||
|---|---|---|---|---|
| Name of software | Prediction | Score | Prediction | Score |
| PolyPhen-2 | Benign | 0.025 | Possibly damaging | 0.972 |
| PMut | Neutral | 0.4499 | Pathological | 0.6236 |
| SIFT | Tolerated | 0.55 | Not Tolerated | 0.24 |
Figure 4.

Prediction for binding of splicing enhancer: an outcome of rs73203482 mutation.
Haplotype analysis
Although SNPs have shown association with CHD, we further checked whether any haplotypes also show association with CHD. We did not find any block of associated markers in control samples (Figure 5A) but we found one LD block in VSD samples (Figure 5B). We also did not find any LD block in TOF samples (Additional file 1: Figure S2). We found that one LD block showed strong associations in VSD (p-value; GG: 1.906e-6, GT: 7e-4, TT: 6.01e-5) (Figure 5D, Additional file 1: Table S3). Same LD block also showed association in 10000 permutations (p-value; GG: 8e-4, GT: 2.9e-3, TT: 1.31e-2) (Figure 5C, Additional file 1: Table S4).
Figure 5.

Haplotype analysis. A. Haplotype frequency distribution of control samples. B. Linkage disequilibrium in VSD samples. C. Permutation based analysis for haplotype association. D. Haplotype analysis of VSD samples and its allelic distributions in cases and controls.
Comparison of allele frequency with 1000 genome project data
We compared the allele frequency of associated markers with 1000 genome project data. There was no significant difference in the allele frequency of control samples and Asian population samples of 1000 genome project (Additional file 1: Figure S3). While we observed differences between patient samples of VSD, TOF and ASD in comparison to control samples. Only three markers rs73203482, rs804280 and rs61277615 showed difference in distribution of allele frequency between disease samples and control samples (Figure 6).
Figure 6.
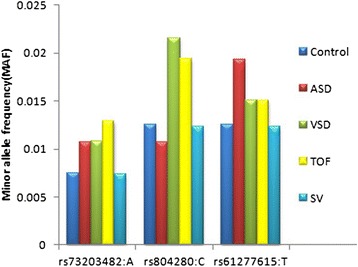
Distribution of minor allele frequency of associated GATA4 variations in ASD, VSD, TOF, SV, control sample.
3′ UTR mutations and their effect on microRNA-target interactions
Exon 7 of GATA4 consists of 1,708 bp the majority (1,525 bp) being untranslated. We identified 5 dbSNPs in 3′-UTR region of GATA4 in the study subjects; that include 2400T>C (rs884662), 2415T>C (rs904018), 2446C>G (rs12825), 3139A>T (rs12458) and 3404C>G (rs3203358). We did RNA folding studies for 1500 bp of 3′-UTR of GATA4 gene that include all of the above SNPs independently using DNA Star software, but we did not find any change (data not shown). We then checked for the microRNA and 3′-UTR target interaction through a bioinformatics tool. We found interaction of 440 numbers of microRNAs with desired UTR sequence. We found change in free energy difference of microRNA binding between wild type and mutant UTR regions. The value of microRNA-target interactions, ΔΔG was found to be equal to the difference between the free energy gained by the binding of the microRNA to the target. As a rough rule of thumb, sites having ΔΔG values below −10 were found likely to be functional in endogenous microRNA expression levels. While the free energy change (ΔΔG) of wild-type UTR region was more than −10, it completely shifted to +10 value for all 3′UTR mutant (Additional file 1: Figure S4A and 4B).
