

Abstract
The nitric oxide (NO) and cyclooxygenase (COX) pathways share a number of similarities. Nitric oxide is the mediator generated from the NO synthase (NOS) pathway, and COX converts arachidonic acid to prostaglandins, prostacyclin, and thromboxane A2. Two major forms of NOS and COX have been identified to date. The constitutive isoforms critically regulate several physiological states. The inducible isoforms are overexpressed during inflammation in a variety of cells, producing large amounts of NO and prostaglandins, which may underlie pathological processes. The cross-talk between the COX and NOS pathways was initially reported by Salvemini and colleagues in 1993, when they demonstrated in a series of in vitro and in vivo studies that NO activates the COX enzymes to produce increased amounts of prostaglandins. Those studies led to the concept that COX enzymes represent important endogenous “receptor” targets for amplifying or modulating the multifaceted roles of NO in physiology and pathology. Since then, numerous studies have furthered our mechanistic understanding of these interactions in pathophysiological settings and delineated potential clinical outcomes. In addition, emerging evidence suggests that the canonical nitroxidative species (NO, superoxide, and/or peroxynitrite) modulate biosynthesis of prostaglandins through non-COX-related pathways. This article provides a comprehensive state-of-the art overview in this area.
Keywords: nitric oxide, superoxide, peroxynitrite, prostaglandins, cyclooxygenase-1, cyclooxygenase-2
prostaglandins (pgs) and nitric oxide (NO) are prominent local mediators that participate in the homeostatic modulation of many cellular functions. Their constitutive biosynthesis and pulsed release in nanomolar concentrations occur via well-identified bioenzymatic complexes [cyclooxygenase (COX) for PGs and nitric oxide synthase (NOS) for NO], which act on specific substrates [arachidonic acid (AA) and l-arginine, respectively]. This basal release of NO and PGs exerts a protective role in many pathophysiological conditions: vascular diseases (via enhanced vasodilatation and antiplatelet activity), gastric lesions (via activation of gastroprotective processes), erectile dysfunction (via enhanced vasodilatation), and learning and memory processing (via potentiation of neuronal plasticity). Therefore, the clinical use of PG derivatives and NO donors has been proposed in the treatment of such disorders due to their ability of restoring basal levels of PGs and NO. One notable example is the organic nitrate esters that release NO after enzymatic bioconversion and have been used for more than a century in the treatment of myocardial ischemia. In both acute and inflammatory settings, NO and PGs are released simultaneously in large amounts, due mainly to the activation of inducible enzymes releasing NO and PGs in micromolar concentrations. Release of PGs and NO in such large amounts, plus evidence that both PGs and NO overproduction, may be detrimental for cell survival, have driven many experiments over the last two decades to identify the possible participation of both mediators in the pathogenesis of multiple diseases. In particular, overt production of PGs and NO is now known to occur in tissues affected by the inflammatory processes of rheumatic diseases, chronic degenerative disorders, central neurodegenerative processes associated with brain ischemia, as well as in neuroinflammatory diseases (e.g., multiple sclerosis, demyelinization, HIV-related brain disorders, and Alzheimer's disease). These results have led to the development of novel and more selective COX and NOS inhibitors for the protection of inflamed tissues. Evidence over the last 18 years has shown that there is constant “cross-talk” between NO and PG release at many levels, its central feature being modulation of molecular mechanisms that regulate PG-generating pathway. The final effect of this modulatory activity is complex. Endogenous NO, as well as NO-donors, stimulate or suppress the COX pathway depending on basal NO released, the cell type in which PG biosynthesis occurs, and the intensity of the stimulus employed for PG release. In addition, NO conjugates with superoxide anion (O2·−, SO) to produce a potent proinflammatory and cytotoxic agent, peroxynitrite (ONOO−, PN), which also modulates COX enzymes. Taken together, these findings support an active reciprocal modulation of NO and PG-generating systems as a clinical rationale for using traditional NO donors and for developing novel therapeutic agents that target nitroxidative stress (herein defined as stress caused by NO, SO, and/or PN) and the prostaglandin pathway.
Nitric Oxide Synthase and Cyclooxygenase Enzymes
Nitric oxide is generated via the oxidation of the terminal guanidino nitrogen of l-arginine by NOS (96, 97) (Fig. 1). In biological systems, NO is used as a generic term to encompass three redox-related species. The redox-related species of NO include NO·, which can modulate ferrous iron (Fe2+) containing proteins by direct coordination to their Fe2+ centers; such NO· can be cytotoxic by reacting with superoxide anion to produce peroxynitrite. Further oxidation of NO· species generates nitrogen dioxide (NO2), dinitrogen trioxide (N2O3), dinitrogen tetroxide (N2O4), nitrite (NO2−), nitrate (NO3−), and others (152). The second important redox species of NO, nitrosonium ion (NO+), can nitrosylate the thiol groups of various proteins, a modification that may serve important regulatory functions during pathophysiological states (35). Nitrosonium ion has an extremely short half-life (∼10−10 s) in solution at physiological pH and binds extremely rapidly to thiol groups. Following such binding, the resulting –SNO adducts maintain reactive “nitrosonium” characteristics and can transfer NO+ to other thiols in a process called transnitrosylation (62, 72, 148). The third important redox-related species of NO, nitronyl (NO−) anion, was reported to have a similar reactivity to NO+ leading to S-nitrosylation (59); however, the normal or pathophysiological relevance of this species is unclear.
Fig. 1.
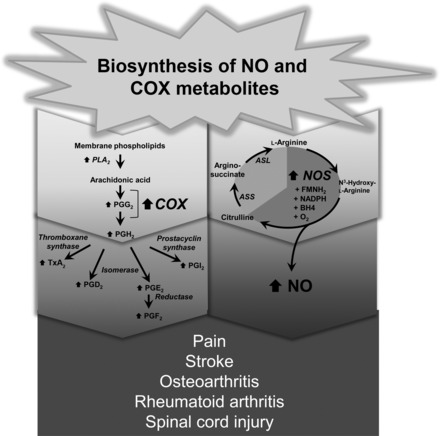
Biosynthesis of nitric oxide and products of arachidonic acid metabolism. COX, cyclooxygenase; NO, nitric oxide; PLA, phospholipase A; PGG2, prostaglandin G2; PGH2, prostaglandin H; TxA, thromboxane; ASS, argininosuccinate synthase; ASL, argininosuccinate lyase.
Three major isoforms of NOS have been identified on the basis of their relatively distinct primary amino acid sequences (only 50–60% identity), tissue and cellular distribution, and mode of regulation (96, 97). Two NOS isoforms are calcium/calmodulin dependent and are constitutively expressed (cNOS): endothelial-derived NOS, and neuronally derived NOS (nNOS) (96, 97). The third NOS isoform is cytokine-inducible and calcium/calmodulin-independent (iNOS) (96, 97). The distinct properties of each NOS isoform have important implications since it is the magnitude, duration, and the cellular sites of NO production that determines its overall physiological or pathophysiological effect. For example, release of NO from cNOS is localized and acts transiently. Nitric oxide released under these circumstances plays a crucial role in the cardiovascular and renal system, where it controls organ blood flow distribution, inhibits the aggregation and adhesion of platelets to the vascular wall, limits leukocyte adhesion and smooth muscle cell proliferation, promotes diuresis and natriuresis with the kidney (96, 97) and is involved in neurotransmission (10, 103). Most of these actions are mediated through the binding of NO to Fe2+ in the heme prosthetic group of soluble guanylate cyclase, which catalyzes the conversion of GTP to cyclic GMP (96, 97). In contrast, iNOS is expressed in a wide variety of host defense and other cells in response to inflammatory stimuli, such as endogenous cytokines and bacterial lipopolysaccharide endotoxin (LPS), resulting in a delayed (hours) but prolonged synthesis of high levels of NO. It is now well accepted that while NO released from iNOS is biologically appropriate, it is simultaneously involved in many pathological events (96, 97) (Fig. 1). The importance of Fe2+ in mediating the functions of NO is also apparent when examining its cytotoxic effects, which are commonly observed when NO is produced in much larger quantities by stimulated macrophages, hepatocytes, and other cells. NO produced via such high-output systems inhibits proliferation of intracellular pathogens and tumor cells. These effects can be explained by the reactivity of NO· with Fe2+ in the [Fe-S] centers of several important macromolecules, including aconitase and complex I and complex II of the electron transport chain (32, 50). The high affinity of NO· for Fe2+ probably results in both the removal of Fe2+ from [Fe-S] centers and the formation of nitrosyl Fe2+ species within [Fe-S] proteins. Thus, NO· exerts its function through its interaction with the Fe2+ of heme and nonheme-iron-centered proteins.
Although NO+ can interact with thiol groups of proteins to form S-nitrosothiols (72) in a nonenzymatically catalyzed S-nitrosylation reaction, there appears to be some specificity in this reaction. First, not all proteins containing free cysteines are likely to be S-nitrosylated, and not every available cysteine in a target protein is S-nitrosylated. For example, the ryanodine receptor contains 87 cysteines, but only 13 are normally targeted to be S-nitrosylated (178). Second, it has been postulated that there is a primary consensus sequence for S-nitrosylation that requires acidic or basic residues adjacent to target cysteines (149). However, this presumptive acid-base consensus motif can also arise as a result of a target protein's three-dimensional structure, as shown by analysis of the crystal structure of caspase-3, whose active cysteine is S-nitrosylated by NO in vivo (51). More than a hundred proteins have been identified as targets of S-nitrosylation associated with modification of their functions or structures including COX (35, 148).
Virtually all cells except erythrocytes can produce PGs from arachidonic acid that occurs at high concentrations among phospholipids of the cell membrane (Fig. 1). Prostaglandins are members of a lipid family that are enzymatically derived from polyunsaturated fatty acids with 20-carbon chains and four cis-double bonds (20:4) via the COX pathway. Prostaglandins are also known as “prostanoids”, which include classical prostaglandin, as well as the more specialized moieties thromboxane and prostacyclin (87, 144). The existence of biologically active PGs was first described in 1930 when Kurzrok and Lieb showed that fresh semen led to rhythmic contraction of human myometrium in vitro (65). Shortly thereafter, other groups independently confirmed the presence of an active substance derived from prostate glands that caused smooth muscle contraction and named it “prostaglandin” (41, 87, 169). Continued research has revealed that prostaglandins are a mixture of biologically active substances such as PGE or PGF, but ironically, these originate in the seminal vesicle and not the prostate gland (7).
One of the most important discoveries in the PG field occurred when Vane showed that the major anti-inflammatory effects of aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) was due to their transient or irreversible inhibition of cyclooxygenase (165). Prostaglandins are formed by the action of prostaglandin synthase in a two-step conversion of arachidonic acid. First, the COX enzyme converts arachidonic acid to a cyclic endoperoxide (PGG2), followed by peroxidase cleavage of the PGG2 peroxide to yield the endoperoxide (PGH2) (105). These unstable intermediate products of arachidonic acid generated by COX are rapidly converted to other prostaglandins (PGE2, PGF2, TxA2, PGI2) by specific isomerases (Fig. 2). (105). COX was first purified as a 140-kDa homodimer from sheep seminal vesicles, which proved to be a prodigious source of the protein (28, 85, 90, 164) and led subsequently to COX being cloned from the same tissue (28, 85). With the availability of the cDNA encoding the protein, as well as COX-specific antibodies, numerous studies have evaluated the distribution, expression, and regulation of COX both in vitro and in vivo. Initially, COX was considered a single isoform that produced PGs in most tissues and cell types. However, a number of studies have illustrated that COX activity is increased in certain inflammatory states, being induced in cells exposed to proinflammatory cytokines and growth factors in vitro (3, 27, 135). Following these observations, extensive research led to the discovery of two COX isoforms that initially were called prostaglandin H synthase-1 and prostaglandin H synthase-2 but are now commonly referred to as COX-1 (constitutive enzyme) and COX-2 (inducible enzyme) (142). These are encoded by two different genes; the human Ptgs2 (COX-2) is a small (8.3 kb) immediate early gene on chromosome 1, while Ptgs1 (COX-1) is a much longer 22-kb gene on chromosome 9. Human COX-1 and COX-2 are homodimers sharing 60% identity in sequence (110, 124). Both isoforms are composed of three regions; an epidermal growth factor binding domain, the membrane binding domain, and the COX catalytic domain containing the cyclooxygenase and peroxygenase active sites on either side of a heme prosthetic group that is important for both activities. The peroxidase active site is on the opposite side of membrane binding domain and consists primarily of a heme prosthetic group (38, 124). When arachidonic acid binds to the cyclooxygenase site, this substrate sits at the end of channel and carbon-13 of arachidonic acid is placed near a crucial amino acid for cyclooxygenase reaction, Tyr-385 (Fig. 2). However, mutating Tyr-385 does not impact the midpoint potential of heme in COX-2, further suggesting that the peroxidase site is independent of the cyclooxygenase site. Moreover, crystallographic and biochemical data showed that the two active sites are physically separated, as fatty acids do not interfere with peroxidase catalysis (64).
