

Abstract
Vascular permeability is a hallmark of several disease states including acute lung injury (ALI). Endocytosis of VE-cadherin, away from the interendothelial junction (IEJ), causes acute endothelial barrier permeability. A novel protein, p18, anchors to the endosome membrane and plays a role in late endosomal signaling via MAPK and mammalian target of rapamycin. However, the fate of the VE-cadherin-positive endosome has yet to be elucidated. We sought to elucidate a role for p18 in VE-cadherin trafficking and thus endothelial barrier function, in settings of ALI. Endothelial cell (EC) resistance, whole-cell ELISA, and filtration coefficient were studied in mice or lung ECs overexpressing wild-type or nonendosomal-binding mutant p18, using green fluorescent protein as a control. We demonstrate a protective role for the endocytic protein p18 in endothelial barrier function in settings of ALI in vitro and in vivo, through enhanced recycling of VE-cadherin-positive early endosomes to the IEJ. In settings of LPS-induced ALI, we show that Src tethered to the endosome tyrosine phosphorylates p18 concomitantly with VE-cadherin internalization and pulmonary edema formation. We conclude that p18 regulates pulmonary endothelial barrier function in vitro and in vivo, by enhancing recycling of VE-cadherin-positive endosomes to the IEJ.—Chichger, H., Duong, H., Braza, J., Harrington, E. O. p18, a novel adaptor protein, regulates pulmonary endothelial barrier function via enhanced endocytic recycling of VEcadherin.
Keywords: endosome, adherensjunction, endocytosis, lunginjury
The annual incidence of acute lung injury (ALI), a disease associated with acute hypoxemic respiratory failure, has been projected to reach 335,000 by 2030 with 44% of cases leading to mortality (1). Increased vascular permeability is a hallmark of ALI and is responsible for the characteristic formation of pulmonary edema observed in patients. Disruption of the pulmonary endothelial barrier, leading to increased vascular permeability, is due, in part, to the breakdown of endothelial cell (EC)-cell contacts. Cell-cell contacts are comprised of tight junctions (TJs) and adherens junctions (AJs) at the interendothelial junction (IEJ). Extracellular calcium chelation, or the addition of antibodies against VE-cadherin, blocks AJ formation and increases EC permeability concomitant with TJ disorganization (2, 3). Thus, VE-cadherin, the major AJ component in ECs, is vital to preserve vascular barrier function (4–6).
The AJ is formed via VE-cadherin–mediated interactions between neighboring ECs. The cytoplasmic tail of VE-cadherin links to the actin cytoskeleton, via the armadillo proteins, p120-catenin and β-catenin. β-Catenin provides linkage between the AJ and the actin cytoskeleton through interactions involving α-catenin (7). p120-Catenin, in turn, regulates cadherin stability at the membrane by masking an endocytic signal on the cadherin cytoplasmic tail to prevent cadherin internalization (3, 8–11). Antibodies against VE-cadherin disrupt endothelial junctions (12), leading to an increased migration of leukocytes into the inflamed tissue (13). The stabilization of VE-cadherin at the AJ, through genetic manipulation of VE-cadherin-catenin interactions, attenuates pulmonary vascular leakage in an LPS-induced in vivo model of ALI (14). Thus, regulation of the pulmonary endothelium through maintenance of VE-cadherin at cell-cell contacts is vital to promote barrier function and may offer therapeutic value in the treatment of ALI.
Enhanced phosphorylation and internalization of VE-cadherin, and the resulting rise in vascular permeability, have been established in ECs following VEGF treatment (14, 15). VEGF binding to its receptor, VEGFR, results in a β-arrestin– and Src-induced signaling cascade leading to phosphorylation of the cytoplasmic tail of VE-cadherin, at tyrosine residues 665/667 (16), resulting in VE-cadherin internalization. This study demonstrated VE-cadherin to be localized to early endosomes; however, the fate of these endosomes is not known (16).
Upon internalization, endocytosed materials are delivered to early endosomes, where they are sorted and either recycled to the cell surface or targeted for degradation (17). The membrane adaptor protein p18 [also named Pdro, p27kip1-release factor, and late endosomal/lysosomal adaptor, MAPK, and mammalian target of rapamycin (mTOR) activator 1] was recently identified and shown to be highly conserved and ubiquitously expressed in vertebrates (18). p18 binds to the outer membrane of the endosome through N-terminal acylation and possesses 2 canonical dileucine motifs responsible for targeting the early endosome to the late endosome or recycling endosome (17). At the late endosome, p18 anchors the Rag-GTPase “Ragulator” complex to mTORC1, facilitating activation of the complex and controlling lysosomal maturation (19). As such, depletion of p18 has been shown to dramatically disrupt late endosomal trafficking within the cell by 1) promoting the redistribution of the late endosome to the cell periphery, thus preventing trafficking to the lysosome (20), and 2) by causing increased accumulation of late endosomes within the cell, thus impairing late endosome-lysosome fusion (21). In addition, p18 tethered to the late endosome serves as an anchor for the p14-MEK partner 1 (MP1)-MEK-ERK signaling pathway, potentially to regulate the dynamics of the late endosome (20). Less is known about p18 at the early endosome; however, p18 ablation causes accumulation of early endosomal cargo, such as integrin β1 and transferrin receptor, in endosomes within the cell, rather than promoting an active recycling of the proteins to the cell surface (20).
