

Abstract
Background
Mutations in the POU3F4 gene cause X-linked deafness type 3 (DFN3), which is characterized by inner ear anomalies.
Methods
Three Turkish, one Ecuadorian, and one Nigerian families were included based on either inner ear anomalies detected in probands or X-linked family histories. Exome sequencing and/or Sanger sequencing were performed in order to identify the causative DNA variants in these families.
Results
Four novel, c.707A>C (p.(Glu236Ala)), c.772delG (p.(Glu258ArgfsX30)), c.902C>T (p.(Pro301Leu)), c.987T>C (p.(Ile308Thr)), and one previously reported mutation c.346delG (p.(Ala116ProfsX26)) in POU3F4, were identified. All mutations identified are predicted to affect the POU-specific or POU homeo domains of the protein and co-segregated with deafness in all families.
Conclusions
Expanding the spectrum of POU3F4 mutations in different populations along with their associated phenotypes provides better understanding of their clinical importance and will be helpful in clinical evaluation and counseling of the affected individuals.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-015-0149-2) contains supplementary material, which is available to authorized users.
Keywords: Molecular Diagnosis, Mutation Detection, POU3F4, Sequencing, X-linked deafness
Background
Hearing loss is the most common sensory deficit with a prevalence of approximately 1 in 1000 newborns [1]. Genetic factors account for at least 50% of cases with congenital or prelingual-onset hearing loss in developed countries. Among the genetic forms, previous studies have indicated that deafness is transmitted with an inheritance pattern consistent with autosomal recessive in 75-77%, autosomal dominant in 15-20%, and X-linked in 2-3% of cases [2,3]. To date, five loci (DFNX1-4, and 6) and four genes, PRPS1 (MIM 311850) for DFNX1 [4], POU3F4 (MIM 300039) for DFNX2 [5], SMPX (MIM 300226) for DFNX4 [6,7], and COL4A6 (MIM 303631) for DFNX6 have been identified for X-linked hearing loss [8].
DFNX2 (also referred to as Nance deafness) was originally described by Nance et al. in 1971 as an X-linked condition characterized in males by profound mixed deafness, vestibular abnormalities and congenital fixation at the stapes with perilymphatic gusher [9]. Computerized tomography studies in patients with DFNX2 showed abnormal dilatation of the internal acoustic canal and abnormal communication between the internal acoustic canal and inner ear compartments. Subsequently molecular analysis revealed mutations in POU3F4 (POU domain, class III, transcription factor 4- NM_000307.4) [10].
POU3F4 belongs to a superfamily of POU domain transcription factors comprised of a POU-specific domain and a POU-homeodomain, both of which influence DNA binding and specificity. Previous studies showed that the POU-specific part of the protein binds to DNA and regulates the downstream of the target genes [11]. The POU3F4 gene is expressed in the developing neural tube during embryogenesis [11,12], and in a variety of other organs including the inner ear [13-17]. Fifty two mutations in POU3F4 have been reported as the cause of DFNX2 since the initial report (Additional file 1: Table S1) [18]. These variants include intragenic mutations and partial or complete deletions of the gene, as well as chromosomal deletions, inversions, and duplications [19]. Here we report four novel and a previously reported mutations in the POU3F4 gene identified in a multiethnic cohort of five families.
Methods
Subjects
This study was approved by the local Intitutional Review Board at the University of Miami (USA), the Ethics Commitee of Ankara University Medical School (Turkey), Bioethics Committee of FFAA (HE-1) in Quito (Ecuador), and the Joint Ethics Commitee of University of Ibadan/University College Hospital Ibadan (Nigeria). A signed informed consent form was obtained from each participant or in the case of a minor, from parents. In addition we obtained consent for publication of individual patient data from all of the study participants.
Diagnosis of sensorineural hearing loss was established via standard audiometry in a sound-proof room according to current clinical standards. A physical examination was performed on at least one hearing-impaired individual from each family including a detailed clinical evaluation and otoscopy. Affected individuals did not have delays in gross motor development. Neither did they have balance problems, vertigo, dizziness, or spontaneous and positional nystagmus. Tandem walking was normal and Romberg test was negative. Three Turkish families (295, 572, 667) were included based on inner ear anomalies detected in probands. One Ecuadorian (1225) and one Nigerian (1535) families were included based on X-linked family histories and physical examination that was negative for additional syndromic finding. Temporal bone CT scans were not available in probands from Ecuador and Nigeria.