Fig. 2.
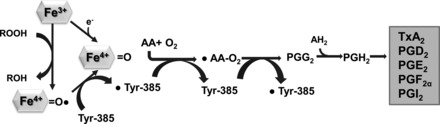
The scheme of COXs enzyme reaction mechanism. COX enzymes produce PGH2 by two-step chemical reactions: cyclooxygenase and peroxidase. PGH2 is further converted to other prostanoid by tissue-specific enzymes. Tyr, tyrosine; AA, amino acid; AH2, nonspecific oxidizing agent that can donate 2 hydrogens.
Despite the structural similarity of the COX isoforms, their expression patterns and localization are very distinct. Constitutively expressed COX-1 is present in tissues such as the stomach, gut, or kidney, where PGs play a cytoprotective role in maintaining normal physiological processes. By contrast, COX-2 is expressed in many stimulated host defense cells at the site of inflammation, leading to robust production of proinflammatory PGs (142) (Fig. 1). Consistent with COX-2's induction during inflammation, selective inhibition of COX-2 is both anti-inflammatory and anti-nociceptive (142). In addition, several groups have shown that upregulated COX-2 expression is involved in brain ischemia and that COX-2 inhibitors are neuroprotective (142). Such evidence led to the suggestion that COX-1 is homeostatically protective, whereas COX-2 may not be. However, this dichotomy is probably simplistic. For example, deletion of COX-1 in mice did not cause an increased susceptibility to gastric ulcer (67), while COX-2 inhibitors have been shown to attenuate the healing process (173). COX-1 is also expressed in quiescent inflammatory cells (18), whereas COX-2 is constitutively expressed in certain tissues, including brain and kidney, where it exerts protective effects (47, 67, 71, 180). Induced COX-2 has also been observed in endothelial cells; its inhibition with COX-2 inhibitors that spare COX-1 function in platelets has been held responsible for certain cardiovascular side effects observed with the usage of COX-2-specific inhibitors under chronic inflammatory conditions (100, 145). Moreover, utilizing a variety of COX inhibitors and sophisticated structure-functional analyses, it has been shown that COX enzymes form structural homodimers, and yet exhibit functional heterodimers, as inhibition of only one catalytic site is sufficient to block homodimer enzymatic activity, and binding of various fatty acids in a catalytic site can alter their enzymatic activities (118, 141, 168, 182–184). Furthermore, simultaneous expression of two isoforms of COXs led to an interesting observation by Yu et al. (182), demonstrating that COX-1 and COX-2 can form a heterodimer, which appears to play a critical role in the transformation that occurs in the blood vessels of newly born mice, implicating more complex roles and regulation of COX enzymes in biology.
Therefore, the NOS and COX systems are often coexpressed, share a number of similarities, and play fundamental roles in similar pathophysiological conditions (Fig. 3) (177). Increasing evidence suggest that there is considerable “cross-talk” between COX and NOS and that modulation of each respective pathway impacts the outcome of a physiological or pathological response. The interaction between NO- and PG-generating machinery occurs at multiple levels (92). Indeed, NO may interfere directly with COX expression and PG biosynthesis. On the other hand, AA and its metabolites generated by COX isoforms may also interfere with NO biosynthesis. Furthermore, expression of NOS seems to correlate with PG receptors and intracellular messengers, such as cAMP, generated by activation of GPCRs (92). Moreover, both NO and PGs interact with their own respective biosynthetic pathway by modulating molecular events underlying NOS and COX expression. Finally, some NO donors combined with several NSAIDs (nitroaspirin and nitroflurbiprofen, for example) have recently been synthesized and used in the treatment of many inflammatory and noninflammatory disorders, suggesting the possible beneficial effect of drugs acting on NO and PGs simultaneously (92).
Fig. 3.
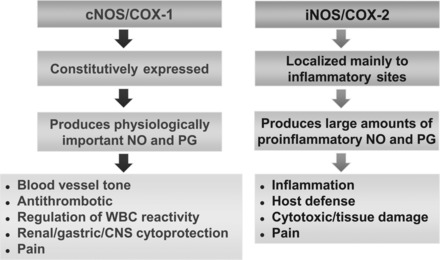
Similarities between the nitric oxide synthase (NOS) and COX pathway. WBC, white blood cell; CNS, central nervous system.
What Are the Molecular Mechanisms Involved in the Regulation of Cyclooxygenase Pathway by NO and Related Nitroxidative Species
In 1993, Salvemini et al. (128) reported that NO increased the production of PGs at least in part by activating COX-1 and COX-2, findings that were subsequently corroborated by many groups in various in vitro and in vivo models (92). Using microsomal sheep vesicles (a rich source of COX-1), they demonstrated that exogenous administration of NO augments COX-1 activity, as well as murine recombinant COX-1 enzyme, leading to exaggerated increase in PGE2 formation (Fig. 4) (127, 128). In addition to COX-1, NO activates COX-2. Indeed, using IL-1β-stimulated fibroblasts as a cellular model to explore the effects of exogenous NO on COX-2 activity, Salvemini and colleagues (127, 128) showed that exposure of IL-1β-stimulated fibroblasts to either NO gas or NO donors, such as sodium nitroprusside and glyceryl trinitrate, increased COX-2 activity several-fold and increased production of PGE2 (Fig. 5) (127, 128). The effects of NO were cGMP-independent since methylene blue, an inhibitor of the soluble guanylyl cyclase (97), did not block the effects of NO (127, 128). These findings were soon confirmed by others (see, for example, Refs. 22, 53, 89, 91, 170, 174). The ability of NO to directly activate COX-2 was supported by the evidence that NO increases the activity of purified recombinant COX-2 enzyme (127, 128). Having observed that NO activates COX-1 and COX-2 enzymes, the group addressed whether COX-2 activity can be modulated by endogenously produced NO. Murine RAW-264.7 macrophages were stimulated with LPS to induce iNOS and COX-2 enzymes and produce large amounts of NO and PGs. As expected, inhibition of iNOS activity by nonselective and selective NOS inhibitors attenuated the release of NO from these cells (127, 128). However, the critical observation was that when NO release was inhibited, there was a simultaneous inhibition of PG release (127, 128). These results suggested that endogenously released NO from macrophages exerted a stimulatory action on COX-2 to enhance the production of PGs independently of the known effects of NO on the soluble guanylyl cyclase. As an extension, release of PGE2 by astroglial cells pretreated with N-methyl-d-aspartate (NMDA) is driven by activation of the l-arginine-NO pathway, an effect potentially relevant in pathophysiological mechanisms in which glutamatergic neurotransmission is involved (91). In addition, spontaneous release of PGE2 by hypoxic astrocytes occurs mainly via IL-1β-dependent activation of iNOS (93). Finally, the release of PGE2 by astroglial cells pretreated with IL-1β and TNF-α is due to enhanced COX-2 expression via activation of iNOS since release was blocked in the presence of a NOS inhibitor. These results may be relevant toward gaining a better understanding of the mechanisms underlying neuroimmune disorders.
Fig. 4.
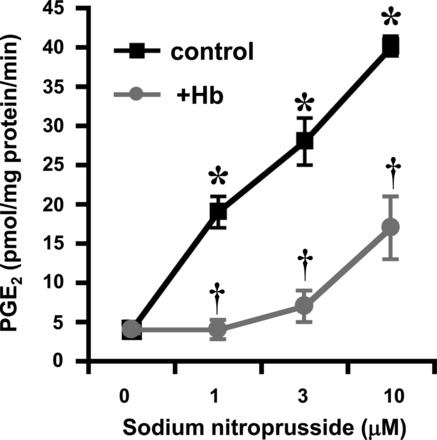
Activation of COX-1 by NO. Sodium nitroprusside increase COX-1 activity in sheep seminal vesicles, as evidenced by a dose-dependent increase in PGE2 release from arachidonic acid; hemoglobin (Hb) attenuates the effects of sodium nitroprusside (SNP) consistent with an NO-mediated activation of COX-1. Results are expressed as the means ± SE of seven experiments and analyzed by Student's unpaired t-test. *P < 0.01 compared with results obtained in the absence of SNP and †P < 0.01 compared with results obtained in the presence of SNP but absence of Hb. Experimental design is detailed in Refs. 127 and 128.
Fig. 5.
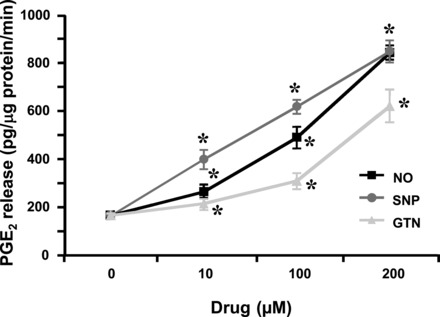
Activation of COX-2 by NO. Coincubation of NO, SNP, or glyceryl trinitrate (GTN) with human fetal fibroblasts expressing COX-2 increased the production of PGE2 by arachidonic acid. Results are the mean ± SE of seven experiments and analyzed by Student's unpaired t-test. *P < 0.01 compared with results obtained in the absence of the NO or the NO donor. Experimental design is detailed in Refs. 127 and 128.
When Salvemini et al. (128) proposed that NO increased PG formation by activating the COX enzymes, the group did not have a mechanistic explanation for how this occurred. So what have we learnt since 1993? It is unlikely that NO activates COX by binding directly to its heme prosthetic group, and several mechanisms have been proposed and depicted in Fig. 6. A first possibility is that NO acts as an antioxidant. Indeed, COX activity also provides a source of superoxide anion, and it has been postulated that superoxide could be involved in the autoinactivation of COX enzymes (33). NO interacts with superoxide and thereby limits the amounts of the superoxide radical that are normally necessary for auto-inactivation (43). Therefore, NO may augment COX activity by removing superoxide and acting as an antioxidant to prevent the auto-inactivation of COX. A second possibility is NO-mediated formation of nitrosothiols, based upon the proposal by Hajjar et al. (46) that S-nitrosothiol may lead to NO-mediated activation of COX as it occurs in an heme-independent manner. On the other hand, Marnett et al. (78) investigated the role of free cysteines in COX-1 and showed that Cys313 and Cys540 mutations with serines alone inhibited its enzyme activity implying that NO-mediated Cys modification may interfere with COX-1 activity. Interestingly, both groups independently reported that SNAP, a NO chemical donor, could not directly activate COX-1 (66, 162). Thus confusion exists over whether S-nitrosylation of COX-1 leads to its activation. A third possibility is NO-induced generation of PN, the spontaneously generated product of SO and NO (5). Using purified COX-1 and COX-2 enzymes in vitro as well as excised sheep seminal vesicles, Landino et al. (66) reported that PN increased COX-1 and COX-2 activity. PN also stimulated COX-1 activity in aortic smooth muscle cells (161, 162). The mechanisms by which PN can activate COX enzymes remain undefined, but they could involve oxidative inactivation (78) or modification of key amino acids residues in the polypeptide backbone (2). Peroxynitrite can also oxidatively modify the COX substrate arachidonic acid, yielding F2-isoprostanes (76, 98). Peroxynitrite could block the activity of prostacyclin synthase and thereby attenuate the production of prostacyclin (189). A fourth possibility first proposed by Snyder and colleagues (58) is that iNOS itself binds to COX-2 and causes its S-nitrosylation at residue Cys-526, thereby enhancing its catalytic activity. This interaction and modification are specific for COX-2 as iNOS does not interact with COX-1). In particular, Kim et al. (58) observed more than one S-nitrosylated cysteine residue in LPS/IFN-γ-activated RAW264.7 cells and subsequently mutated each cysteine to a serine. In this way, they identified Cys-523, located near the catalytic domain, as the only cysteine that does not affect constitutive enzyme activity; when Cys-523 was mutated, NO no longer activated COX-2. Since Cys-523 is located near the catalytic site of COX-2, we speculated that this mutation leads to a slight conformational change in COX-2 that affects its enzyme kinetics (58). Indeed, Kim et al. (58) subsequently performed viscosity studies examining enzyme activity in various concentrations of sucrose and showed that release of product is accelerated in the presence of NO donor. However, it is not possible to exclude a potential involvement of any other cysteines for NO-mediated activation of COX-2 as mutations of other cysteines alone affected the basal level enzyme activity of COX-2. Given that NO has a high affinity for heme, it is unclear how NO specifically S-nitrosylates the cysteines of COX-2 rather than interacting with its heme moiety. This may be partially explained by the fact that S-nitrosylation of COX-2 by iNOS requires physical interaction between the two proteins, specifically between the subunit-binding site of iNOS and the catalytic domain of COX-2 (Fig. 7A). Hence, Kim and coworkers speculated that NO generated from iNOS is simply in direct proximity of the S-nitrosylation target residue in COX-2 for immediate delivery and modification. Indeed, disruption of iNOS/COX-2 binding diminishes S-nitrosylation and activation of COX-2 in LPS-induced RAW264.7 cells (Fig. 7B). These studies elegantly demonstrated how NO generated from iNOS modulates COX-2 in macrophages but also raises additional questions. First, this interaction does not explain how exogenous administration of NO or NO donors increases PG generation in vitro. Indeed, a recent study by Tsikas and Niemann (159) showed that chemical donors of NO are unlikely to modify cysteines of recombinant COX-1/2 proteins in vitro, further corroborating importance of protein-protein interaction between iNOS/COX-2 for NO-mediated modification and activation of COX-2. Second, do both COX-2 in homodimer need to be S-nitrosylated to enhance its activity, as it has been shown that only one site of dimer is catalytically functional at any given time (183). We also do not know whether NO generated from iNOS can modulate COX-1 by either direct S-nitrosylation or transnitrosylation from COX-2 if a heterodimer is formed. Moreover, Goodwin et al. (42) demonstrated that removal of superoxide generated by LPS-activated RAW264.7 cells inhibited prostaglandin formation to the same extent as NOS inhibitors. Whether these effects are mediated by SO or PN remains unknown. While the immediate speculation might be that be PN-mediated nitrotyrosine formation of COX-2, Cross's group showed that PN can also generate S-nitrosothiol (163). Therefore, further studies will be necessary to identify the form of NO-mediated modification of COX-2 in activated macrophages. S-nitrosylation of COX-2 does not seem to be limited to macrophages. In contrast to other tissues, COX-2 in the brain is constitutively expressed. We showed that COX-2 is S-nitrosylated in neuronal cells by nNOS (156). To our surprise, nNOS binding is mediated by its PDZ domain (postsynaptic density 95, PSD-85; discs large, Dlg; zonula occludens-1, ZO-1; a common structural domain of 80–90 amino-acids found in the signaling proteins of various organisms), differing from the domain near the catalytic region that mediates iNOS binding to COX-2 (58, 156). We also showed that a physical interaction between nNOS and COX-2 is required for S-nitrosylation of COX-2 and that disruption of binding attenuated NMDA-mediated excitotoxicity, as did a selective COX-2 inhibitor (156).