Despite studies investigating VE-cadherin phosphorylation and barrier permeability, the trafficking of VE-cadherin within the ECs or the subsequent effects on the pulmonary vasculature is not known. In the study presented here, we demonstrate elevated VE-cadherin levels and cadherin binding at the AJ and, thus, enhanced pulmonary barrier function in vitro and in vivo, by overexpression of the novel endosomal adaptor protein p18. These protective effects were independent of mTOR-MAPK inhibitors but abrogated following overexpression of p18N39, a p18 mutant lacking the endosome-binding region. We propose that p18 attenuates LPS-induced barrier disruption by associating with VE-cadherin-positive endosomes and promoting the recycling of VE-cadherin to the cell surface allowing the formation of functional AJs at the IEJ. Thus, our work suggests that VE-cadherin trafficking in the ECs is a key regulator of EC barrier function.
MATERIALS AND METHODS
Cell lines and reagents
All materials were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. Rat lung microvascular endothelial cells (LMVECs; Vec Technologies, Rensselaer, NY, USA) were maintained in MCDB-131 (Vec Technologies) and used between passages 3 and 11.
LPS (endotoxin) from Escherichia coli serotype 011:B4 was obtained from Enzo Life Sciences (Plymouth Meeting, PA, USA). Pseudomonas aeruginosa strain 103 (PA103) was a kind gift from Dr. Troy Stevens (University of South Alabama, Mobile, AL, USA). PolyJet reagent and Protein G Agarose beads were purchased from SignaGen Laboratories (Gaithersburg, MD, USA) and Thermo Fisher Scientific (Rockford, IL, USA), respectively.
The vector encoding wild-type (WT) p18 [green fluorescent protein (GFP)-p18wt] and mutant p18, lacking endosome-binding region (p18N39), were kind gifts from Shigeyuki Nada (20, 21) (Department of Oncogene Research, Osaka University, Osaka, Japan). Phosphorylated GFP (pGFP-C1) was obtained from Clontech (Mountain View, CA, USA). Rat p18 and nonsilencing (ns) small interfering RNA (siRNA) duplexes were obtained from OriGene (Rockville, MD, USA). Antibodies directed against VE-cadherin, Src, early endosome antigen 1 (EEA1), lysosomal-associated membrane protein 1 (LAMP1), phosphorylated MEK1/MEK2 (Ser218/Ser222), MEK, p70, and actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). p120-Catenin and β-catenin antibodies were obtained from BD Biosciences (San Jose, CA, USA). Pan phospho-tyrosine antibody (clone 4G10) was obtained from Millipore (Billerica, MA, USA). GFP antibody was purchased from Invitrogen (Grand Island, NY, USA). Phosphorylated mTOR (Ser2448), p70 (Thr389), p38 (Thr180/Tyr182), ERK1/ERK2 (Thr202/Tyr204), unphosphorylated mTOR, p38 and ERK1/ERK2 antibodies, and ERK1/ERK2 inhibitor U0126 were all purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies directed against p18 were a kind gift from Sergie Manié (22) (Cancer Research Center of Lyon, INSERM UMR 1052, Lyon, France) and Shigeyuki Nada (20).
Liposome preparation
For production of liposomes, dimethyldioctadecyl-ammonium bromide and cholesterol were mixed in chloroform, and the mixture was rotated under constant vacuum pressure and temperature in a rotary evaporator (23, 24). Liposomes were dissolved in sterile 5% glucose solution and combined with GFP or GFP-p18 cDNA (50 μg) and injected into the retrobulbar sinus of the orbit of 8- to 10-wk-old anesthetized C57 BL/6 mice (25–30). Mice were anesthetized with continuous isoflurane inhalation (2–5%), and adequacy of anesthetic was assessed by pedal reflex. cDNA was overexpressed for 48 h prior to experiment end point. Confirmation of cDNA overexpression was assessed by EC isolation and immunoblot analysis, as previously described (31). In brief, ECs were isolated from the lung homogenate of transfected mice, using CD31-conjugated magnetic beads purified through a magnetic column and measured by immunoblot analysis for GFP overexpression and VE-cadherin expression.
In vivo models of ALI
There were 2 models of inflammatory injury used to assess pulmonary vascular dysfunction via inflammatory lung injury in vivo: the Gram-negative endotoxin LPS, found in patients infected with Gram-negative bacteria (32), or the live Gram-negative bacteria P. aeruginosa (PA103) (33).
LPS or vehicle (saline) was administered to nonanesthetized, adult 8- to 10-wk-old C57BL/6 mice via a single injection (5 mg/kg i.p.). At 24 h following intraperitoneal injection of LPS or vehicle into mice, lungs were either removed for homogenization or isolated and perfused for filtration coefficient (kf) measurements (31, 34, 35). Isolated lungs were removed en bloc, ventilated, and perfused. The kf was determined using the rate of weight gain during the final 2 min following an ∼8 cm H2O increase in venous pressure (high hydrostatic challenge) for 15 min divided by the change in capillary pressures induced by the double-occlusion technique. The kf value was normalized to 100 g wet lung mass, empirically derived as 0.00472 body mass (36).