Discussion
Genetics and environmental factors play important role in the etiology of CHD. It has been shown that fetal heart development is regulated by a group of highly conserved transcription factors [35]. GATA4 is one of the transcription factors that plays a major role in the regulation of several other cardiac proteins, including atrial natriuretic peptide, brain natriuretic peptide, cardiac troponin C, cardiac troponin I, muscarinic acetylcholine receptor, and slow myosin heavy chain. Small changes in the level of GATA4 protein expression can dramatically influence cardiac development and embryonic survival [36]. GATA4 has been identified in familial and sporadic cardiac septal defects. Although several studies from different countries have established the association of GATA4 mutation with CHD [11,37], very few studies have been conducted in India. Therefore, we conducted this study to find genetic link with GATA4 mutation and correlate genotype with phenotype from subjects with ASD, VSD, TOF and single ventricle.
Analysis of all the exons of GATA4 revealed 19 mutations, out of which 2 were in promoter region, 9 were in intronic region, 3 were in coding region (that include 2 missense mutations and one synonymous mutation) and 5 were in 3′ UTR. The GATA4 mutation frequencies from different studies in VSD were as follows; in Americans 1.67% (2/120) [10], Chinese 1.22% (1/82) [11], Americans 0.80% (5/628) [38], Germans 16.7% (1/6) [12], and Chinese 0.48% (1/210) [8] whereas in case of TOF the frequencies were; Americans 7.69% (2/26) [10] and Chinese 8.33% (1/12) [11]. Since, Dravidian (Indian) population has different genetic architecture [39,40], we observed high frequency of rs73203482 in TOF, rs804280 in VSD, rs61277615 in ASD in comparison to ASD, VSD, SV and control samples. The associations of rs804280 with VSD and TOF, rs73203482 with TOF and rs61277615 with ASD are interesting findings (Additional file 1: Figure S2). These markers may have major contribution in CHD disease in Dravidian population but need to be validated in large number of samples. GATA4 is a hyper mutable protein according to previously published data [41]. Currently, approximately 111 non-synonymous mutations have been found in the GATA4 protein, including the mutations identified in this study. We identified two missense mutations i.e., Pro394Thr, and Asp425Asn and one synonymous mutation Gly214Gly in the coding region of CHD patients. The amino acid that changes due to novel mutation is conserved throughout the species. All three softwares PolyPhen (score 0.97), Pmut (score 0.8625) and SIFT (score 0.27) showed that missense mutation Asp425Asn is probably damaging, pathological and not tolerated but other missense mutation Pro394Thr is benign, not pathological and tolerated. Novel mutation, Asp425Asn maps to the C-terminal domain of GATA4, and is important for the trans-activation of downstream targets [42].
We observed one mutation in GATA4 gene promoter region (−490 to 100 bp). This exists in homozygous condition in; two ASD patients, one VSD patient and one TOF patient. Further analysis of this mutation in the parents of the above patients revealed the presence of heterozygous mutation. Due to high rate of consanguineous marriages among South Indian populations, this recessive mutation is much more common in the study populations, compared to other populations. We analyzed transcription factor binding with the wild and mutant type GATA4 gene and found that one transcription factor strongly binds to the mutant but not with the wild type. We also found one mutation near the promoter region, specifically in the splicing region. Position of this mutation is interesting as it may affect the splicing of GATA4 gene. Alternative first exons are located several kilobases upstream of the classic GATA4 transcription initiation site suggesting that their expression is being driven from novel upstream promoters. Some of these first exons are conserved across species suggesting alternative promoter usage to be likely an important regulatory mechanism for controlling the tissue- and cell-specific expression of the GATA4 gene in humans and other mammalian species. GATA4 first exons are used alternatively and rarely in association with one another. The rs73203482, which is present near to splicing site of exon 1 (ENST00000335135) and in intron 1 has shown to affect binding of SFRS6, a splicing factor (Figure 4). This splicing factor has a crucial role in site selection in alternative splicing.