Fig. 6.
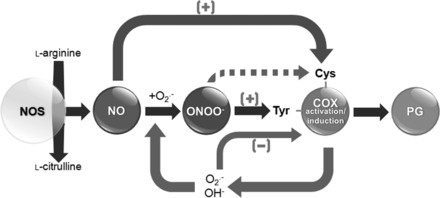
Reciprocal regulation of PG biosynthesis by nitroxidative stress. NO can directly modify/S-nitrosylate cysteine(s) in COX by direct protein-protein interaction between NOS and COX and activate it. COX also produces superoxide, which is speculated to be responsible for the autoinactivation of the COX enzymes. Rapid interaction between superoxide and NO reduces the levels of superoxide and produces PN, which can target either heme or tyrosine residue(s) in COX. PN may interact with Fe in the heme group of COX and forms a radical intermediate product, which can accelerate the enzyme reaction while PN-mediated modification of Tyr-385 in COX can inactivate the activity of the enzyme. In those conditions, in which both the NOS and COX systems coexist and are coactivated, there can be a nitroxidative-mediated modulation in the production of PG resulting in beneficial or detrimental effects, such as increased inflammation and tissue injury.
Fig. 7.

An interaction between inducible NOS (iNOS) and COX-2 is required to mediate NO-induced PGE2 production. A: COX-2 and iNOS are simultaneously in activated macrophages by LPS treatment. COX-2 and iNOS are also coimmunoprecipitated by COX-2 antibody. B: dominant negative (DN; 484–604 amino acid based on COX-2 sequence) peptide, which is a minimum region required to bind to iNOS and inhibit endogenous protein-protein interaction significantly reduced PGE2 production.
In addition to effects on COX-2 enzyme activity, NO increases PG production from macrophages by acting post-transcriptionally or posttranslationally to increase their intracellular COX-2 protein levels (170). Studies using human osteoarticular chrondrocytes revealed that NO released from sodium nitroprusside induced cell death, an event associated with DNA fragmentation, caspase-3 activation, down-regulation of Bcl-2, overexpression of COX-2, and increased release of PGE2 (107). These events were abolished by blocking either the mitogen-activated protein (MAP) kinase pathway with the MAP kinase inhibitor PD98059, or the p38 kinase with inhibitor SB202190, as well as COX-2 activity, with the selective inhibitor NS-398. Although PGE2 alone had no effect on cell death, it did sensitize chrondrocytes to sodium nitroprusside-mediated death. These results suggest that NO activates both the extracellular signal-related protein kinase and/or the p38 kinase pathway, which, in turn, induce COX-2 and subsequent PGE2 release. The latter may then sensitize human osteoarticular chondrocytes to an incipient NO-induced cell death (107). Similarly, interactions between NO and COX-2 and the signaling pathway involved were investigated in primary cultured rabbit cortical thick ascending limb cells (cTAL) (17). In these cells, immunoreactive COX-2 and vasodilatory prostaglandins were increased by exogenous application of the NO donor, S-nitroso-N-acetyl-d,l-penicillamine, and were associated with increased expression of phosphorylated p38. In addition, the administration of a low-salt diet to wild-type mice increased immunoreactive COX-2 and nNOS in the macula densa and surrounding cTAL of their kidneys, while the same diet did not significantly elevate COX-2 expression in nNOS−/− mice. Collectively, the results demonstrate that in cTAL, NO can increase COX-2 expression in the macula densa through p38-dependent signaling pathways via activation of NF-κB (17). NO is also necessary for maintaining prolonged COX-2 gene expression and sustained PGE2 biosynthesis (117). NO-induced alterations in COX-2 gene expression were not related to changes in COX-2 mRNA (117, 155), and thus, the mechanism through which NO influences COX gene expression is not defined. Liu et al. (74) showed that NO can increase COX-2 mRNA levels via the β-catenin/TCF pathway leading to activation of the polyomavirus enhancer activator 3 (PEA3) transcription factor. Furthermore, NO also interacts with various other pathways that can influence COX expression, notably, the cAMP/PKA/CREB and JNK/Jun/ATF2 signaling cascades (74, 115). Recent evidence suggests that intracellular concentrations of NO and PGs may activate or suppress certain inflammatory cells by modulating endogenous biosynthesis and NOS/COX enzymes. Indeed, in human fetal microglial cells, very low concentrations of constitutive NO from cNOS are required to activate COX expression (55). Furthermore, low concentrations of NO enhance prostanoid formation, while higher concentrations downregulate the COX-2 pathway (151). Thus, endogenous or exogenous NO plays a crucial role in the regulation of COX activity, although the mechanism of this effect is still unclear. Evidence exists that in astroglia and other cells that possess both cNOS and iNOS, as well as in cells expressing only inducible iNOS, both NO and NO donors are able to regulate the expression of the constitutive and inducible COX enzymes (for review, see Refs. 20 and 21). In particular, NO released under basal conditions by cNOS maintains iNOS in a nonactivated state through the inhibition of the NF-κB signaling, which, in turn, regulates the iNOS transcriptional mechanisms. NF-κB represents one of the most relevant signaling pathways that mediate iNOS expression following many extracellular stimuli, including bacterial endotoxin and endogenous inflammatory cytokines (9, 11, 49, 80). When a low concentration of endogenous or exogenous NO is released, NF-κB remains inactive (157). Many factors may contribute in this NO-related inhibition of NF-κB, including the direct effect of NO on binding of NF-κB to its promoter response element, without affecting the activation and translocation of NF-κB (26, 83). Moreover, NO inhibits NF-κB binding to DNA through S-nitrosylation of the Cys-62 residue of the p50 subunit (26). Recent data show that NO interacts with NF-κB by stabilizing its endogenous inhibitor IκB-α. In particular, NO and NO− donors stabilize IκB-α by preventing its degradation from NF-κB and also increase the mRNA expression of IκB-α without affecting the mRNA expression of NF-κB subunits p65 or p50 (116). Recently, we have shown that agents such as tianeptine prevented an early downregulation of IκB- α (30 min) induced by HIV-1 coating gp120 in astrocytes needed for the activation of the classical NF-κB pathway and iNOS and COX2 induction (56). Thus, in cells whose iNOS is activated by extracellular inducers of NF-κB, NO may exert an early inhibitory role. Then, an increasing amount of NO combines with SO to generate PN, favoring derepression of NF-κB (possibly via nitration of IKK-β), resulting in the activation of iNOS. Since NF-κB is also a potent inducer of COX-2, and the action of NO on COX-2 is mediated by inhibition of NF-κB in mesangial cells (29), it is likely that these mechanisms may underlie some of the reported inhibitory effects of NO in prostaglandin biosynthesis.
Finally, COX enzymes are not the only target that can be modified by PN. Prostacyclin synthase is selectively inhibited by PN at low concentrations (IC50 = 50 nM), while thromboxane A2 synthase is activated at low concentrations too (187, 189). In particular, prostacyclin synthase contains one Cys at position 469, which is involved in heme binding, and a spectrophotometric study showed that this thiolate ligand at the fifth coordination position of the heme iron is not affected by PN. It appears that nitration of tyrosine residue 430 in prostacyclin synthase interferes the metal center of the active site and inhibits its activity (188, 189). iNOS-mediated regulation of prostaglandin production is not limited to COX enzymes. Xu et al. (179) showed that iNOS activates cytosolic phospholipase A2α (cPLA2α) by S-nitrosylation. In this situation, cPLA2α does not directly interact with iNOS, but rather forms ternary complex with COX-2 and iNOS. Hence, induction of COX-2 is a crucial step for S-nitrosylation and activation of cPLA2α (179).
In addition to this body of evidence showing the positive regulation of COX by NO, other reports support the idea that NO can inactivate COX under certain conditions. Indeed, Minghetti et al. (88) showed that LPS-induced COX-2 expression in microglial cells and their subsequent PG release are enhanced by co-incubation with NOS inhibitors. On the other hand, these results were confirmed by evidence that exogenous NO donors applied exogenously to LPS-treated microglial cells inhibit their PG release, an effect driven by reduced expression of COX-2 (44). This has also been found in other cell types, including vascular endothelial cells (30), rat Kupffer cells (147), and the J774 macrophage cell line (151). In addition, COX activation by inducible stimuli is driven in some models by mechanisms that do not seem to involve NO formation. This has been shown to occur, for instance, in astroglial cells treated with IFN-γ, where the elevation of PG levels and the expression of COX-2 have been found to not be affected by NOS activation (52).
At this stage, the relative roles of NO and PN on COX activation and/or induction remain to be defined, although there is no doubt that both species are involved at least in vitro. The relevance for PN as an activator/inducer of the COX enzymes in vivo has not been confirmed, mainly because selective PN decomposition catalysts (PNDCs) have not been available until now. Selective PNDCs have been designed and synthesized by Neumann and colleagues (31, 121, 122) for a recent series of elegant experiments. Studies are currently planned by our groups to use these probes as pharmacological tools to tease apart the roles of PN on PG biosynthesis. We have already demonstrated their use as potent anti-inflammatory agents, which are also able to block pain of various etiologies (31, 121, 122).
NO Pathway Is Modulated by COX-Derived Products
Products of the COX pathway may inhibit or increase the release of NO. High levels of PGI2 inhibit LPS-stimulated iNOS induction in J774.2 macrophages or murine peritoneal macrophages without affecting the enzyme once expressed (92). In rat Kupffer cells stimulated with endotoxin, inhibition of endogenously produced PG by indomethacin resulted in a concomitant inhibition of NO release (92) and conversely, indomethacin did not inhibit NO release in mouse RAW 264.7 cells nor J774.2 macrophages (128). Thus, there are clearly cell-specific responses to PG exposure. The mechanism of action of PGs on the NOS pathway has been attributed in most instances to their activation of endogenous adenylate cyclases with subsequent increase in cyclic AMP (cAMP) levels. Increases in intracellular cAMP have been suggested as mediating either a stimulatory or an inhibitory role of PG on NOS pathway. For instance, in rat vascular smooth muscle, PKA activators such as forskolin or dibutyryl cAMP increases iNOS mRNA, as well as the formation of NO (92). Other studies have found an inhibitory influence of PGE2 on nNOS expression. Thus, in cultured macula densa cells, PGE2 significantly reduced nNOS mRNA expression. In COX-2−/− mice, nNOS mRNA expression in kidney was upregulated throughout the postnatal periods. The induction of nNOS protein expression and NOS activity in COX-2−/− mice were localized to macula densa cells. These findings reveal that the absence of either COX-2 or nNOS is associated with suppressed renin secretion (15). Furthermore, the inhibitory effect of PGE2 on nNOS mRNA levels is a novel interaction between NO- and PG-mediated pathways of renin regulation (15).