Alternatively, PA103 or PBS vehicle was administered via a single intratracheal injection [107 colony-forming units (CFUs)]. At 4 h following PA103 administration, wet and dry lung weights were taken.
All animal experimental protocols were approved by the Institutional Animal Care and Use Committees of the Providence Veterans Affairs Medical Center and Brown University and comply with the Health Research Extension Act and U.S. Public Health Service policy.
Whole-cell indirect ELISA
Cell surface levels of VE-cadherin were measured by whole-cell ELISA as previously described (3). LMVECs were transiently transfected and seeded onto 96-well plates. At 48 h posttransfection, cells were treated with VEGF (50 ng/ml) for 30 min, or LPS (1 μg/ml) for 6 h. Following treatment, cells were rinsed with PBS, fixed in 1% paraformaldehyde at room temperature for 10 min, and blocked in 2% bovine serum albumin (BSA) for 1 h at 37°C. Cells were incubated with antibody specific to the extracellular domain of VE-cadherin (N-14, sc 31017) in 1% BSA for 2 h at 37°C. Following incubation with the appropriate horseradish peroxidase-conjugated secondary antibody, chemiluminescence was measured at 20 s exposure times using a microplate reader (GenTech Scientific, Arcade, NY, USA).
Endothelial monolayer permeability
Changes in endothelial monolayer permeability were assessed using the electrical cell impedance sensor technique (Applied Biophysics, Troy, NY, USA). LMVECs were transiently transfected with cDNA encoding GFP-p18wt, GFP-p18N39, or GFP, using PolyJet reagent. Alternatively, LMVECs were transiently transfected with p18 siRNA duplex (100 or 300 nM), or ns, scrambled control, using the Amaxa (Allendale, NJ, USA) electroporation technique. Transfected cells were cultured for 48 h on collagen-coated electric cell-substrate impedance sensing arrays. Following a baseline read period, monolayers were treated with VEGF (50 ng/ml), LPS (1 μg/ml), or rapamycin (10 nM), and resistance was measured over time. cDNA overexpression or siRNA knockdown was confirmed at 48 h posttransfection via immunoblot analysis.
Methylthiazolyl tetrazolium assay
EC viability was measured using methylthiazolyl tetrazolium (MTT) assay by quantifying the reduction of soluble yellow tetrazolium dye into insoluble purple formazan (37, 38). LMVECs were transiently transfected with cDNA encoding GFP-p18wt or GFP and cultured for 48 h followed by incubation with MTT (5 mg/ml) for 4 h at 37°C. Cells were then incubated with 0.04–0.1 N HCl in isopropanol for 15 min. The absorbance was measured using a microplate reader (GenTech Scientific) at a wavelength of 570 nm with background readings at 630 and 690 nm.
Immunoprecipitations and immunoblotting
LMVECs were transiently transfected with cDNA encoding GFP-p18 or GFP, using PolyJet reagent and treated with LPS (1 μg/ml) or vehicle for 6 h. Alternatively, LMVECs were pretreated with Src kinase inhibitor PP2 (12 µM) for 30 min, followed by treatment with LPS (1 μg/ml) for 6 h. Cells were lysed with RIPA buffer and subjected to immunoprecipitation or immunoblot analysis.
Individual lobes of mouse lungs were homogenized in buffer [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.9), 1.5 mM NaCl, 0.25 M sucrose, 0.2 mM EDTA, 200 mM PMSF, 0.5 mM DTT, and 1.5 mM MgCl2] for 2 min and subjected to immunoprecipitation analysis. Lysate (500 μg) or homogenate (1.5 mg) was immunoprecipitated with VE-cadherin antibody overnight (4°C). Immunocomplexes were incubated with Protein G Agarose beads for 4 h, washed, and resuspended in Laemmli buffer. Purified immunocomplexes, as well as original lysates, were subjected to immunoblot analysis.
Immunoblot analyses were performed on 7.5% (mTOR) and 10% SDS-PAGEs using primary antibody dilutions of 1:1000, except vinculin (1:5000), and secondary antibody dilutions of 1:5000.
Endosome isolations
Following treatment with LPS (1 μg/ml) for 6 h, LMVECs were suspended in isolation buffer [10 mM HEPES (pH 7.2), 100 mM KCl, 1 mM EDTA, and 25 mM sucrose] and passed through a 22-gauge syringe needle 15 times. To obtain a crude endosome isolation (39), preparations were centrifuged (3000 × g, 10 min, 4°C), and subsequent supernatants (500 μg) were rotated with anti-EEA1 or LAMP1 antibody (3 μg) overnight at 4°C. Immunocomplexes were treated as above (immunoprecipitations) followed by immunoblot analysis.
Immunofluorescence studies
LMVECs were seeded onto collagen-coated glass coverslips to confluence, followed by exposure to LPS (1 μg/ml) for 6 h. Cells were washed in PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate. Cells were blocked in 5% donkey serum in PBS, followed by incubation in primary antibody for 1 h at 37°C. Cells were rinsed and incubated with the appropriate fluorescently tagged secondary antibody for 1 h at 37°C. Coverslips were mounted using ProLong Gold Antifade Reagent (Life Technologies, Grand Island, NY, USA) with DAPI, images were captured using a laser-scanning confocal microscope (LSM 700; Zeiss, Jena, Germany), and colocalization quantification was performed using Zen 2011 (Zeiss).