DNA Sequencing
Genomic DNA from total 5 affected and 8 unaffected family members was extracted from peripheral blood using standard methods. In probands from families 295, 572, 667, and 1535, the single exon of POU3F4 was PCR-amplified and Sanger-sequenced. 20 ng of genomic DNA were used for PCR amplification. PCR products were purified by Qiagen MinElute PCR Purification Kit and directly sequenced with a Beckman Coulter CEQ2000XL or ABI 3730XL automated sequencers. In the proband of family 1225, exome sequencing was performed via Illumina HiSeq 2000 by using a previously published protocol [20]. Segregation of the identified variants was evaluated in the available family members of all families. DNA sequencing data are stored in a secure internal database, which are available upon request to researchers wishing to use them for research purposes only.
Bioinformatics Analysis
For in silico analysis of the missense mutations, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), SIFT (http://sift.jcvi.org/), and Mutation Taster (http://www.mutationtaster.org/) were used for the prediction of the effect of amino acid substitutions on protein function. In addition 3D structures were modeled on the Swiss-Model website (http://swissmodel.expasy.org) which generates modeling for the target protein based on a sequence alignment between the target protein and a suitable template structure. These models were visualized and delineated with RasMol (v. 2.7.5) software (http://rasmol.org/).
Results and discussion
Five different POU3F4 mutations were detected in the families (Table 1 and Additional file 2: Figure S1, S2, S3, S4 and S5). Detected variants co-segregated with hearing loss in all families. All four novel mutations were predicted to affect the POU domains of the protein. The novel NM_000307.4:c.772delG (p.(Glu258ArgfsX30)) mutation truncates the protein within POU-specific domain. Similarly, NM_000307.4:c.346delG (p.(Ala116ProfsX26)), which has been previously reported in a Korean family with severe deafness [21], leads to a truncation that eliminates both POU domains.
Table 1.
Clinical and genetic information of the probands in the affected families
| Family 295 | Family 572 | Family 667 | Family 1225 | Family 1535 | |
|---|---|---|---|---|---|
| Country origin | Turkey | Turkey | Turkey | Ecuador | Nigeria |
| POU3F4 | c.772delG | c.346delG | c.707A>C | c.902C>T | c.987T>C |
| Mutation | p.(Glu258ArgfsX30) | p.(Ala116Profsx26) | p.(Glu236Ala) | p.(Pro301Leu) | p.(Ile308Thr) |
| Age of onset | Congenital | <5 years | <5 years | <5 years | <5 years |
| Pattern of inheritance | Multiplex-X linked | Simplex | Simplex | Multiplex-X linked | Multiplex-X linked |
| Hearing loss type | Sensorineural | Sensorineural | Sensorineural | Mixed | Mixed |
| Hearing loss severity | Moderate to severe | Severe | Profound | Severe | Severe |
| Laterality | Bilateral | Bilateral | Bilateral | Bilateral | Bilateral |
| Inner Ear Anomaly | Basal turns of cochlea are incompletely separated from IAC which was dilated in the lateral end; modioli are absent | Dilated IAC, absence of modioli, interscalar septum is present | Bulbous enlargement of IAC; modioli are absent | N/A | N/A |
| Intervention | Hearing aids | Cochlear implant | Hearing aids | Hearing aids | Hearing aids |
In silico analysis for the three missense mutations, NM_000307.4:c.707A>C (p.(Glu236Ala)), c.902C>T (p.(Pro301Leu)), c.987T>C (p.(Ile308Thr)), predicted the effect of amino acid substitutions on protein function classified as pathogenic (PolyPhen-2: probably damaging; SIFT: damaging; Mutation Taster: disease causing). Structural analysis of the p.Glu236Ala mutation showed that the wild-type glutamate residue has a possible stabilizing relationship with the lysine residue (Lys202) residing within the alpha helix across the glutamate. The p.Glu236Ala mutation introduces a non-polar amino acid (alanine) instead of an acidic residue and possibly interferes with the stability between the secondary structures of the protein (Figure 1A). Proline has a distinctive cyclic structure, which provides a conformational rigidity. For that reason, it can commonly be found at turns in protein structures, as seen here in the POU homeodomain. In the p.Pro301Leu mutation, the structure of the turn changes becoming shorter (Figure 1B). The wild-type isoleucine at position 308 is part of an alpha-helix in the POU homeodomain. The p.Ile308Thr mutation introduces a threonine residue with a hydroxide group and, according to the model, the hydroxide group causes a conformational change in the arginine residue at 323 (Figure 1C). All three missense mutations are located in the two POU domains of the protein (Figure 1D). None of the identified variants was present in the Exome Variant Server (http://evs.gs.washington.edu/EVS/), 1000 Genomes, dbSNP, Leiden Open Variation Database (LOVD), or Deafness Variation Database (DVD). These variants were also absent in our next-generation sequencing database that contains data of more than 2000 samples from different ethnicities including Turkish (>200), Latin American (>200), and Nigeria (>100).