In the present study, we also observed several mutations at the 3′UTR region of GATA4. We evidenced five 3′ UTR mutations from patients with ASD, VSD and TOF. All the five mutations have been already reported by different investigators [43]. However, Kertesz et al. experimentally showed that mutations at 3′UTR substantially reduce microRNA-mediated translational repression, with effects comparable to those of mutations that disrupt sequence complementarity. They devise a bioinformatics tool for microRNA-target interaction that computes the difference between the free energy gained from the formation of the microRNA-target duplex and the energetic cost of un-pairing the target to make it accessible to the microRNA. We used the same software to study the functional role of the 3′UTR mutation. However, none of the study showed any functional study of these mutations for the pathogenesis of CHD. Here, we used bioinformatics tools to find out if these mutations in 3′UTR cause any alteration of GATA4 function. The 3′-UTR of GATA4 is relatively long and likely contains regulatory elements essential for the regulation and transport of the mRNA transcript [44]. Accumulating evidence suggests that the 3′-UTR of mRNA is involved in the control of nuclear transport, polyadenylation status, sub-cellular targeting as well as rates of translation and degradation of mRNA by altering RNA secondary structure. One of the reasons for alteration of RNA secondary structure is due to aberrant RNA folding [45]. To confirm the alteration of secondary structure of RNA due to mutations in 3′ UTR, we performed RNA folding studies, but we did not observe any changes in RNA folding between wild type and mutants (data not shown). We approached a parameter free model for miRNA–target interactions (Pita algorithm). Previously, this bioinformatics tool was used to find the difference of miRNA–target interactions among mutated gene [31]. Using this tool, we predicted the thermodynamic changes during miRNA–mRNA duplex formation. Our results showed that in wild GATA4, a pool of microRNA binds strongly with the mRNA. However, this binding affinity of miRNA was altered due to the presence of 3′UTR mutations. Our study strongly predicts that all 3′UTR GATA4 mutations observed in CHD patients may alter the transcript level in diseased heart and affect the embryonic development of heart.
Conclusion
Our study identified c.620C>T mutation in GATA4 is associated with CHD in South India. Although this mutation reported in healthy individuals of other population. For the first time we are reporting its association with CHD in Indian population. Out of four phenotypes we studied, genetic variations were found to be associated with ASD (rs61277615), VSD (rs804280) and TOF (rs73203482, rs804280). c.620C>T either independently or combinly associated with CHD. Hence GATA4 is an important marker for CHD in India, particularly South India.
Acknowledgement
The authors are thankful to Department of Biotechnology (DBT) for providing Ramalingaswami Fellowship to SKB, Senior Research Fellowship (SRF) to SM from Indian Council of Medical Research (ICMR). K.T. was supported by a Network project grant (CARDIOMED-BSC0122) from the Council of Scientific and Industrial Research (CSIR), Government of India.
Additional file
List of primers used for amplification and sequencing of GATA4. Table S2. The list of transcription factors and their binding score with the wild type and mutant sequence. Table S3. Haplotype based association analysis of block 1 for VSD samples. Table S4. Permutation based analysis for haplotype association: 10000 permutations. Figure S1. Hardy-Weinberg equilibrium P-value. Figure S2. Linkage disequilibrium in TOF samples. Figure S3. Distribution of minor allele frequency of observed SNPs in control and 1000 genome project samples (ASN: Asian,CHB: Han Chinese in Bejing, China, CHS: Southern Han Chinese, JPT: Japanese in Tokyo, Japan,, EUR : European, AMR: American). Figure S4. A. Free energy change (ΔΔG) for Micro RNA binding with wild-type of 3′UTR region. B. Free energy change (ΔΔG) for Micro RNA binding with mutant of 3′UTR region.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SM has carried out the sample collection, DNA isolation, PCR and sequencing, genotype and sequence analysis, sequencing alignment and drafted the manuscript. NS has carried out the statistical part and helped to draft the manuscript. KSM has participated in the selection of studied patients and carried out diagnosis of patient. KT has participated in the study design, statistical analysis and drafted the manuscript. SKB has conceived the study, participated in its design, coordination and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Saidulu Mattapally, Email: mattapallysaidulu@gmail.com.
Sheikh Nizamuddin, Email: snizam@ccmb.res.in.