Statistical analysis
For 3 or more groups, differences among the means were tested for significance in all experiments by ANOVA with Tukey’s range significance difference test. For 2 groups, differences among the means were tested for significance by the Mann-Whitney U variance test followed by the t test. Significance was reached when P < 0.05. Values are the mean ± SD.
RESULTS
p18 elevates endothelial barrier function through preservation of VE-cadherin at the IEJ
Although endocytic trafficking of cadherins has been established to influence permeability in the epithelium, regulation of the endothelium by VE-cadherin trafficking has yet to be determined. Thus, we first established whether the endosomal adaptor protein p18 plays a role in endothelial monolayer permeability in vitro by overexpressing the WT p18 conjugated to GFP (GFP-p18wt) protein in microvascular ECs. Equivalent numbers of LMVECs were transiently transfected with GFP-p18wt or GFP cDNA in vitro. Time course studies demonstrate increases in monolayer resistance in p18-overexpressing cells at 15 h after transfection, concomitant with protein overexpression (Supplemental Fig. S1). Maximum overexpression of GFP and GFP-p18wt was observed at 48 h following transfection; thus, remaining experiments were all performed at this time point (Supplemental Fig. S1). Overexpression of GFP-p18wt and GFP in transfected ECs was confirmed by immunoblot analysis (Fig. 1A, inset). Densitometry quantification showed that equivalent levels of GFP were expressed in LMVECs transiently transfected with GFP-p18wt [0.62 ± 0.13 relative chemiluminescence unit (r.c.u.)] and GFP (0.69 ± 0.18 r.c.u.) cDNA. Endothelial monolayer resistance of LMVECs overexpressing GFP-p18wt was significantly increased compared to the barrier resistance of GFP-overexpressing cells (Fig. 1A). Conversely, siRNA knockdown of endogenous p18 in LMVECs, using increasing concentrations of siRNA (Fig. 1B, inset), significantly increased endothelial monolayer permeability compared to ns siRNA (Fig. 1B). We noted that p18 overexpression in LMVECs exhibited no significant effect on cell viability, as measured by reduction of soluble yellow tetrazolium dye into insoluble purple formazan using MTT assay (data not shown). To further assess the protective role of p18 overexpression on the endothelial monolayer, we measured the endothelial barrier resistance of LMVECs overexpressing GFP-p18wt or GFP in the presence of the barrier-disruptive agent VEGF. Treatment with VEGF caused a significant acute increase in endothelial monolayer permeability within 30 min of exposure in GFP-overexpressing LMVECs, which was completely blocked in cells overexpressing GFP-p18wt protein (Supplemental Fig. S2).
Figure 1.
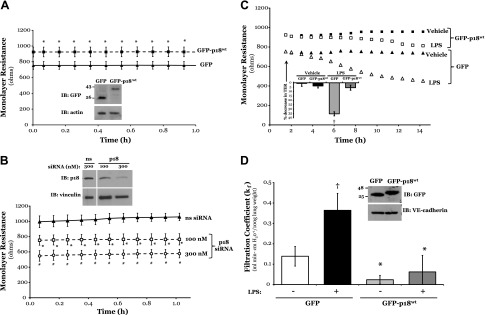
p18wt overexpression elevates endothelial barrier function and attenuates LPS disruption of the pulmonary endothelial barrier in vitro and in vivo. Equivalent numbers of LMVECs were transiently transfected with GFP (triangle symbol) or GFP-p18wt (square symbol) cDNA (A and C), or ns (300 nM, triangle symbol) or p18 (100 nM, square symbol; 300 nM, circle symbol) siRNA (B). A–C) Following 48 h, changes in endothelial monolayer resistance were measured using electric cell-substrate impedance sensing, under baseline conditions (A, B) and in the presence (open shape) and absence (solid shape) of LPS (1 µg/ml) (C). Overexpression of cDNA and knockdown of endogenous protein were confirmed by immunoblot (IB) analysis of lysates from transiently transfected cells, with an antibody specific to GFP (A, inset) and p18 (B, inset). Arrow indicates addition of LPS. A representative tracing and percent drop in transendothelial resistance at 10.5 h following LPS exposure (B, inset) are shown. Data are presented as the mean ± SD (n = 5–6). *P < 0.05 vs. GFP; †P < 0.05 vs. vehicle. D) Adult (8–10 wk) C57/BL6 mice were injected with liposomes containing cDNA encoding GFP or GFP-p18wt. At 24 h after injection, mice were injected with vehicle (saline) or LPS (5 mg/kg, i.p.) for a further 24 h, and vascular permeability in the intact lung was assessed by measuring the capillary kf of the ex vivo lung. In parallel, to prove the overexpression of GFP-p18wt cDNA in lung endothelium, ECs were isolated from the lung homogenate of transfected mice, using CD31-conjugated magnetic beads, purified through a magnetic column, and measured by immunoblot analysis for p18 overexpression and VE-cadherin expression (inset). A representative image of the blot is shown. Data are presented as the mean ± SD (n = 6–7). *P < 0.05 vs. GFP or ns duplex; †P < 0.05 vs. vehicle; #P < 0.05 vs. p18 siRNA 100 nM.