Figure 1.
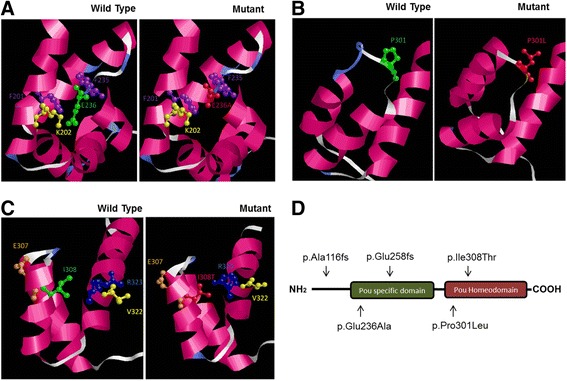
In silico analysis and molecular modeling of the wild-type and mutant POU3F4 proteins. The p.Glu236Ala (p.E236A) mutation possibly interferes with the stability between the secondary structures of the protein (A). The p.Pro301Leu (p.P301L) mutation affects the structure of the turn and the turn becomes shorter (B). The hydroxide group introduced by the p.Ile308Thr (p.I308T) mutation causes a conformational change in the Arginine residue at 323 (C). Schematic illustration of POU3F4 protein labeled with the five mutations identified in this study (D).
The phenotype of Nance deafness occurs because of the loss of function mutations in the POU3F4 gene, which encodes a transcription factor consisting of two highly conserved domains [22]. The 76-78 amino acids that contain the POU-specific domain cooperate with the POU-homedomain to enhance the binding affinity and specificity of DNA binding [16]. Hemizygous males and heterozygous females carrying POU3F4 mutations present with different clinical features. The hearing loss affects males with rapid progression to severe or profound deafness within the prelingual period. Temporal bone CT scans of males with POU3F4 mutations show inner ear anomalies. Males have increased perilymphatic pressure, which is responsible for gusher during stapes surgery. The female carriers of POU3F4 mutations usually show no or slight hearing loss [23]. In this study only males were affected while carrier females did not present with any clinical findings. Affected males’ middle and inner ear anomalies were within the expected spectrum.
POU3F4 gene product is localized in the nucleus and has a critical role in inner ear morphogenesis. Recent studies showed that POU3F4 mutations might cause a shift of Pou3f4 nuclear localization to the cytoplasm [24]. All mutations we identified affect a POU domain suggesting that loss of DNA binding capability may be a common pathological mechanism for function [21].
Conclusions
In the present study we conclude that mutations in POU3F4 cause X-linked hearing loss in different populations. It is especially important that the Ecuadorian and Nigerian families were studied solely based on their inheritance pattern and found to have mutations in POU3F4, suggesting that it is a common cause of X-linked hearing loss worldwide. We recommend that diagnostic molecular laboratories prioritize developing a POU3F4 test that would be offered even when the results of CT scans were not available. Identification of a causative mutation for deafness provides families with a chance to make informed reproductive decisions.
Acknowledgement
This study was supported by the National Institutes of Health - National Institute on Deafness and Other Communication Disorders grant R01DC012836 to MT and the University of Ibadan Tertiary Education Trust Fund to AOL.
Abbreviations
- CT
Computerized tomography
- IAC
Internal Acoustic Canal
- N/A
Not Available
Additional files
Reported POU3F4 mutations.