Kona Samba Murthy, Email: konasmurthy@gmail.com.
Kumarasamy Thangaraj, Email: thangs@ccmb.res.in.
Sanjay K Banerjee, Email: skbanerjee@thsti.res.in.
References
- 1.Marelli AJ, Mackie AS, Ionescu-Ittu R, Rahme E, Pilote L. Congenital heart disease in the general population: changing prevalence and age distribution. Circulation. 2007;115(2):163–172. doi: 10.1161/CIRCULATIONAHA.106.627224. [DOI] [PubMed] [Google Scholar]
- 2.Garg V. Insights into the genetic basis of congenital heart disease. CMLS. 2006;63(10):1141–1148. doi: 10.1007/s00018-005-5532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ransom J, Srivastava D. The genetics of cardiac birth defects. Semin Cell Dev Biol. 2007;18(1):132–139. doi: 10.1016/j.semcdb.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451(7181):943–948. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 5.Bentham J, Bhattacharya S. Genetic mechanisms controlling cardiovascular development. Ann N Y Acad Sci. 2008;1123:10–19. doi: 10.1196/annals.1420.003. [DOI] [PubMed] [Google Scholar]
- 6.Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, Garg V. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol. 2009;326(2):368–377. doi: 10.1016/j.ydbio.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A, Nakazawa M, et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A. 2005;135(1):47. doi: 10.1002/ajmg.a.30684. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Fang M, Liu XY, Xin YF, Liu ZM, Chen XZ, Wang XZ, Fang WY, Liu X, Yang YQ. A novel GATA4 mutation responsible for congenital ventricular septal defects. Int J Mol Med. 2011;28(4):557–564. doi: 10.3892/ijmm.2011.715. [DOI] [PubMed] [Google Scholar]
- 9.Yang YQ, Gharibeh L, Li RG, Xin YF, Wang J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, et al. GATA4 loss-of-function mutations underlie familial tetralogy of fallot. Hum Mutat. 2013;34(12):1662–1671. doi: 10.1002/humu.22434. [DOI] [PubMed] [Google Scholar]
- 10.Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat. 2006;27(3):293–294. doi: 10.1002/humu.9410. [DOI] [PubMed] [Google Scholar]
- 11.Peng T, Wang L, Zhou SF, Li X. Mutations of the GATA4 and NKX2.5 genes in Chinese pediatric patients with non-familial congenital heart disease. Genetica. 2010;138(11–12):1231–1240. doi: 10.1007/s10709-010-9522-4. [DOI] [PubMed] [Google Scholar]
- 12.Posch MG, Perrot A, Schmitt K, Mittelhaus S, Esenwein EM, Stiller B, Geier C, Dietz R, Gessner R, Ozcelik C, et al. Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am J Med Genet A. 2008;146A(2):251–253. doi: 10.1002/ajmg.a.32042. [DOI] [PubMed] [Google Scholar]
- 13.Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A, Dallapiccola B. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet. 2005;42(2):e16. doi: 10.1136/jmg.2004.026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424(6947):443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 15.Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet. 2001;28(3):276–280. doi: 10.1038/90123. [DOI] [PubMed] [Google Scholar]
- 16.Moskowitz IP, Kim JB, Moore ML, Wolf CM, Peterson MA, Shendure J, Nobrega MA, Yokota Y, Berul C, Izumo S, et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell. 2007;129(7):1365–1376. doi: 10.1016/j.cell.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 17.Pierpont ME, Basson CT, Benson DW, Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):3015–3038. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 18.Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL, American Heart Association Council on Cardiovascular Disease in the Y Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):2995–3014. doi: 10.1161/CIRCULATIONAHA.106.183216. [DOI] [PubMed] [Google Scholar]
- 19.Zhao QM, Ma XJ, Jia B, Huang GY. Prevalence of congenital heart disease at live birth: an accurate assessment by echocardiographic screening. Acta Paediatr. 2013;102(4):397–402. doi: 10.1111/apa.12170. [DOI] [PubMed] [Google Scholar]