Endothelial monolayer permeability has been shown to correlate with increased internalization of VE-cadherin (16). Thus, we next assayed the levels of VE-cadherin at the cell surface, following overexpression of GFP or GFP-p18wt. We observed a significant increase in the expression of VE-cadherin on the plasma membrane of LMVECs overexpressing GFP-p18wt, compared with GFP, as assessed by whole-cell ELISA (Supplemental Fig. S3). To determine whether these changes in VE-cadherin surface expression were a result of increased total VE-cadherin levels, whole-cell lysates from LMVECs overexpressing GFP or GFP-p18wt were subjected to immunoblot analysis of VE-cadherin. We observed no significant change in total expression of VE-cadherin in cDNA-overexpressing cells under any conditions (Supplemental Fig. S3, inset). These data suggest that overexpression of WT p18 enhances endothelial barrier function and attenuates VEGF-induced barrier disruption via preservation of VE-cadherin levels at the IEJ.
p18 overexpression attenuates LPS-induced barrier permeability in vitro and in vivo through preservation of the AJ
To test whether the protective actions of GFP-p18wt overexpression were universal or solely protective to VEGF-induced barrier dysfunction, we next tested the edemagenic agent LPS. A dose-dependent increase in permeability was observed with increasing concentrations of LPS from 0.01 to 1 μg/ml, with 1 μg/ml resulting in the most significant decrease in EC monolayer resistance (Supplemental Fig. S4). Following treatment with LPS, LMVECs transiently transfected with GFP displayed a significant sustained increase in permeability at 7.5 h following exposure (Fig. 1C). However, in EC overexpressing GFP-p18wt protein, the effect of LPS was significantly attenuated (Fig. 1C). To determine if the in vitro observations were also noted in vivo, we examined whether the overexpression of GFP-p18wt within the intact lung was associated with enhanced pulmonary vasculature integrity. Cationic liposomes encapsulating cDNA encoding GFP-p18wt or GFP were injected into the retro-orbital vein of C57/BL6 mice and injected with LPS at 24 h posttransfection. Following a further 24 h period, protein overexpression was confirmed in ECs isolated from the lung and subjected to immunoblot analysis of p18 (Fig. 1D, inset). We observed that p18wt overexpression caused a significant reduction in the kf values of isolated, perfused lungs (Fig. 1D), suggesting a tighter endothelial barrier at baseline. We further noted that the mice overexpressing GFP-p18wt in the pulmonary vasculature displayed significant protection against LPS-induced pulmonary edema, as compared to mice overexpressing GFP exposed to LPS (Fig. 1D).
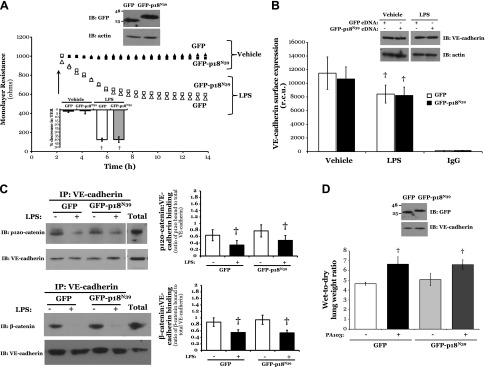
To characterize how p18 overexpression enhances endothelial monolayer resistance in vitro and in vivo under both baseline and barrier-disruptive conditions, we next assessed the effect of p18 on VE-cadherin protein expression at the IEJ. We studied the levels of catenin-binding and, thus, AJ-forming VE-cadherin at the cell surface following overexpression of p18. A significant increase was observed in the expression of VE-cadherin on the plasma membrane surface of LMVECs overexpressing GFP-p18wt, compared with GFP (Fig. 2A). Following 6 h exposure to LPS, GFP-overexpressing cells exhibited a significant 22.2 ± 6.9% decrease in VE-cadherin expression at the cell surface, which was not observed in GFP-p18wt-overexpressing cells (Fig. 2A). Previous studies have established that the association of VE-cadherin with catenin strengthens the AJ (40). Thus, we next examined the interaction of VE-cadherin with the AJ proteins β-catenin and p120-catenin in GFP-p18wt-overexpressing LMVECs in the presence and absence of LPS. Upon exposure to LPS, we observed a decrease in catenin:cadherin protein associations in GFP-overexpressing cells, an effect that was not observed in cells overexpressing GFP-p18wt (Fig. 2B). The preservation of catenin:cadherin protein interactions, following LPS treatment, was also observed in lungs from mice overexpressing p18 in the pulmonary vasculature, when compared to GFP (Fig. 2C). Taken together, these data suggest an important role for p18 in protecting the pulmonary vasculature from ALI-induced barrier disruption through the maintenance of VE-cadherin expression at the cell surface, thus preserving vascular integrity via enhanced function at the AJ.
Figure 2.