COX activation, in turn, modulates l-arginine-NO pathway, and it has been shown that COX inhibition decreases NOS activity in human platelets (13). Both COX and NOS pathways regulate platelet function; TxA2 induces platelet aggregation, whereas NO inhibits it. Inhibition of COX by two different agents, aspirin and indomethacin, markedly attenuates NOS activity, as determined directly by the measurement of l-citrulline and nitrite formation and indirectly by the measurement of cGMP accumulation in platelets. The inhibitory effects of aspirin and indomethacin on NOS activity were reversed by exogenous Ca2+, as well as by a functional TxA2 mimetic, whereas a selective thromboxane A2 synthase inhibitor and the TxA2 endoperoxide receptor blocker exerted qualitatively similar effects as COX inhibitors on NOS activity. These effects were not associated with a change in NOS protein expression in platelets, suggesting that the effects of COX inhibitors are mediated, at least in part, via TxA2 inhibition and Ca2+ mobilization in platelets (13). Because Ca2+ is a key regulator of NOS activity, it is not surprising that a reduction in intracellular Ca2+ by aspirin, indomethacin, or a selective TxA2 inhibitor results in decreased NOS activity (13). Thus, mobilization of intracellular Ca2+ is a key step in the regulation of platelet function, and inhibition of NOS and that Ca2+ mobilization by COX inhibitors may serve as a regulatory step in the interaction between the COX and NOS pathways.
When analyzed collectively, these diverse experimental data on the inhibitory/stimulatory effects of PG on the NOS pathways are controversial and insufficient to draw any potential conclusion on functional relevance.
Physiological Implications of the Modulation of the Cyclooxygenase Pathway by Nitroxidative Species
The activation of COX-1 by intracellular or extracellular NO released from cNOS has important consequences in normal physiological conditions. Production of small amounts of NO and PGs from the constitutive enzymes regulates various physiological processes, including platelet aggregation, white blood cell adhesion, blood vessel tone, cytoprotection in the kidney and in the intestinal mucosa, renal nephron transport, tubuloglomerular feedback, and renin release. The main difference between NO and PGs in mediating these effects lies at the level of their respective intracellular transduction mechanisms. Thus, NO activates soluble guanylate cyclases to increase cGMP, while PGs activate adenylate cyclase to increase cAMP. Another interesting effect of NO acting upon COX-1 may be the complex regulation of neuropeptide release. Norepinephrine mediates the release of luteinizing hormone-releasing hormone (LHRH) from its terminals, a process known to require increased release of PGE2 (123). In an elegant study, Rettori et al. (123) demonstrated that LHRH release from norepinephrine-stimulated hypothalamic slices was prevented by NOS inhibition and showed that this inhibition was due to inhibition of norepinephrine-mediated PGE2 release by the NOS inhibitors. These results raise the possibility that NO through its ability to activate COX, may mediate exocytosis of secretory granules for other neuropeptides that are known to be regulated by PGE2. NO-driven COX-1 activation also affects reproduction in that NO modulates uterine motility (36) by activating COX-1 and releasing PGE2. Moreover, the recent observation of the dual inhibition of NO and PG by nonselective NOS inhibitors may well explain the clinically deleterious effects caused by these drugs in kidneys and the gastrointestinal tract, where both NO and PG are considered to be cytoprotective. As initially discussed by Wallace (173), there is ample evidence that selective inhibitors of COX-2 produce less gastric damage than standard NSAIDs when administered acutely to healthy animals. However, most patients taking standard NSAIDs do not develop clinically significant gastric injury, although a subset of patients is more susceptible to the gastric-damaging actions of these drugs (54). Moreover, the presence of both NOS isoforms in the vasculature raises the question of which is the predominant cause of the increased production of vasodilatory PGs that are critical to preserving renal blood flow during volume depletion and dehydration stress. Inhibition of this homeostatic response accounts for the most common renal side effects associated with nonselective NSAID therapy (102). In particular, it has been demonstrated that combinations of NSAIDs, taken in large doses over a prolonged period, cause renal papillary necrosis and interstitial scarring (63). Such patients develop chronic renal failure and are susceptible to the subsequent development of uroepithelial tumors. It is not clear what comprises the mechanism of NSAID toxicity; theories include the effects of COX inhibition and direct toxicity owing to elevated concentration of the drugs in the medulla, anoxia, and metabolic effects (8). During endotoxemia, PGE2 derived from COX-1 and COX-2 apparently exerts an inhibitory effect on gastric iNOS, despite no change in iNOS protein or mRNA. Thus, this PGE2 may represent a cytoprotective mechanism for iNOS, since upregulation of iNOS and generation of toxic byproducts, such as PN is deleterious (160, 176). As an extension to these studies, Franco and Talamini (37) reported that COX-2 inhibition in human gastric biopsies during Helicobacter pylori infection decreased the production of PGE2 and NO, but iNOS inhibitors did not modulate PGE2 generation. It is, therefore, possible that inhibition of NO production following COX inhibition contributes to gastrointestinal issues associated with the use of both selective and nonselective COX inhibitors (171, 173).
The findings that NO activates COX-1 enzyme has uncovered additional mechanisms by which NO donors might exert beneficial clinical effects. As one important example, in the presence of an intact endothelium, antiplatelet NO normally keeps blood vessels dilated, and in conjunction with PGI2, maintains the nonthrombogenic nature of the endothelium. Disease states that damage the endothelium or alter the normal properties of the endothelial cells lead to a decrease in the production of NO, tipping the balance in favor of constriction, thrombosis, and vasoocclusion.
Diseases associated with locally impaired release of NO include coronary artery spasms that occur either spontaneously (Prinzmetal angina) or in the context of atherosclerosis, myocardial ischemia, and stroke (166). Theoretically this imbalance can be restored by exogenous NO donors, which should be uniquely beneficial in conditions with endothelial dysfunction because they preempt a failing endogenous system. Since NO donors do not require an intact endothelium to be effective, they will restore vascular dilation and suppress the tendency to platelet aggregation. Although the vascular effects of NO donors have been well documented, these compounds are also endowed with antiplatelet effects and antithrombotic properties. NO donors inhibit platelet adhesion and aggregation in vitro and in vivo (24, 95, 130), increase bleeding time in humans (70), and synergize with PGI2 (61, 95). Furthermore, NO donors potentiate the activity of thrombolytic factors in providing further protection against vascular occlusion (61). NO donors that release NO spontaneously are far more potent inhibitors of platelet function in vitro compared with those requiring bioconversion. Platelets do not have the enzyme to convert organic nitrates to NO, and therefore, in vitro, they will respond only to very high doses of nitrates (1). The fact that endothelial cells can metabolize organic nitrates, such as glyceryl trinitrate (GTN), as efficiently as smooth muscle cells (6, 34, 129), is important because it provides the key to understand how organic nitrates inhibit platelet function in vivo, whereas they generally display weak anti-platelet effects in vitro (6, 75, 129). The findings that smooth muscle cells and endothelial cells significantly increase the in vitro antiplatelet effects of GTN (where the dose of GTN required to inhibit platelet aggregation in vitro approximates that used in vivo) led to the proposal that the increased effectiveness of organic nitrates in vivo as antiplatelet agents is due to conversion to NO by cells of the vascular tree, such as smooth muscle cells and endothelial cells (6, 34, 129). In this respect, endothelial cells may be more important because of their potentially direct contact with platelets. NO donors also promote the release of PGI2 in vivo and in vitro from endothelial cells (25, 69, 84, 138). The latter effect presumably reflects the ability of NO donors to activate the cyclooxygenase pathway directly, a phenomenon associated with a marked release of prostaglandins (128).
Pathological Implications of Modulating the Cyclooxygenase Pathway by Nitroxidative Species
In vivo studies reveal that regulation of COX by NO is a powerful mechanism by which NO modulates the course of the inflammatory response. Thus, iNOS and COX-2 are induced in a number of inflammatory models, including rabbit hydronephrotic kidney (131, 139), endotoxin-induced septic shock (132, 150), inflammation (45, 57, 86, 126, 134, 136, 137, 167, 186), ischemia-reperfusion injuries, and pain (82, 104, 134, 146). In these settings, inhibition of NO inhibits both NO and PG release, and the anti-inflammatory potency of the iNOS inhibitors correlates with their respective ability to block both NO and PGs. For instance, in the acute inflammation of carrageenan-induced paw edema in rats, inhibition of edema with N6-(1-imimoethyl)-l-lysine (l-NIL) correlates with dose-dependent inhibition of NO synthesis and formation of proinflammatory PGs (134). That the proinflammatory role of NO involves prostaglandins has also been demonstrated by injection of sodium nitroprusside to elicit edema in the footpad of rats, a response that is attenuated or blocked by NSAIDS (137). Therefore, the effects of endogenously released NO on COX-2 are mimicked by exogenous NO. In acute pancreatitis, the increased production of PGs was inhibited by NOS inhibitors, indicating the participation of NO in this process (19). Furthermore, injection of carrageenan into the preformed air pouch of a rat induces an inflammatory response characterized by iNOS and COX-2 induction, white blood cell infiltration, edema, and protein leakage into the pouch (126). Selective inhibition of iNOS (or other iNOS inhibitors) by l-NIL or aminoguanidine inhibited not only NO but also PG production. All other parameters of inflammation were attenuated, and histological examination of the pouch lining from animals that were dosed in vivo with the l-NIL revealed a lack of inflammation (126). Additional studies also reported a cross talk between iNOS and COX-2 in the lung of rats given endotoxin; release of PGI2 from lungs harvested from endotoxin-treated rats in the presence of an iNOS inhibitor was significantly attenuated compared with control animals (136) Importantly, targeted deletion of iNOS in mice decreases COX-2 activity (79) clearly strengthening the tie between NO and PN and eicosanoid biosynthesis. Injection of NO donors, such as sodium nitroprusside or nitroglycerin, into the air pouch activates COX-2, resulting in profound increase in PGE2 release (126). Treatment of diabetic rats with nitro-l-arginine methyl ester (l-NAME) prevents the up-regulation of renal COX-2 and the associated increase in renal PG release in response to arachidonic acid, suggesting that NO or a product of NO may contribute to the induction of COX-2 in diabetes (14). Such induction of COX-2 in diabetes may contribute to renal functional and structural changes associated with this condition, as inhibition of COX-2 reduces GFR in the diabetic rat (60) and other markers of renal injury in hypertensive diabetic rats (12). In this content, it is well documented that COX-2 and nNOS are coexpressed in macula densa cells, and their coexpression is stimulated in a number of high-renin states. In particular, plasma renin activity was significantly reduced in nNOS−/− mice of an otherwise mixed genetic background and in COX-2−/− mice with either BALB/c or C57/BL6 backgrounds (112). Additional findings further implicate important interactions between the NO and COX-2 systems in the regulation of arteriolar tone and the renin-angiotensin system by the macula densa (16, 48).
The interaction between iNOS-derived NO and COX-2 has raised the possibility that some deleterious effects of ischemia-induced iNOS expression are mediated through COX-2 reaction products, in brain and bladder, for example (81, 101, 106). The important interaction between iNOS-derived NO and COX-2 in cerebral ischemia was described for the first time by Iadecola and colleagues (106). Although iNOS-positive neutrophils were found in close spatial proximity to COX-2-positive neurons, inhibition of iNOS by aminoguanidine was found to attenuate COX-2 reaction products only in the ischemic region (106). Furthermore, the authors demonstrated that postischemic PGE2 production is reduced in iNOS-/- mice despite normal COX-2 mRNA and protein expression (106). These observations led them to propose that in the ischemic brain, NO produced by iNOS drives COX-2 activity and increases its catalytic output (106). In a subsequent study, Nagayam et al. (101) assessed the relative contribution of iNOS and COX-2 to ischemic brain damage. They reasoned that if the pathogenic effects of iNOS-derived NO and COX-2 are mediated through largely independent mechanisms, then inhibition of COX-2 in iNOS-null mice (iNOS−/−) should confer a protection greater than that attained by iNOS deletion alone. Conversely, if iNOS-derived NO mediates some of its deleterious effects through COX-2, then inhibiting COX-2 in iNOS−/− mice should not provide added protection. Using the selective COX-2 inhibitor, NS398, the authors reported that inhibition of COX-2 attenuates ischemic brain damage in wild-type mice but not in iNOS−/− mice, thus supporting the hypothesis that COX-2 activation is an important pathogenic factor in the damage produced by iNOS-derived NO (101). In a model of bladder ischemia-reperfusion (I/R), NO derived from up-regulated iNOS after I/R increased COX-2-derived PG via a cGMP-independent mechanism. Thus, NO-mediated activation of COX-2 may be an important mechanism for the modulation of bladder function after I/R injury (81). Another interesting interaction between the NOS and COX pathways occurs in pain. For example, inhibiting the antihyperalgesic effects of NOS inhibitors in models of inflammation is associated with inhibition of both NO and PGs. NOS inhibitors (73) have also been shown to attenuate spinal release of excitatory amino acids, such as glutamate and PGE2, following NMDA receptor activation (146). In addition, Matsui et al. (82) demonstrated that activation of Toll-like receptor 4 in spinal cord microglial cells released both NO and PGE2 following activation of p38 kinase. This was attenuated by NOS inhibitors, while COX inhibitors had no effect on NO (82).