- 20.Okubo A, Miyoshi O, Baba K, Takagi M, Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N, et al. A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet. 2004;41(7):e97. doi: 10.1136/jmg.2004.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281(5373):108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 22.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–1900. doi: 10.1016/S0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 23.Deeparani T, Pillai MR, Elavazhagan T. Detection of MTHFR C677T and A1298C Gene Polymorphism in Congenital Heart Disease. Middle-East J Scientific Res. 2009;4(2):127–132. [Google Scholar]
- 24.McGregor TL, Misri A, Bartlett J, Orabona G, Friedman RD, Sexton D, Maheshwari S, Morgan TM. Consanguinity mapping of congenital heart disease in a South Indian population. PLoS One. 2010;5(4):e10286. doi: 10.1371/journal.pone.0010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramegowda S, Kumar A, Savitha MR, Krishnamurthy B, Doddaiah N, Ramachandra NB. Missense mutation G296S in GATA4 is not responsible for cardiac septal defects. Indian J Human Genet. 2007;13(1):30–32. doi: 10.4103/0971-6866.32032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinesh SM, Lingaiah K, Savitha MR, Krishnamurthy B, Narayanappa D, Ramachandra NB. GATA4 specific nonsynonymous single-nucleotide polymorphisms in congenital heart disease patients of Mysore, India. Genet Test Mol Biomarkers. 2011;15(10):715–720. doi: 10.1089/gtmb.2010.0278. [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 28.Thangaraj K, Singh L, Reddy AG, Rao VR, Sehgal SC, Underhill PA, Pierson M, Frame IG, Hagelberg E. Genetic affinities of the Andaman Islanders, a vanishing human population. Current Biol. 2003;13(2):86–93. doi: 10.1016/S0960-9822(02)01336-2. [DOI] [PubMed] [Google Scholar]
- 29.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15(16):2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- 31.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39(10):1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 32.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 33.Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76(3):449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.R., Development., Core., Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- 35.Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407(6801):221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 36.Pu WT, Ishiwata T, Juraszek AL, Ma Q, Izumo S. GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev Biol. 2004;275(1):235–244. doi: 10.1016/j.ydbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Reamon-Buettner SM, Borlak J. GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J Med Genet. 2005;42(5):e32. doi: 10.1136/jmg.2004.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E. GATA4 sequence variants in patients with congenital heart disease. J Med Genet. 2007;44(12):779–783. doi: 10.1136/jmg.2007.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reich D, Thangaraj K, Patterson N, Price AL, Singh L. Reconstructing Indian population history. Nature. 2009;461(7263):489–494. doi: 10.1038/nature08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41(2):187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang E, Sun S, Qiao B, Duan W, Huang G, An Y, Xu S, Zheng Y, Su Z, Gu X, et al. Identification of functional mutations in GATA4 in patients with congenital heart disease. PLoS One. 2013;8(4):e62138. doi: 10.1371/journal.pone.0062138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Posch MG, Perrot A, Berger F, Ozcelik C. Molecular genetics of congenital atrial septal defects. Clin Res Cardiol. 2010;99(3):137–147. doi: 10.1007/s00392-009-0095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reamon-Buettner SM, Cho SH, Borlak J. Mutations in the 3′-untranslated region of GATA4 as molecular hotspots for congenital heart disease (CHD) BMC Med Genet. 2007;8:38. doi: 10.1186/1471-2350-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hesketh J. 3′-Untranslated regions are important in mRNA localization and translation: lessons from selenium and metallothionein. Biochem Soc Trans. 2004;32(Pt 6):990–993. doi: 10.1042/BST0320990. [DOI] [PubMed] [Google Scholar]
- 45.Conne B, Stutz A, Vassalli JD. The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat Med. 2000;6(6):637–641. doi: 10.1038/76211. [DOI] [PubMed] [Google Scholar]