p18wt overexpression attenuates LPS-induced AJ disruption and internalization of VE-cadherin. A, B) Equivalent numbers of LMVECs were transiently transfected with GFP (open bars) or GFP-p18wt (closed bars) cDNA. Following 48 h, cells were treated with LPS for 6 h. A) Cell surface expression was determined with whole-cell indirect ELISA using chemiluminescence. Expression of VE-cadherin in whole-cell lysates was assessed by immunoblot analysis of lysates from transiently transfected cells with an antibody specific to VE-cadherin. Nonspecific binding was assayed using IgG and subtracted from values. In parallel, to assess total cell levels of VE-cadherin, LMVECs were harvested, and VE-cadherin expression was measured by immunoblot analysis. Blots were stripped and reprobed for actin as a loading control. Data are presented as the mean ± SD (n = 6). *P < 0.05 vs. GFP; †P < 0.05 vs. vehicle. B) Cell lysates were immunoprecipitated (IP) with an antibody specific to VE-cadherin. Immunocomplexes and total lysate were resolved via SDS-PAGE, transferred to Immobilon membrane, and probed with antibodies specific to p120-catenin or β-catenin. Membranes were then stripped and reprobed for VE-cadherin. Representative immunoblots are shown. Data are presented as the mean ± SD (n = 3). *P < 0.05 vs. GFP; †P < 0.05 vs. vehicle. C) Adult (8–10 wk) C57/BL6 mice were injected with liposomes containing cDNA encoding GFP or GFP-p18wt. At 24 h after injection, mice were injected with LPS (5 mg/kg i.p.) for a further 24 h, and lung homogenates were immunoprecipitated with an antibody specific to VE-cadherin and assessed as (B). Representative immunoblots are shown. Data are presented as the mean ± SD (n = 3). *P < 0.05 vs. GFP; †P < 0.05 vs. vehicle.
Effect of p18 on AJs and barrier function in vitro and in vivo is dependent on the ability to bind the endosome
We next sought to understand whether the protective effects of p18wt overexpression on the pulmonary vascular barrier are mediated by its function at the endosome. p18 anchors to the endosome lipid raft membrane via its N terminus to play a role in endosomal trafficking (20). Thus, we overexpressed cDNA encoding a p18 mutant that lacks the first 39 residues (i.e., the endosomal-binding motif) at the N terminus (GFP-p18N39) in LMVECs in the presence and absence of LPS and assessed endothelial barrier function and VE-cadherin cell surface expression. Following treatment with LPS, LMVECs transiently transfected with GFP-p18N39 similarly display enhanced endothelial barrier permeability as noted in GFP-overexpressing cells (Fig. 3A). Likewise, the decrease in cell surface expression of VE-cadherin and catenin:cadherin protein associations, observed following LPS treatment in LMVECs transiently transfected with GFP, was also noted in cells overexpressing GFP-p18N39 (Fig. 3B, C). Mice overexpressing both GFP and GFP-p18N39 in the pulmonary vasculature displayed significantly increased lung edema formation, as noted by an increased wet:dry lung weight ratio, following 4 h exposure to P. aeruginosa (PA103) (Fig. 3D). Thus, these data demonstrate that the mechanism through which p18 enhances the AJ and maintains endothelial barrier integrity in vitro and in vivo is dependent on its ability to bind the endosome.
Figure 3.
Binding of p18 to the endosome is necessary for an effect on enhanced barrier function and VE-cadherin expression at the AJ. A) Equivalent numbers of LMVECs were transiently transfected with GFP (triangle symbol) or GFP-p18N39 (square symbol) cDNA. Following 48 h, changes in endothelial monolayer resistance were measured using ECIS in the presence (open symbol) or absence (closed symbol) of LPS (1 µg/ml). A representative tracing and percent drop in transendothelial resistance at 10.5 h following LPS exposure (inset) are shown. Arrow indicates addition of LPS. Overexpression of cDNA was confirmed by immunoblot analysis of lysates from transiently transfected cells, with an antibody specific to GFP (inset). Arrow indicates addition of LPS (n = 6). †P < 0.05 vs. vehicle. B) Equivalent numbers of LMVECs were transiently transfected with GFP (open bars) or GFP-p18N39 (closed bars) cDNA. Following 48 h, LMVECs were exposed to LPS (1 µg/ml, 6 h), and cell surface expression was determined with whole-cell indirect ELISA using chemiluminescence. Expression of VE-cadherin in whole-cell lysates was assessed by immunoblot analysis of lysates from transiently transfected cells with an antibody specific to VE-cadherin (inset). Blots were stripped and reprobed for actin as a loading control. n = 7–8. †P < 0.05 vs. vehicle. C) Cell lysates were immunoprecipitated (IP) with an antibody specific to VE-cadherin and subjected to immunoblot analysis as in Fig. 2B. Representative immunoblots are shown. Data are presented as the mean ± SD (n = 3). †P < 0.05 vs. vehicle. D) Adult (8–10 wk) C57/BL6 mice were injected with liposomes containing cDNA encoding GFP or GFP-p18N39. At 44 h after injection, mice were injected with PA103 (107 CFUs, intratracheal) for a further 4 h, and lung weights were measured before and after a 72 h drying period. Data are presented as the mean ± SD (n = 4–5). †P < 0.05 vs. vehicle.