A specific area of interest in which the simultaneous modulation of NO and COX has been recently investigated is the development and prevention of neoplasias, notably colon cancer. Colorectal carcinoma remains a major cause of cancer patient mortality worldwide. The development, progression, and metastasis of this carcinoma follow a well-defined series of steps, apparently including a dependence on local NO concentration and overexpression of cyclooxygenase enzymes (111, 119, 120, 154). In particular, at low levels, NO increases tumor growth and development, while higher levels of NO (above optimal concentrations) may express cytotoxic or cytostatic effects (140). In addition, the degree of tumor malignancy is also linked to the amount and activity of NOS proteins (39), and the expression of NOS may promote metastatic behavior of tumor cells. Moreover, the level of nitrite and nitrate in serum or culture supernatants can be significantly reduced by application of l-NAME, a competitive inhibitor of NOS. Its activity is based on structural similarity to l-arginine, a substrate for NOS, and competitive inhibition of l-arginine metabolization. It has been shown that l-NAME exerts anti-invasive and antimetastatic effects on many cancers, including colon carcinoma (181). On the other hand, COX-2 has been suggested to play an important role in carcinogenesis and opens new anticancer therapeutic possibilities (40, 185). Furthermore, evidence exists that NS398 may in a dose-dependent manner inhibit proliferation and induce death in many carcinomas, including colon cancer (99). COX-2-derived bioactive prostaglandins, especially PGE2, have a direct effect on cancer cells by increasing their motility and metastatic potential. However, PGE2 is one of the main proinflammatory factors, which is elevated in colorectal cancers and promotes their development by inducing cell proliferation and inhibiting apoptosis (68, 153, 175). Therefore, selective inhibitors of COX-2 may also block PGE2 generation, thus minimizing its colorectal tumor-promoting effects. Recent reports also demonstrated cross-talk between NO, NOS levels, and COX expression in cancer cells (111, 114, 115). Selective inhibition of COX-2 and PGE2 production has been shown to influence also NO synthesis in the tumor microenvironment and may represent an important goal for prevention or therapy of colon cancers. In particular, l-arginine applied in experimental settings exerts presumptively antitumor effects by reducing tumor size and incidence and by retarding tumor growth and metastasis (109), producing beneficial effects in chemically induced colorectal carcinomas by influencing host immune system functions (77). Experimental supplementation with this amino acid also leads to increased NO formation by the oxidative deaminase pathway. In turn, an increased local NO level may induce cytostasis by inhibiting hyperproliferation of tumor cells or may be cytotoxic to tumor cells by the formation of toxic peroxynitrite (77). l-arginine was also recently found able to decrease PGE2 levels in cocultures of tumor spheroids with colonic epithelium. These results suggest an important role of NO in chemoprevention of tumor-epithelial cell interactions rather than its effect on tumor-stromal development, activity, and viability. In addition, NO was found to inhibit COX-2 enzymatic activity by reacting with iron in the enzyme's heme group. Moreover, l-NAME, an inhibitor of NO synthases, which decreased the NO level and motility of tumor cells under in vitro experimental settings, has a relatively weak effect on COX-2 level. These apparently contradictory results reflect that any NO and COX-2 relationship depends upon cell types that have been used, their direct interactions, the kind of inhibitor applied, and the local microenvironmental conditions. These elements regulate NO effects on COX-2 and PGE2 synthesis in normal as well as in cancer cell lines. Indeed, it has recently been reported that mucosal PGE2 in noncancer cell lines is decreased by SC-560, a COX-1 selective inhibitor (143), but not by l-NAME (108). On the other hand, l-NAME reduces PGE2 generation by attenuating COX-2 expression (113), which may limit the invasion and migration of tumor cells (181). Thus, consistent cross-talk among NO, COX-2, and PGE2 exists in colorectal cancer cell lines. This relationship is rather unidirectional where NO level influences COX-2 and PGE2 levels. Moreover, any imbalances in NO level caused by exogenous factors influence COX-2 and PGE2 amounts depending on the kind of cells, their reciprocal interactions, and the local conditions of the microenvironment. The knowledge of these effects may be useful in limiting progression and invasion of colon carcinoma.
Perspectives and Significance
Compelling data have been generated over the last 18 yr supporting the concept that NO is involved in the regulation of COX pathway and vice versa. In particular, basal release of both NO and PGs appears to be vital in the maintenance of a basal “protective” homeostasis in both vascular and nonvascular tissues against exogenous, as well as endogenous insults. This occurs via inducible NOS and COX enzymes that when activated during inflammatory states, release micromolar concentration of both mediators. On the other hand, disruption of NOS/COX cross-talk with the simultaneous release of large quantities of NO and PGs represents key events in tissue damage occurring in a diverse array of inflammatory and degenerative processes. Such a process may have significant implications for the regulation of key pathophysiological events, including blood flow and oxygen delivery, smooth muscle cell modulation in vascular and nonvascular tissues, immune response, and neuron/glia interaction. Furthermore, the finding that COX inhibition via NSAIDs has a significant effect on vascular tone during hypoxia only after prior inhibition of NOS with l-NAME, suggests a compensatory response by PGs to maintain the vasodilator response to hypoxia, which is tightly NO dependent.
A similar conclusion can be drawn when evaluating the role of NO and PGs in neurodegenerative processes. In brain tissues, in which cellular populations coexist that express both constitutive and iNOS and COX enzymes (e.g., microglial cells and astrocytes), early stages of HIV-related brain damage are characterized by down-regulation of constitutive NOS and COX that, under basal conditions, tend to counteract the massive diffusion of HIV-related neurotoxins into brain tissue. Under these conditions, it is likely that the inhibition of both NOS and COX aggravates the neurodegenerative process and enhances neuronal cell death. Conversely, the overproduction of free oxyradical species, combined with elevated levels of NO, leads to generation of PN that then modulates NF-κB via IKK-B, leading to “intensive” activation of iNOS and COX-2. This activation seems to be the key event in generating toxic concentration of both mediators, as shown by evidence that the combined effect of NO donors and NSAID, at this stage, exerts a beneficial effect on HIV-related cognitive dysfunction (56, 94). That this represents a more general reactive model for the cooperation of NOS and COX enzymes is clearly expressed by changes of protective enzymes in astroglial cells accompanying NOS/COX interaction. For example, glutamine synthase, the enzyme that depletes the synaptic space of secreted glutamate by converting this mediator into nontoxic glutamine, is regulated in the same way by NO and PGs. In particular, neurotoxic insults (e.g., HIV coating gp120 glycoprotein), which work by modulating basal NO/PGs release, also decrease glutamine synthase expression via this mechanism, an effect restored by NO-NSAIDs. When large quantities of NO/PGs are generated via iNOS and COX-2, GS is unable to remove right quantities of glutamate from periastroglial space leading to apoptotic cell (56). Thus, maintaining adequate basal concentrations of NO/PGs mechanisms serves not only to keep iNOS and COX inhibited, but also to protect specific tissues against toxic insults, including oxidative stress. This is relevant not only for the understanding of pathophysiological events involving NO/PGs mechanisms but also for the choice of right treatment to be performed based on NO/PG modulation.
It remains to be better understood how the protective effect of NO/PGs basal synthesis mechanisms is disrupted under pathological conditions, which then lead to widespread activation of toxic mechanisms. In this respect, the formation of PN seems to represent a key event in the shift of NO/PGs mechanisms into toxic behavior.
Current data indicate that NO modulates eicosanoid production at several levels, and it is increasingly recognized that more than one enzyme in the arachidonic acid pathway can be affected by NO and/or PN. At this stage, it is difficult to determine whether effects of NO on PG production are mediated by NO itself or its byproduct, PN; currently available NOS inhibitors block both NO and PN formation (73, 152). Clearly, PN can activate COX-1 and COX-2 and can induce COX-2 (73, 152). In this regard, we recently reported that intraplantar administration of PN in rats causes peripheral sensitization and pain following activation of COX-1 and COX-2, as well as the NF-κB-induction of COX-2 leading to increased formation of PGE2. In this model we found that the decomposition of elevated PN levels using a PNDC, such as FeTMPyP [Fe(III)5,10,15,20-tetrakis(N-methylpyridinium-4-yl)porphyrin)] (125, 133), significantly blocked pain (104). Importantly, such PNDCs potently synergized with NSAIDs, such as ibuprofen and NS398, in blocking pain and inflammation. These exciting results suggest that such a combination treatment paradigm may provide an alternative strategy to safely and effectively manage pain states that are currently poorly controlled or limited by the side-effect profile of selective and nonselective COX inhibitors (104). Regrettably, current PNDCs are rather nonselective, showing equal catalytic activities toward both PN and SO (4). Thus, current data cannot decipher each species' contribution in the normal regulation of PG biosynthesis, nor the clinically important pathophysiological processes underlying a host of acute and chronic diseases and pain states. Use of a “SO-sparing” PNDC recently described by Neumann and colleagues (31, 121, 122) will provide a means to define the relative contribution of SO and PN, and thus NO, providing invaluable insight on the importance of these species in the regulation of PG biosynthesis.
Therefore, detailed mechanisms by which NO regulates prostaglandin production and vice versa remains poorly understood, partly because of the interaction between these two pathways occurs at multiple levels, the complexity of NO redox chemistry (23, 92), and the lack of selective probes to decipher pathways in vivo.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.S., S.F.K., and V.M. prepared figures; D.S., S.F.K., and V.M. drafted manuscript; D.S., S.F.K., and V.M. edited and revised manuscript; D.S., S.F.K., and V.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We would like to thank Dr. Andrew J. Lechner (Department of Pharmacological and Physiological Science, Saint Louis University School of Medicine, St. Louis, MO), Dr. William L. Neumann (Department of Pharmaceutical Sciences, School of Pharmacy, Southern Illinois University Edwardsville, Edwardsville, IL) and Dr. Daniel L. Simmons (Department of Chemistry and Biochemistry, Brigham Young University, Provo, UT) for editorial help and for providing pertinent insight and suggestions to our review article.