Data of the Family 295. Chromatograms showing wild type (1A), hemizygous NM_000307.4:c.772delG (p.(Glu258ArgfsX30)) mutation in the affected male (1B) and c.772delG (p.(Glu258ArgfsX30)) mutation in the female carrier (1C). The pedigree contains two affected male siblings (1D). Figure S2. Data of the family 667. Chromatograms showing wild type (2A), hemizygous NM_000307.4:c.707A>C (p.(Glu236Ala)) mutation in the affected male (2B) and c.707A>C (p.(Glu236Ala)) mutation in the female carrier (2C). One affected male was in the family (2D). Figure S3. Data of the family 572. Chromatograms showing wild type (3A), hemizygous NM_000307.4:c.346delG (p.(Ala116Profsx26)) mutation in the affected male (3B) and c.346delG (p.(Ala116Profsx26)) mutation in the female carrier (3C). One affected male was in the family (3D). Figure S4. Data of the family 1225. Chromatograms showing wild type (4A), hemizygous NM_000307.4:c.902C>T (p.(Pro301Leu)) mutation in the affected male (4B) and c.902C>T (p.(Pro301Leu)) mutation in the female carrier (4C). The pedigree contains five affected males and is consistent with X-linked recessive inheritance (4D). Figure S5. Data of the family 1535. Chromatogram showing hemizygous NM_000307.4:c.987T>C (p.(Ile308Thr)) mutation in the affected male (5A). There are three affected males and the pedigree is consistent with X-Linked recessive inheritance (5B).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed with interpretation of the results. GB, AS, KOY, MT contributed the genetic data analysis and manuscript writing. AOL, SF, AI, LS, IM, PM, RV, GM, RP, MT worked in patient recruitment and obtaining samples. SB and MT have designed the study and given final approval of the version to be published. All authors read and approved the final manuscript.
Contributor Information
Guney Bademci, Email: g.bademci@med.miami.edu.
Akeem O Lasisi, Email: akeemlasisi@gmail.com.
Kemal O Yariz, Email: koyariz@gmail.com.
Paola Montenegro, Email: paom14@gmail.com.
Ibis Menendez, Email: IMenendez1@med.miami.edu.
Rodrigo Vinueza, Email: genmolhg1@gmail.com.
Rosario Paredes, Email: charoparedes@yahoo.fr.
Germania Moreta, Email: germaniamoreta@yahoo.com.
Asli Subasioglu, Email: asuzak78@hotmail.com.
Susan Blanton, Email: SBlanton@med.miami.edu.
Suat Fitoz, Email: sfitoz@yahoo.com.
Armagan Incesulu, Email: armaganincesulu@yahoo.com.
Levent Sennaroglu, Email: lsennaroglu@gmail.com.
Mustafa Tekin, Email: mtekin@med.miami.edu.
References
- 1.Morton CC, Nance WE. Newborn hearing screening a silent revolution. N Engl J Med. 2006;354:2151–64. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 2.Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet. 1993;46:486–91. doi: 10.1002/ajmg.1320460504. [DOI] [PubMed] [Google Scholar]
- 3.Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Han D, Li J, Han B, Ouyang X. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am J Hum Genet. 2010;86:65–71. doi: 10.1016/j.ajhg.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bitner-Glindzicz M, Turnpenny P, Höglund P, Kääriäinen H, Sankila EM, van der Maarel SM, de Kok YJ, Ropers HH, Cremers FP, Pembrey M, et al. Further mutations in Brain 4 (POU3F4) clarify the phenotype in the X-linked deafness, DFN3. Hum Mol Genet. 1995;4:1467–9. doi: 10.1093/hmg/4.8.1467. [DOI] [PubMed] [Google Scholar]
- 6.Schraders M, Haas SA, Weegerink NJ, Oostrik J, Hu H, Hoefsloot LH, Kannan S, Huygen PL, Pennings RJ, Admiraal RJ, Kalscheuer VM, Kunst HP, Kremer H. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am J Hum Genet. 2011;88:628–34. doi: 10.1016/j.ajhg.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huebner AK, Gandia M, Frommolt P, Maak A, Wicklein EM, Thiele H, Altmüller J, Wagner F, Viñuela A, Aguirre LA, Moreno F, Maier H, Rau I, Giesselmann S, Nürnberg G, Gal A, Nürnberg P, Hübner CA, del Castillo I, Kurth I. Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss. Am J Hum Genet. 2011;88:621–7. doi: 10.1016/j.ajhg.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rost S, Bach E, Neuner C, Nanda I, Dysek S, Bittner RE, Keller A, Bartsch O, Mlynski R, Haaf T, Müller CR, Kunstmann E. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur J Hum Genet. 2014;22:208–15. doi: 10.1038/ejhg.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nance WE, Setleff R, McLeod A, Sweeney A, Cooper C, McConnell F. X-Linked Mixed Deafness with Congenital Fixation of the Stapedial Footplate and Perilyphatic Gusher. Birth Defects. 1971;7:64. [PubMed] [Google Scholar]