Effect of p18 on in vitro barrier function and VE-cadherin at the AJ is independent of MAPK-mTOR signaling
p18 has previously been observed to regulate mTOR activation, via anchoring the mTOR-activating (Ragulator) complex to mTORC1 (19), and MEK/ERK activation, potentially via tethering p14-MP1-MEK complex to the late endosome (20). Thus, we next sought to assess the effect of p18 overexpression on MAPK signaling, assessed by phosphorylation of p38, ERK1/ERK2, and the upstream ERK kinase, MEK1/MEK2, or mTOR activation, measured by phosphorylation of mTOR and its downstream target, p70 S6K.
Exposure of LMVECs to the mTOR inhibitor, rapamycin, resulted in reduced phosphorylation of MEK1/MEK2, p70 S6 kinase, and mTOR in LMVECs overexpressing either GFP or GFP-p18wt (Fig. 4A, B). Overexpression of GFP-p18wt significantly increased p38 phosphorylation but exerted no effect on ERK1/ERK2 phosphorylation (Fig. 4A and Supplemental Fig. S5). Also, GFP-p18wt overexpression had no effect on the rapamycin-induced decrease in MEK1/MEK2 phosphorylation (Fig. 4A). Interestingly, LMVECs overexpressing GFP-p18wt demonstrate increased phosphorylation, and attenuated LPS-induced dephosphorylation, of mTOR, and its downstream target p70 S6K (Fig. 4B and Supplemental Fig. S5).
Figure 4.
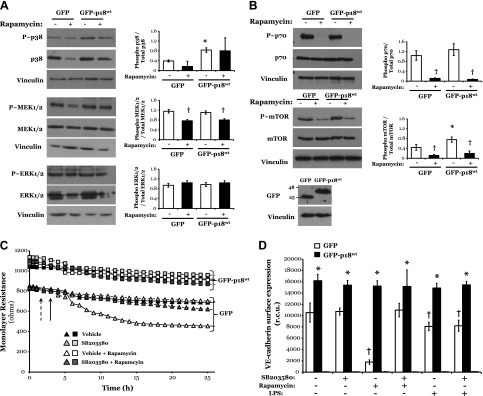
p18wt overexpression-induced p38 and mTOR activation does not influence endothelial barrier function and VE-cadherin expression at the AJ. A, B) Equivalent numbers of LMVECs were transiently transfected with GFP or GFP-p18wt cDNA. Following 24 h, LMVECs were treated in the presence (closed bars) or absence (open symbol) of rapamycin (10 nM) for a further 24 h. Phosphorylation of p38, MEK1/MEK2, ERK1/ERK2 (A), or mTOR and p70 (B) was assessed in whole-cell lysates by immunoblot analysis with an antibody specific to each phosphorylated protein. Blots were stripped and reprobed for total protein expression and vinculin as a loading control. Representative blots are shown. Data are presented as the mean ± SD (n = 3). *P < 0.05 vs. GFP; †P < 0.05 vs. vehicle. C) Equivalent numbers of LMVECs were transiently transfected with GFP (triangle symbol) or GFP-p18WT (square symbol) cDNA. Following 48 h, changes in endothelial monolayer resistance were measured using ECIS in the presence of SB20358 (10 nM) pretreatment (gray symbols) or vehicle (DMSO) (open or closed symbol). LMVECs were then treated with rapamycin (10 nM) or vehicle (DMSO). A representative tracing is shown. Dashed arrow indicates addition of SB203580; unbroken arrow indicates addition of rapamycin. Overexpression of cDNA was confirmed by immunoblot analysis of lysates from transiently transfected cells, with an antibody specific to GFP (inset) (n = 3). D) Equivalent numbers of LMVECs were transiently transfected with GFP (open bar) or GFP-p18wt (closed bar) cDNA. Following 48 h, LMVECs were preincubated with p38 inhibitor SB203580 (10 nM, 30 min) or vehicle followed by exposure to LPS (1 µg/ml, 6 h) or rapamycin (10 nM, 24 h), and cell surface expression was determined with whole-cell indirect ELISA using chemiluminescence. Data are presented as the mean ± SD (n = 4–5). *P < 0.05 vs. GFP; †P < 0.05 vs. vehicle.
We next studied whether the role of p18 on VE-cadherin surface expression and endothelial barrier function is mediated by p38 or mTOR activation. Following treatment with rapamycin, LMVECs transiently transfected with GFP-p18wt display enhanced endothelial barrier resistance and increased VE-cadherin surface expression, compared to the diminished barrier function and reduced surface VE-cadherin observed in GFP-overexpressing cells (Fig. 4C, D). Interestingly, GFP-overexpressing LMVECs exhibit significantly reduced VE-cadherin cell surface expression following treatment with the MEK-ERK inhibitor, U0126, but not the p38 inhibitor, SB203580 (Fig. 4D and Supplemental Fig. S6). However, these effects of ERK inhibition were attenuated by the overexpression of GFP-p18wt, with VE-cadherin cell surface levels maintained ∼2-fold higher than those observed in GFP-overexpressing LMVECs (Fig. 4C, D).