REFERENCES
- 1. Ahlner J, Andersson RG, Torfgard K, Axelsson KL. Organic nitrate esters: clinical use and mechanisms of actions. Pharmacol Rev 43: 351–423, 1991. [PubMed] [Google Scholar]
- 2. Alvarez B, Ferrer-Sueta G, Freeman BA, Radi R. Kinetics of peroxynitrite reaction with amino acids and human serum albumin. J Biol Chem 274: 842–848, 1999. [DOI] [PubMed] [Google Scholar]
- 3. Bailey JM, Muza B, Hla T, Salata K. Restoration of prostacyclin synthase in vascular smooth muscle cells after aspirin treatment: regulation by epidermal growth factor. J Lipid Res 26: 54–61, 1985. [PubMed] [Google Scholar]
- 4. Batinic-Haberle I, Reboucas JS, Spasojevic I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antiox Redox Signal 13: 877–918, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87: 1620–1624, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benjamin N, Dutton JA, Ritter JM. Human vascular smooth muscle cells inhibit platelet aggregation when incubated with glyceryl trinitrate: evidence for generation of nitric oxide. Brit J Pharmacol 102: 847–850, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bergstrom S, Sjovall J. The isolation of prostaglandin. Acta Chem Scand 11: 1086, 1957. [Google Scholar]
- 8. Bing RJ. Cyclooxygenase-2 inhibitors: is there an association with coronary or renal events? Curr Atheroscler Rep 5: 114–117, 2003. [DOI] [PubMed] [Google Scholar]
- 9. Bonaiuto C, McDonald PP, Rossi F, Cassatella MA. Activation of nuclear factor-κB by β-amyloid peptides and interferon-gamma in murine microglia. J Neuroimmunol 77: 51–56, 1997. [DOI] [PubMed] [Google Scholar]
- 10. Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem 63: 175–195, 1994. [DOI] [PubMed] [Google Scholar]
- 11. Chao CC, Lokensgard JR, Sheng WS, Hu S, Peterson PK. IL-1-induced iNOS expression in human astrocytes via NF-κB. Neuroreport 8: 3163–3166, 1997. [DOI] [PubMed] [Google Scholar]
- 12. Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol 87: 2851–2857, 2002. [DOI] [PubMed] [Google Scholar]
- 13. Chen L, Salafranca MN, Mehta JL. Cyclooxygenase inhibition decreases nitric oxide synthase activity in human platelets. Am J Physiol Heart Circ Physiol 273: H1854–H1859, 1997. [DOI] [PubMed] [Google Scholar]
- 14. Chen YJ, Li J, Quilley J. Effect of inhibition of nitric oxide synthase on renal cyclooxygenase in the diabetic rat. Eur J Pharmacol 541: 80–86, 2006. [DOI] [PubMed] [Google Scholar]
- 15. Cheng HF, Wang CJ, Moeckel GW, Zhang MZ, McKanna JA, Harris RC. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of diabetes and hypertension. Kidney Intl 62: 929–939, 2002. [DOI] [PubMed] [Google Scholar]
- 16. Cheng HF, Wang JL, Zhang MZ, McKanna JA, Harris RC. Nitric oxide regulates renal cortical cyclooxygenase-2 expression. Am J Physiol Renal Physiol 279: F122–F129, 2000. [DOI] [PubMed] [Google Scholar]
- 17. Cheng HF, Zhang MZ, Harris RC. Nitric oxide stimulates cyclooxygenase-2 in cultured cTAL cells through a p38-dependent pathway. Am J Physiol Renal Physiol 290: F1391–F1397, 2006. [DOI] [PubMed] [Google Scholar]
- 18. Choi SH, Langenbach R, Bosetti F. Genetic deletion or pharmacological inhibition of cyclooxygenase-1 attenuate lipopolysaccharide-induced inflammatory response and brain injury. FASEB J 22: 1491–1501, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Closa D, Hotter G, Prats N, Bulbena O, Rosello-Catafau J, Fernandez-Cruz L, Gelpi E. Prostanoid generation in early stages of acute pancreatitis: a role for nitric oxide. Inflammation 18: 469–480, 1994. [DOI] [PubMed] [Google Scholar]
- 20. Colasanti M, Persichini T. Nitric oxide: an inhibitor of NF-κB/Rel system in glial cells. Brain Res Bull 52: 155–161, 2000. [DOI] [PubMed] [Google Scholar]
- 21. Colasanti M, Suzuki H. The dual personality of NO. Trends Pharmacol Sci 21: 249–252, 2000. [DOI] [PubMed] [Google Scholar]
- 22. Corbett JA, Kwon G, Turk J, McDaniel ML. IL-1β induces the coexpression of both nitric oxide synthase and cyclooxygenase by islets of Langerhans: activation of cyclooxygenase by nitric oxide. Biochemistry 32: 13767–13770, 1993. [DOI] [PubMed] [Google Scholar]
- 23. Cuzzocrea S, Salvemini D. Molecular mechanisms involved in the reciprocal regulation of cyclooxygenase and nitric oxide synthase enzymes. Kidney Int 71: 290–297, 2007. [DOI] [PubMed] [Google Scholar]
- 24. De Caterina R, Giannessi D, Crea F, Chierchia S, Bernini W, Gazzetti P, L'Abbate A. Inhibition of platelet function by injectable isosorbide dinitrate. Am J Cardiol 53: 1683–1687, 1984. [DOI] [PubMed] [Google Scholar]
- 25. De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest 96: 60–68, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. delaTorre A, Schroeder RA, Bartlett ST, Kuo PC. Differential effects of nitric oxide-mediated S-nitrosylation on p50 and c-jun DNA binding. Surgery 124: 137–141; discussion 141–132, 1998. [PubMed] [Google Scholar]
- 27. DeWitt DL. Prostaglandin endoperoxide synthase: regulation of enzyme expression. Biochim Biophys Acta 1083: 121–134, 1991. [DOI] [PubMed] [Google Scholar]
- 28. DeWitt DL, Smith WL. Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc. Natl. Acad. Sci. USA 85: 1412–1416, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diaz-Cazorla M, Perez-Sala D, Lamas S. Dual effect of nitric oxide donors on cyclooxygenase-2 expression in human mesangial cells. J Am Soc Nephrol 10: 943–952, 1999. [DOI] [PubMed] [Google Scholar]
- 30. Doni MG, Whittle BJ, Palmer RM, Moncada S. Actions of nitric oxide on the release of prostacyclin from bovine endothelial cells in culture. EurJ Pharmacol 151: 19–25, 1988. [DOI] [PubMed] [Google Scholar]
- 31. Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, Dagostino C, Ryerse J, Rausaria S, Kamadulski A, Neumann WL, Salvemini D. Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci 32: 6149–6160, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drapier JC, Hibbs JB., Jr Differentiation of murine macrophages to express nonspecific cytotoxicity for tumor cells results in l-arginine-dependent inhibition of mitochondrial iron-sulfur enzymes in the macrophage effector cells. J Immunol 140: 2829–2838, 1988. [PubMed] [Google Scholar]
- 33. Egan RW, Paxton J, Kuehl FA., Jr Mechanism for irreversible self-deactivation of prostaglandin synthetase. J Biol Chem 251: 7329–7335, 1976. [PubMed] [Google Scholar]
- 34. Feelisch M, Kelm M. Biotransformation of organic nitrates to nitric oxide by vascular smooth muscle and endothelial cells. Biochem Biophys Res Commun 180: 286–293, 1991. [DOI] [PubMed] [Google Scholar]
- 35. Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med 15: 391–404, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Franchi AM, Chaud M, Rettori V, Suburo A, McCann SM, Gimeno M. Role of nitric oxide in eicosanoid synthesis and uterine motility in estrogen-treated rat uteri. Proc Natl Acad Sci USA 91: 539–543, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franco L, Talamini G. Cross-talk between inducible nitric oxide synthase and cyclooxygenase in Helicobacter-pylori-induced gastritis. Medical principles and practice. Intl. J. Kuwait Univ., Health Science Centre 18: 477–481, 2009. [DOI] [PubMed] [Google Scholar]
- 38. Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu Rev Biophys Biomol Struct 32: 183–206, 2003. [DOI] [PubMed] [Google Scholar]
- 39. Gavilanes J, Moro MA, Lizasoain I, Lorenzo P, Perez A, Leza JC, Alvarez-Vicent JJ. Nitric oxide synthase activity in human squamous cell carcinoma of the head and neck. Laryngoscope 109: 148–152, 1999. [DOI] [PubMed] [Google Scholar]
- 40. Giercksky KE. COX-2 inhibition and prevention of cancer. Best Practice Res Clin Gastroenterol 15: 821–833, 2001. [DOI] [PubMed] [Google Scholar]
- 41. Goldblatt MW. Properties of human seminal plasma. J Physiol 84: 208–218, 1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goodwin DC, Gunther MR, Hsi LC, Crews BC, Eling TE, Mason RP, Marnett LJ. Nitric oxide trapping of tyrosyl radicals generated during prostaglandin endoperoxide synthase turnover. Detection of the radical derivative of tyrosine 385. J BiolChem 273: 8903–8909, 1998. [DOI] [PubMed] [Google Scholar]
- 43. Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320: 454–456, 1986. [DOI] [PubMed] [Google Scholar]
- 44. Guastadisegni C, Minghetti L, Nicolini A, Polazzi E, Ade P, Balduzzi M, Levi G. Prostaglandin E2 synthesis is differentially affected by reactive nitrogen intermediates in cultured rat microglia and RAW 264.7 cells. FEBS Lett. 413: 314–318, 1997. [DOI] [PubMed] [Google Scholar]
- 45. Haider A, Lee I, Grabarek J, Darzynkiewicz Z, Ferreri NR. Dual functionality of cyclooxygenase-2 as a regulator of tumor necrosis factor-mediated G1 shortening and nitric oxide-mediated inhibition of vascular smooth muscle cell proliferation. Circulation 108: 1015–1021, 2003. [DOI] [PubMed] [Google Scholar]
- 46. Hajjar DP, Lander HM, Pearce SF, Upmacis RK, Pomerantz KB. Nitric oxide enhances prostaglandin-H synthase-1 activity by a hemeindependentmechanism: evidence implicating nitrosothiols. J Am Chem Soc 117: 3340–3346, 1995. [Google Scholar]
- 47. Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol 70: 357–377, 2008. [DOI] [PubMed] [Google Scholar]
- 48. Harris RC, Cheng H, Wang J, Zhang M, McKanna JA. Interactions of the renin-angiotensin system and neuronal nitric oxide synthase in regulation of cyclooxygenase-2 in the macula densa. Acta Physiol Scan 168: 47–51, 2000. [DOI] [PubMed] [Google Scholar]
- 49. Hartlage-Rubsamen M, Lemke R, Schliebs R. Interleukin-1β, inducible nitric oxide synthase, and nuclear factor-κB are induced in morphologically distinct microglia after rat hippocampal lipopolysaccharide/interferon-gamma injection. J Neurosci Res 57: 388–398, 1999. [PubMed] [Google Scholar]
- 50. Henry Y, Lepoivre M, Drapier JC, Ducrocq C, Boucher JL, Guissani A. EPR characterization of molecular targets for NO in mammalian cells and organelles. FASEB J 7: 1124–1134, 1993. [DOI] [PubMed] [Google Scholar]
- 51. Hess DT, Matsumoto A, Nudelman R, Stamler JS. S-nitrosylation: spectrum and specificity. Nat Cell Biol 3: E46–E49, 2001. [DOI] [PubMed] [Google Scholar]
- 52. Hewett SJ. Interferon-gamma reduces cyclooxygenase-2-mediated prostaglandin E2 production from primary mouse astrocytes independent of nitric oxide formation. J Neuroimmunol 94: 134–143, 1999. [DOI] [PubMed] [Google Scholar]
- 53. Inoue T, Fukuo K, Morimoto S, Koh E, Ogihara T. Nitric oxide mediates interleukin-1-induced prostaglandin E2 production by vascular smooth muscle cells. Biochem Biophys Res Commun 194: 420–424, 1993. [DOI] [PubMed] [Google Scholar]
- 54. Jacobsen RB, Phillips BB. Reducing clinically significant gastrointestinal toxicity associated with nonsteroidal antiinflammatory drugs. Annals Pharmacother 38: 1469–1481, 2004. [DOI] [PubMed] [Google Scholar]
- 55. Janabi N, Chabrier S, Tardieu M. Endogenous nitric oxide activates prostaglandin F2 alpha production in human microglial cells but not in astrocytes: a study of interactions between eicosanoids, nitric oxide, and superoxide anion (O2−) regulatory pathways. J Immunol 157: 2129–2135, 1996. [PubMed] [Google Scholar]
- 56. Janda E, Visalli V, Colica C, Aprigliano S, Musolino V, Vadala N, Muscoli C, Sacco I, Iannone M, Rotiroti D, Spedding M, Mollace V. The protective effect of tianeptine on Gp120-induced apoptosis in astroglial cells: role of GS and NOS, and NF-κB suppression. Brit J Pharmacol 164: 1590–1599, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kanematsu M, Ikeda K, Yamada Y. Interaction between nitric oxide synthase and cyclooxygenase pathways in osteoblastic MC3T3–E1 cells. J Bone Min Res 12: 1789–1796, 1997. [DOI] [PubMed] [Google Scholar]
- 58. Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 310: 1966–1970, 2005. [DOI] [PubMed] [Google Scholar]
- 59. Kim WK, Choi YB, Rayudu PV, Das P, Asaad W, Arnelle DR, Stamler JS, Lipton SA. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO. Neuron 24: 461–469, 1999. [DOI] [PubMed] [Google Scholar]
- 60. Komers R, Lindsley JN, Oyama TT, Schutzer WE, Reed JF, Mader SL, Anderson S. Immunohistochemical and functional correlations of renal cyclooxygenase-2 in experimental diabetes. J Clin Invest 107: 889–898, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Korbut R, Lidbury PS, Vane JR. Prolongation of fibrinolytic activity of tissue plasminogen activator by nitrovasodilators. Lancet 335: 669, 1990. [DOI] [PubMed] [Google Scholar]
- 62. Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol 12: 1094–1100, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kovacevic L, Bernstein J, Valentini RP, Imam A, Gupta N, Mattoo TK. Renal papillary necrosis induced by naproxen. Pediatr Nephrol 18: 826–829, 2003. [DOI] [PubMed] [Google Scholar]
- 64. Kulmacz RJ, van der Donk WA, Tsai AL. Comparison of the properties of prostaglandin H synthase-1 and -2. Progr Lipid Res 42: 377–404, 2003. [DOI] [PubMed] [Google Scholar]