- 10.de Kok YJ, van der Maarel SM, Bitner-Glindzicz M, Huber I, Monaco AP, Malcolm S, Pembrey ME, Ropers HH, Cremers FP. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995;267:685–8. doi: 10.1126/science.7839145. [DOI] [PubMed] [Google Scholar]
- 11.Mathis JM, Simmons DM, He X, Swanson LW, Rosenfeld MG. Brain 4: a novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J. 1992;11:2551–2561. doi: 10.1002/j.1460-2075.1992.tb05320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Moine C, Young WS., 3rd RHS2, a POU domain-containing gene, and its expression in developing and adult rat. Proc Natl Acad Sci USA. 1992;89:3285–3289. doi: 10.1073/pnas.89.8.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dominov JA, Miller JB. POU homeodomain genes and myogenesis. Dev. Genet. 1996;19:108–118. doi: 10.1002/(SICI)1520-6408(1996)19:2<108::AID-DVG2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 14.Hussain MA, Lee J, Miller CP, Habener JF. POU domain transcription factor brain 4 confers pancreatic alpha-cell-specific expression of the proglucagon gene through interaction with a novel proximal promoter G1 element. Mol Cell Biol. 1997;17:7186–7194. doi: 10.1128/mcb.17.12.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phippard D, Heydemann A, Lechner M, Lu L, Lee D, Kyin T, Crenshaw EB., 3rd Changes in the subcellular localization of the Brn4 gene product precede mesenchymal remodeling of the otic capsule. Hear Res. 1998;120:77–85. doi: 10.1016/S0378-5955(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 16.Phippard D, Lu L, Lee D, Saunders JC, Crenshaw EB., 3rd Targeted mutagenesis of the POU-domain gene Brn4/Pou3f4 causes developmental defects in the inner ear. J Neurosci. 1999;19:5980–5989. doi: 10.1523/JNEUROSCI.19-14-05980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arellano B, Ramírez Camacho R, García Berrocal JR, Villamar M, del Castillo I, Moreno F. Sensorineural hearing loss and Mondini dysplasia caused by a deletion at locus DFN3. Arch Otolaryngol Head Neck Surg. 2000;126:1065–9. doi: 10.1001/archotol.126.9.1065. [DOI] [PubMed] [Google Scholar]
- 18.The Human Gene Mutation Database. (www.hgmd.org) (09/09/2014)
- 19.Cremers FP, Cremers FR, Kremer H. POU3F4 and mixed deafness with temporal defect (DFN3) In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn Errors of Development. New York: Oxford University Press; 2008. pp. 1042–1047. [Google Scholar]
- 20.Diaz-Horta O, Duman D, Foster J, 2nd, Sırmacı A, Gonzalez M, Mahdieh N, Fotouhi N, Bonyadi M, Cengiz FB, Menendez I, Ulloa RH, Edwards YJ, Züchner S, Blanton S, Tekin M. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One. 2012;7:e50628. doi: 10.1371/journal.pone.0050628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee HK, Song MH, Kang M, Lee JT, Kong KA, Choi SJ, Lee KY, Venselaar H, Vriend G, Lee WS, Park HJ, Kwon TK, Bok J, Kim UK. Clinical and molecular characterizations of novel POU3F4 mutations reveal that DFN3 is due to null function of POU3F4 protein. Physiol Genomics. 2009;39:195–201. doi: 10.1152/physiolgenomics.00100.2009. [DOI] [PubMed] [Google Scholar]
- 22.Hagiwara H, Tamagawa Y, Kitamura K, Kodera K. A new mutation in the POU3F4 gene in a Japanese family with X-linked mixed deafness (DFN3) Laryngoscope. 1998;108:1544–7. doi: 10.1097/00005537-199810000-00022. [DOI] [PubMed] [Google Scholar]
- 23.Marlin S, Moizard MP, David A, Chaissang N, Raynaud M, Jonard L, Feldmann D, Loundon N, Denoyelle F, Toutain A. Phenotype and genotype in females with POU3F4 mutations. Clin Genet. 2009;76:558–63. doi: 10.1111/j.1399-0004.2009.01215.x. [DOI] [PubMed] [Google Scholar]
- 24.Parzefall T, Shivatzki S, Lenz DR, Rathkolb B, Ushakov K, Karfunkel D, Shapira Y, Wolf M, Mohr M, Wolf E, Sabrautzki S, de Angelis MH, Frydman M, Brownstein Z, Avraham KB. Cytoplasmic mislocalization of POU3F4 due to novel mutations leads to deafness in humans and mice. Hum Mutat. 2013;34:1102–10. doi: 10.1002/humu.22339. [DOI] [PMC free article] [PubMed] [Google Scholar]