- 65. Kurzrok R, Lieb C. Biochemical studies of human semen. II. Proc Soc Exp BiolMed 28: 268–272, 1930. [Google Scholar]
- 66. Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc Natl Acad Sci USA 93: 15069–15074, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Davis BJ, Lee CA. Disruption of the mouse cyclooxygenase 1 gene. Characteristics of the mutant and areas of future study. Adv Exp Med Biol 407: 87–92, 1997. [PubMed] [Google Scholar]
- 68. Larkins TL, Nowell M, Singh S, Sanford GL. Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer 6: 181, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Levin RI, Jaffe EA, Weksler BB, Tack-Goldman K. Nitroglycerin stimulates synthesis of prostacyclin by cultured human endothelial cells. J Clin Invest 67: 762–769, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lichtenthal PR, Rossi EC, Louis G, Rehnberg KA, Wade LD, Michaelis LL, Fung HL, Patrignani P. Dose-related prolongation of the bleeding time by intravenous nitroglycerin. Anes Anal 64: 30–33, 1985. [PubMed] [Google Scholar]
- 71. Lipsky PE, Brooks P, Crofford LJ, DuBois R, Graham D, Simon LS, Van De Putte LB, Abramson SB. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch Intern Med 160: 913–920, 2000. [DOI] [PubMed] [Google Scholar]
- 72. Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364: 626–632, 1993. [DOI] [PubMed] [Google Scholar]
- 73. Little JW, Doyle T, Salvemini D. Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain. Amino Acids 42: 75–94, 2012. [DOI] [PubMed] [Google Scholar]
- 74. Liu Y, Borchert GL, Phang JM. Polyoma enhancer activator 3, an ets transcription factor, mediates the induction of cyclooxygenase-2 by nitric oxide in colorectal cancer cells. J Biol Chem 279: 18694–18700, 2004. [DOI] [PubMed] [Google Scholar]
- 75. Loscalzo J. N-Acetylcysteine potentiates inhibition of platelet aggregation by nitroglycerin. J Clin Invest 76: 703–708, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lynch SM, Morrow JD, Roberts LJ, 2nd, Frei B. Formation of non-cyclooxygenase-derived prostanoids (F2-isoprostanes) in plasma and low density lipoprotein exposed to oxidative stress in vitro. J Clin Invest 93: 998–1004, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ma Q, Hoper M, Anderson N, Rowlands BJ. Effect of supplemental l-arginine in a chemical-induced model of colorectal cancer. World J Surg 20: 1087–1091, 1996. [DOI] [PubMed] [Google Scholar]
- 78. Markey CM, Alward A, Weller PE, Marnett LJ. Quantitative studies of hydroperoxide reduction by prostaglandin H synthase. Reducing substrate specificity and the relationship of peroxidase to cyclooxygenase activities. J Biol Chem 262: 6266–6279, 1987. [PubMed] [Google Scholar]
- 79. Marnett LJ, Wright TL, Crews BC, Tannenbaum SR, Morrow JD. Regulation of prostaglandin biosynthesis by nitric oxide is revealed by targeted deletion of inducible nitric-oxide synthase. J Biol Chem 275: 13427–13430, 2000. [DOI] [PubMed] [Google Scholar]
- 80. Massa PT, Wu C. Increased inducible activation of NF-κB and responsive genes in astrocytes deficient in the protein tyrosine phosphatase SHP-1. J Interfer Cytokine Res 18: 499–507, 1998. [DOI] [PubMed] [Google Scholar]
- 81. Masuda H, Kawano K, Matsuoka Y, Yokoyama M, Koga F, Saito K, Kihara K, Azuma H. Interactions between inducible nitric oxide synthase and cyclooxygenase-2 in response to ischaemia-reperfusion of rabbit bladder. BJU Int 106: 716–722, 2010. [DOI] [PubMed] [Google Scholar]
- 82. Matsui T, Svensson CI, Hirata Y, Mizobata K, Hua XY, Yaksh TL. Release of prostaglandin E2 and nitric oxide from spinal microglia is dependent on activation of p38 mitogen-activated protein kinase. Anesth Analg 111: 554–560, 2010. [DOI] [PubMed] [Google Scholar]
- 83. Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acids Res 24: 2236–2242, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mehta J, Mehta P, Ostrowski N. Effects of nitroglycerin on human vascular prostacyclin and thromboxane A2 generation. J Lab Clin Med 102: 116–125, 1983. [PubMed] [Google Scholar]
- 85. Merlie JP, Fagan D, Mudd J, Needleman P. Isolation and characterization of the complementary DNA for sheep seminal vesicle prostaglandin endoperoxide synthase (cyclooxygenase). J Biol Chem 263: 3550–3553, 1988. [PubMed] [Google Scholar]
- 86. Miceli F, Tringali G, Tropea A, Minici F, Orlando MT, Lanzone A, Navarra P, Apa R. The effects of nitric oxide on prostanoid production and release by human umbilical vein endothelial cells. Life Sci 73: 2533–3542, 2003. [DOI] [PubMed] [Google Scholar]
- 87. Miller SB. Prostaglandins in health and disease: an overview. Semin Arthritis Rheum 36: 37–49, 2006. [DOI] [PubMed] [Google Scholar]
- 88. Minghetti L, Polazzi E, Nicolini A, Creminon C, Levi G. Interferon-γ and nitric oxide down-regulate lipopolysaccharide-induced prostanoid production in cultured rat microglial cells by inhibiting cyclooxygenase-2 expression. J Neurochem 66: 1963–1970, 1996. [DOI] [PubMed] [Google Scholar]
- 89. Misko TP, Trotter JL, Cross AH. Mediation of inflammation by encephalitogenic cells: interferon gamma induction of nitric oxide synthase and cyclooxygenase 2. J Neuroimmunol 61: 195–204, 1995. [DOI] [PubMed] [Google Scholar]
- 90. Miyamoto T, Ogino N, Yamamoto S, Hayaishi O. Purification of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. J Biol Chem 251: 2629–2636, 1976. [PubMed] [Google Scholar]
- 91. Mollace V, Colasanti M, Rodino P, Lauro GM, Rotiroti D, Nistico G. NMDA-dependent prostaglandin E2 release by human cultured astroglial cells is driven by nitric oxide. Biochem Biophys Res Commun 215: 793–799, 1995. [DOI] [PubMed] [Google Scholar]
- 92. Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol Rev 57: 217–252, 2005. [DOI] [PubMed] [Google Scholar]
- 93. Mollace V, Muscoli C, Rotiroti D, Nistico G. Spontaneous induction of nitric oxide- and prostaglandin E2-release by hypoxic astroglial cells is modulated by interleukin 1β. Biochem Biophys Res Commun 238: 916–919, 1997. [DOI] [PubMed] [Google Scholar]
- 94. Mollace V, Nottet HS, Clayette P, Turco MC, Muscoli C, Salvemini D, Perno CF. Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends Neurosci 24: 411–416, 2001. [DOI] [PubMed] [Google Scholar]
- 95. Mollace V, Salvemini D, Vane J. Studies on the importance of the proposed release of nitric oxide from platelets. Thromb Res 64: 533–542, 1991. [PubMed] [Google Scholar]
- 96. Moncada S, Higgs EA. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J 9: 1319–1330, 1995. [PubMed] [Google Scholar]
- 97. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, pharmacology. Pharmacol Rev 43: 109–142, 1991. [PubMed] [Google Scholar]
- 98. Moore KP, Darley-Usmar V, Morrow J, Roberts LJ., 2nd Formation of F2-isoprostanes during oxidation of human low-density lipoprotein and plasma by peroxynitrite. Circ Res 77: 335–341, 1995. [DOI] [PubMed] [Google Scholar]
- 99. Moreira L, Castells A. Cyclooxygenase as a target for colorectal cancer chemoprevention. Curr Drug Targets 12: 1888–1894, 2011. [DOI] [PubMed] [Google Scholar]
- 100. Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 286: 954–959, 2001. [DOI] [PubMed] [Google Scholar]
- 101. Nagayama M, Niwa K, Nagayama T, Ross ME, Iadecola C. The cyclooxygenase-2 inhibitor NS-398 ameliorates ischemic brain injury in wild-type mice but not in mice with deletion of the inducible nitric oxide synthase gene. J Cerebral Blood Flow Metab 19: 1213–1219, 1999. [DOI] [PubMed] [Google Scholar]
- 102. Nantel F, Meadows E, Denis D, Connolly B, Metters KM, Giaid A. Immunolocalization of cyclooxygenase-2 in the macula densa of human elderly. FEBS Lett 457: 475–477, 1999. [DOI] [PubMed] [Google Scholar]
- 103. Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J 6: 3051–3064, 1992. [PubMed] [Google Scholar]
- 104. Ndengele MM, Cuzzocrea S, Esposito E, Mazzon E, Di Paola R, Matuschak GM, Salvemini D. Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J 22: 3154–3164, 2008. [DOI] [PubMed] [Google Scholar]
- 105. Needleman P, Turk J, Jakschik BA, Morrison AR, Lefkowith JB. Arachidonic acid metabolism. Annu Rev Biochem 55: 69–102, 1986. [DOI] [PubMed] [Google Scholar]
- 106. Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C. Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proc Natl Acad Sci USA 95: 10966–10971, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Notoya K, Jovanovic DV, Reboul P, Martel-Pelletier J, Mineau F, Pelletier JP. The induction of cell death in human osteoarthritis chondrocytes by nitric oxide is related to the production of prostaglandin E2 via the induction of cyclooxygenase-2. J Immunol 165: 3402–3410, 2000. [DOI] [PubMed] [Google Scholar]
- 108. Ohno R, Yokota A, Tanaka A, Takeuchi K. Induction of small intestinal damage in rats following combined treatment with cyclooxygenase-2 and nitric-oxide synthase inhibitors. J Pharmacol Exp Therap 310: 821–827, 2004. [DOI] [PubMed] [Google Scholar]
- 109. Oshikawa T, Okamoto M, Tano T, Uddin Ahmed S, Sasai A, Kan S, Moriya Y, Ryoma Y, Saito M, Sato M. Involvement of nitric oxide in anti-tumor effects of OK-432, a streptococcal anti-tumor immunotherapeutic agent. Intl Immunopharmacol 6: 764–773, 2006. [DOI] [PubMed] [Google Scholar]
- 110. Otto JC, Smith WL. Prostaglandin endoperoxide synthases-1 and -2. J Lipid Mediat Cell Signal 12: 139–156, 1995. [DOI] [PubMed] [Google Scholar]
- 111. Paduch R, Kandefer-Szerszen M. Nitric Oxide (NO) and Cyclooxygenase-2 (COX-2) cross-talk in co-cultures of tumor spheroids with normal cells cancer microenvironment. 4: 187–198, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Paliege A, Mizel D, Medina C, Pasumarthy A, Huang YG, Bachmann S, Briggs JP, Schnermann JB, Yang T. Inhibition of nNOS expression in the macula densa by COX-2-derived prostaglandin E2. Am J Physiol Renal Physiol 287: F152–F159, 2004. [DOI] [PubMed] [Google Scholar]
- 113. Paoletti AM, Piccirilli S, Costa N, Rotiroti D, Bagetta G, Nistico G. Systemic administration of N omega-nitro-l-arginine methyl ester and indomethacin reduces the elevation of brain PGE2 content and prevents seizures and hippocampal damage evoked by LiCl and tacrine in rat. Exp Neurol 149: 349–355, 1998. [DOI] [PubMed] [Google Scholar]
- 114. Park SW, Lee SG, Song SH, Heo DS, Park BJ, Lee DW, Kim KH, Sung MW. The effect of nitric oxide on cyclooxygenase-2 (COX-2) overexpression in head and neck cancer cell lines. Intl J Cancer 107: 729–738, 2003. [DOI] [PubMed] [Google Scholar]
- 115. Park SW, Sung MW, Heo DS, Inoue H, Shim SH, Kim KH. Nitric oxide upregulates the cyclooxygenase-2 expression through the cAMP-response element in its promoter in several cancer cell lines. Oncogene 24: 6689–6698, 2005. [DOI] [PubMed] [Google Scholar]
- 116. Peng HB, Libby P, Liao JK. Induction and stabilization of IκBα by nitric oxide mediates inhibition of NF-κB. J Bioll Chem 270: 14214–14219, 1995. [DOI] [PubMed] [Google Scholar]
- 117. Perkins DJ, Kniss DA. Blockade of nitric oxide formation down-regulates cyclooxygenase-2 and decreases PGE2 biosynthesis in macrophages. J Leukoc Biol 65: 792–799, 1999. [DOI] [PubMed] [Google Scholar]
- 118. Prusakiewicz JJ, Duggan KC, Rouzer CA, Marnett LJ. Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 48: 7353–7355, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Rao CV, Indranie C, Simi B, Manning PT, Connor JR, Reddy BS. Chemopreventive properties of a selective inducible nitric oxide synthase inhibitor in colon carcinogenesis, administered alone or in combination with celecoxib, a selective cyclooxygenase-2 inhibitor. Cancer Res 62: 165–170, 2002. [PubMed] [Google Scholar]
- 120. Rao CV, Kawamori T, Hamid R, Reddy BS. Chemoprevention of colonic aberrant crypt foci by an inducible nitric oxide synthase-selective inhibitor. Carcinogenesis 20: 641–644, 1999. [DOI] [PubMed] [Google Scholar]
- 121. Rausaria S, Ghaffari MM, Kamadulski A, Rodgers K, Bryant L, Chen Z, Doyle T, Shaw MJ, Salvemini D, Neumann WL. Retooling manganese(III) porphyrin-based peroxynitrite decomposition catalysts for selectivity and oral activity: a potential new strategy for treating chronic pain. J Med Chem 54: 8658–8669, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rausaria S, Kamadulski A, Rath NP, Bryant L, Chen Z, Salvemini D, Neumann WL. Manganese(III) complexes of bis(hydroxyphenyl)dipyrromethenes are potent orally active peroxynitrite scavengers. J Am Chem Soc 133: 4200–4203, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Rettori V, Gimeno M, Lyson K, McCann SM. Nitric oxide mediates norepinephrine-induced prostaglandin E2 release from the hypothalamus. Proc Natl Acad Sci USA 89: 11543–11546, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res 50 Suppl: S29–S34, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Salvemini D, Jensen MP, Riley DP, Misko TP. Therapeutic manipulations of peroxynitrite. Drug News Perspect 11: 204–214, 1998. [PubMed] [Google Scholar]
- 126. Salvemini D, Manning PT, Zweifel BS, Seibert K, Connor J, Currie MG, Needleman P, Masferrer JL. Dual inhibition of nitric oxide and prostaglandin production contributes to the antiinflammatory properties of nitric oxide synthase inhibitors. J Clin Invest 96: 301–308, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Salvemini D, Masferrer JL. Interactions of nitric oxide with cyclooxygenase: in vitro, ex vivo, and in vivo studies. Methods Enzymol 269: 12–25, 1996. [DOI] [PubMed] [Google Scholar]
- 128. Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA 90: 7240–7244, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Salvemini D, Mollace V, Pistelli A, Anggard E, Vane J. Metabolism of glyceryl trinitrate to nitric oxide by endothelial cells and smooth muscle cells and its induction by Escherichia coli lipopolysaccharide. Proc Natl Acad Sci USA 89: 982–986, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Salvemini D, Radziszewski W, Korbut R, Vane J. The use of oxyhaemoglobin to explore the events underlying inhibition of platelet aggregation induced by NO or NO-donors. Brit J Pharmacol 101: 991–995, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Salvemini D, Seibert K, Masferrer JL, Misko TP, Currie MG, Needleman P. Endogenous nitric oxide enhances prostaglandin production in a model of renal inflammation. J Clin Invest 93: 1940–1947, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Salvemini D, Settle SL, Masferrer JL, Seibert K, Currie MG, Needleman P. Regulation of prostaglandin production by nitric oxide; an in vivo analysis. Brit J Pharmacol 114: 1171–1178, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Salvemini D, Wang ZQ, Stern MK, Currie MG, Misko TP. Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc Natl Acad Sci USA 95: 2659–2663, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Salvemini D, Wang ZQ, Wyatt PS, Bourdon DM, Marino MH, Manning PT, Currie MG. Nitric oxide: a key mediator in the early and late phase of carrageenan-induced rat paw inflammation. Brit J Pharmacol 118: 829–838, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sano H, Hla T, Maier JA, Crofford LJ, Case JP, Maciag T, Wilder RL. In vivo cyclooxygenase expression in synovial tissues of patients with rheumatoid arthritis and osteoarthritis and rats with adjuvant and streptococcal cell wall arthritis. J Clin Invest 89: 97–108, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sautebin L, Di Rosa M. Nitric oxide modulates prostacyclin biosynthesis in the lung of endotoxin-treated rats. Eur J Pharmacol 262: 193–196, 1994. [DOI] [PubMed] [Google Scholar]
- 137. Sautebin L, Ialenti A, Ianaro A, Di Rosa M. Modulation by nitric oxide of prostaglandin biosynthesis in the rat. Brit J Pharmacol 114: 323–328, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Schror K, Ahland B, Darius H, Weiss P. Stimulation of vascular PGI2 by organic nitrates and its significance for the antianginal effect. Scan J Clin Lab Invest Suppl 173: 33–40, 1984. [PubMed] [Google Scholar]
- 139. Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA 91: 12013–12017, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Shang ZJ, Li ZB, Li JR. In vitro effects of nitric oxide synthase inhibitor l-NAME on oral squamous cell carcinoma: a preliminary study. Intl J Oral Maxillofac Surg 35: 539–543, 2006. [DOI] [PubMed] [Google Scholar]
- 141. Sidhu RS, Lee JY, Yuan C, Smith WL. Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 49: 7069–7079, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56: 387–437, 2004. [DOI] [PubMed] [Google Scholar]
- 143. Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL, Seibert K, Isakson PC. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci USA 95: 13313–13318, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Smith WL, Marnett LJ, DeWitt DL. Prostaglandin and thromboxane biosynthesis. Pharmacol Ther 49: 153–179, 1991. [DOI] [PubMed] [Google Scholar]
- 145. Solomon DH, Schneeweiss S, Glynn RJ, Kiyota Y, Levin R, Mogun H, Avorn J. Relationship between selective cyclooxygenase-2 inhibitors and acute myocardial infarction in older adults. Circulation 109: 2068–2073, 2004. [DOI] [PubMed] [Google Scholar]
- 146. Sorkin LS, Moore JH. Evoked release of amino acids and prostanoids in spinal cords of anesthetized rats: changes during peripheral inflammation and hyperalgesia. Am J Therapeut 3: 268–275, 1996. [DOI] [PubMed] [Google Scholar]
- 147. Stadler J, Harbrecht BG, Di Silvio M, Curran RD, Jordan ML, Simmons RL, Billiar TR. Endogenous nitric oxide inhibits the synthesis of cyclooxygenase products and interleukin-6 by rat Kupffer cells. J Leukoc Biol 53: 165–172, 1993. [DOI] [PubMed] [Google Scholar]
- 148. Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell 78: 931–936, 1994. [DOI] [PubMed] [Google Scholar]
- 149. Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18: 691–696, 1997. [DOI] [PubMed] [Google Scholar]
- 150. Swaisgood CM, Zu HX, Perkins DJ, Wu S, Garver CL, Zimmerman PD, Iams JD, Kniss DA. Coordinate expression of inducible nitric oxide synthase and cyclooxygenase-2 genes in uterine tissues of endotoxin-treated pregnant mice. Am J Obstetr Gynecol 177: 1253–1262, 1997. [DOI] [PubMed] [Google Scholar]
- 151. Swierkosz TA, Mitchell JA, Warner TD, Botting RM, Vane JR. Co-induction of nitric oxide synthase and cyclo-oxygenase: interactions between nitric oxide and prostanoids. BrJ Pharmacol 114: 1335–1342, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6: 662–680, 2007. [DOI] [PubMed] [Google Scholar]
- 153. Tammali R, Ramana KV, Srivastava SK. Aldose reductase regulates TNF-α-induced PGE2 production in human colon cancer cells. Cancer Lett 252: 299–306, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Tanaka T, Kohno H, Shimada R, Kagami S, Yamaguchi F, Kataoka S, Ariga T, Murakami A, Koshimizu K, Ohigashi H. Prevention of colonic aberrant crypt foci by dietary feeding of garcinol in male F344 rats. Carcinogenesis 21: 1183–1189, 2000. [PubMed] [Google Scholar]
- 155. Tetsuka T, Daphna-Iken D, Miller BW, Guan Z, Baier LD, Morrison AR. Nitric oxide amplifies interleukin 1-induced cyclooxygenase-2 expression in rat mesangial cells. J Clin Invest 97: 2051–2056, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Tian J, Kim SF, Hester L, Snyder SH. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci USA 105: 10537–10540, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Togashi H, Sasaki M, Frohman E, Taira E, Ratan RR, Dawson TM, Dawson VL. Neuronal (type I) nitric oxide synthase regulates nuclear factor-κB activity and immunologic (type II) nitric oxide synthase expression. Proc Natl Acad Sci USA 94: 2676–2680, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tsai AL, Wei C, Kulmacz RJ. Interaction between nitric oxide and prostaglandin H synthase. Arch Biochem Biophys 313: 367–372, 1994. [DOI] [PubMed] [Google Scholar]
- 159. Tsikas D, Niemann J. Nitric oxide, peroxynitrite, S-nitrosothiols and thiols are unlikely to exert their effects on recombinant cyclooxygenase-1 and cyclooxygenase-2 activity in vitro by modifying cysteine moieties. Nitric Oxide Biol Chem 26: 192–194, 2012. [DOI] [PubMed] [Google Scholar]
- 160. Uno K, Iuchi Y, Fujii J, Sugata H, Iijima K, Kato K, Shimosegawa T, Yoshimura T. In vivo study on cross talk between inducible nitric-oxide synthase and cyclooxygenase in rat gastric mucosa: effect of cyclooxygenase activity on nitric oxide production. J Pharmacol Exp Therapeut 309: 995–1002, 2004. [DOI] [PubMed] [Google Scholar]
- 161. Upmacis RK, Deeb RS, Hajjar DP. Oxidative alterations of cyclooxygenase during atherogenesis. Prostaglandins Other Lipid Mediat 80: 1–14, 2006. [DOI] [PubMed] [Google Scholar]
- 162. Upmacis RK, Deeb RS, Hajjar DP. Regulation of prostaglandin H2 synthase activity by nitrogen oxides. Biochemistry 38: 12505–12513, 1999. [DOI] [PubMed] [Google Scholar]
- 163. van der Viet A, Hoen PA, Wong PS, Bast A, Cross CE. Formation of S-nitrosothiols via direct nucleophilic nitrosation of thiols by peroxynitrite with elimination of hydrogen peroxide. J Biol Chem 273: 30255–30262, 1998. [DOI] [PubMed] [Google Scholar]
- 164. Van der Ouderaa FJ, Buytenhek M, Nugteren D, Van Dorp D. Purification and characterisation of prostaglandin endoperoxide synthase from sheep vesicular glands. Biochim Biophys Acta 367: 315, 1977. [DOI] [PubMed] [Google Scholar]
- 165. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231: 232–235, 1971. [DOI] [PubMed] [Google Scholar]
- 166. Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. New England J Med 323: 27–36, 1990. [DOI] [PubMed] [Google Scholar]
- 167. Vane JR, Mitchell JA, Appleton I, Tomlinson A, Bishop-Bailey D, Croxtall J, Willoughby DA. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci USA 91: 2046–2050, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Vecchio AJ, Simmons DM, Malkowski MG. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J Biol Chem 285: 22152–22163, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. von Euler US. On the specific vaso-dilating and plain muscle stimulating substances from accessory genital glands in man and certain animals (prostaglandin and vesiglandin). J Physiol 88: 213–234, 1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. von Knethen A, Brune B. Cyclooxygenase-2: an essential regulator of NO-mediated apoptosis. FASEB J 11: 887–895, 1997. [PubMed] [Google Scholar]
- 171. Wallace JL. Nitric oxide, aspirin-triggered lipoxins and NO-aspirin in gastric protection. Inflammation Allergy Drug Targets 5: 133–137, 2006. [DOI] [PubMed] [Google Scholar]
- 172. Wallace JL, Fiorucci S. A magic bullet for mucosal protection … and aspirin is the trigger! Trends Pharm Sci 24: 323–326, 2003. [DOI] [PubMed] [Google Scholar]
- 173. Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 119: 706–714, 2000. [DOI] [PubMed] [Google Scholar]
- 174. Watkins DN, Garlepp MJ, Thompson PJ. Regulation of the inducible cyclo-oxygenase pathway in human cultured airway epithelial (A549) cells by nitric oxide. Brit J Pharmacol 121: 1482–1488, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wendum D, Masliah J, Trugnan G, Flejou JF. Cyclooxygenase-2 and its role in colorectal cancer development. Virchows Archiv 445: 327–333, 2004. [DOI] [PubMed] [Google Scholar]
- 176. West SD, Suliburk JW, Helmer KS, Mercer DW. Cyclooxygenase-1 suppresses lipopolysaccharide-induced changes in rat gastric inducible nitric oxide synthase. Crit Care Med 36: 572–579, 2008. [PubMed] [Google Scholar]
- 177. Wu KK. Inducible cyclooxygenase and nitric oxide synthase. Adv Pharmacol 33: 179–207, 1995. [DOI] [PubMed] [Google Scholar]
- 178. Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279: 234–237, 1998. [DOI] [PubMed] [Google Scholar]
- 179. Xu L, Han C, Lim K, Wu T. Activation of cytosolic phospholipase A2α through nitric oxide-induced S-nitrosylation. Involvement of inducible nitric-oxide synthase and cyclooxygenase-2. J BiolChem 283: 3077–3087, 2008. [DOI] [PubMed] [Google Scholar]
- 180. Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron 11: 371–386, 1993. [DOI] [PubMed] [Google Scholar]
- 181. Yu LB, Dong XS, Sun WZ, Zhao DL, Yang Y. Effect of a nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester on invasion of human colorectal cancer cell line SL-174T. World J Gastroenterol 11: 6385–6388, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Yu Y, Fan J, Chen XS, Wang D, Klein-Szanto AJ, Campbell RL, FitzGerald GA, Funk CD. Genetic model of selective COX2 inhibition reveals novel heterodimer signaling. Nat Med 12: 699–704, 2006. [DOI] [PubMed] [Google Scholar]
- 183. Yuan C, Rieke CJ, Rimon G, Wingerd BA, Smith WL. Partnering between monomers of cyclooxygenase-2 homodimers. Proc Natl Acad Sci USA 103: 6142–6147, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, Smith WL. Cyclooxygenase allosterism, fatty acid-mediated cross-talk between monomers of cyclooxygenase homodimers. J Biol Chem 284: 10046–10055, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zhang H, Sun XF. Overexpression of cyclooxygenase-2 correlates with advanced stages of colorectal cancer. Am J Gastroenterol 97: 1037–1041, 2002. [DOI] [PubMed] [Google Scholar]
- 186. Zingarelli B, Southan GJ, Gilad E, O'Connor M, Salzman AL, Szabo C. The inhibitory effects of mercaptoalkylguanidines on cyclo-oxygenase activity. Brit J Pharmacol 120: 357–366, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Zou M, Martin C, Ullrich V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol Chem 378: 707–713, 1997. [DOI] [PubMed] [Google Scholar]
- 188. Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest 109: 817–826, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Zou MH, Ullrich V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett 382: 101–104, 1996. [DOI] [PubMed] [Google Scholar]