Thus, these data demonstrate that, whereas p18 influences activation of p38 MAPK and mTOR, the effects of p18 on the maintenance of VE-cadherin at the AJ and enhanced endothelial barrier function are mTOR and MAPK independent.
p18 colocalizes with VE-cadherin-positive endosomes in response to LPS
Following VEGF exposure, VE-cadherin has been observed to internalize into the early endosome (16, 20). Nada et al. (20) noted that the p18 N terminus associates with both early and late endosomes, a function that we show is necessary for p18 to regulate endothelial barrier function (Fig. 4). To understand the mechanism through which p18 maintains VE-cadherin at the cell surface, we next sought to study the effect of LPS on p18 association with the early endosome. Following exposure to either vehicle or LPS, both EEA1 and p18 were localized to endosome-like vesicles within the cytosol (Fig. 5D, H). Following LPS treatment, there was no significant difference in the proportion of p18-positive early endosomes within the ECs (Fig. 5I).
Figure 5.
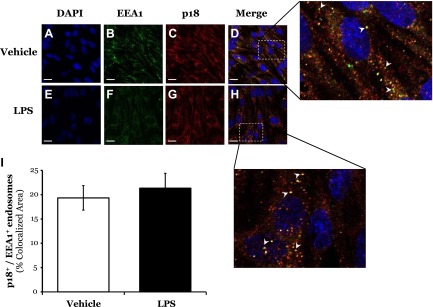
LPS does not affect colocalization of p18 with the early endosome. A–H) LMVECs were grown to confluence on gelatin-coated glass coverslips and treated with vehicle (saline) or LPS (1 μg/ml, 6 h). Cells were then fixed, permeabilized, and immunofluorescently stained for EEA1 and p18, followed by FITC- and Texas Red-labeled secondary antibodies. Images were captured via confocal microscopy at ×600 magnification. Nuclei were stained with DAPI. On enlarged images, white arrowheads indicate areas of p18 colocalization with EEA1. Representative images are shown. Scale bars, 20 μm. I) Colocalization was quantified using Zeiss LSM700 Zen software. Data are presented as the mean ± SD (n = 3).
Our next experiments assessed whether p18 can associate with VE-cadherin-positive endosomes. Upon exposure to vehicle, we observed VE-cadherin expression primarily at the IEJ, whereas p18 expression was punctate within the cytosol (Fig. 6D), similar to EEA1 staining (Fig. 5D). Likewise, overexpression of GFP-p18wt displays similar localization within the cell (data not shown). Following exposure to LPS, AJ disruption was observed concomitant with VE-cadherin internalization into the cytosol (Fig. 6H). In addition, LPS treatment increased the proportion of p18-positive endosomes colocalizing with VE-cadherin (p18+ VE-cad+ endosomes) (Fig. 6I).
Figure 6.
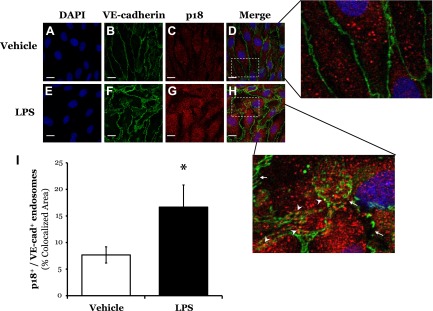
LPS increases colocalization of p18 with VE-cadherin-positive endosomes. A–H) LMVECs were grown to confluence on gelatin-coated glass coverslips and treated with vehicle (saline) or LPS (1 μg/ml, 6 h). Cells were then fixed, permeabilized, and immunofluorescently stained for VE-cadherin and p18, followed by FITC- and Texas Red-labeled secondary antibodies. Images were captured via confocal microscopy at ×600 magnification. Nuclei were stained with DAPI. On enlarged images, white arrows indicate regions of VE-cadherin internalization from the AJ. White arrowheads indicate areas of p18 colocalization with VE-cadherin. Representative images are presented. Scale bars, 20 μm. I) Colocalization was quantified using Zeiss LSM700 Zen software. Data are presented as the mean ± SD (n = 3). *P < 0.05 vs. vehicle.
To further characterize the effect of LPS on VE-cadherin-positive endosomes, we isolated early endosomes, by targeting a protein known to selectively associate with the early endosome, EEA1. Under baseline conditions, we observed the association of VE-cadherin with EEA1, which was significantly increased following exposure to LPS (Fig. 7A, B). Finally, we assessed the effect of p18 on VE-cadherin-positive late endosomes. Interestingly, although overexpression of GFP-p18wt exerted no effect on the expression level of LAMP1, a significant decrease in the association of VE-cadherin with LAMP1-positive endosomes (late endosomes) was noted in p18-overexpressing cells (Supplemental Fig. S7).
Figure 7.

LPS increases the formation of VE-cadherin-positive early endosomes. LMVECs were exposed to LPS (1 μg/ml, 6 h), collected, and subjected to endosome preparation. Early endosomes were immunoprecipitated with an antibody specific to the EEA1 and subjected to immunoblot analysis with an antibody specific to VE-cadherin. Blots were stripped and reprobed for EEA1. A representative blot (A) and densitometry analysis (B) are shown. Data are presented as the mean ± SD (n = 3). *P < 0.05 vs. vehicle.
Taken together, these data suggest that upon LPS exposure, p18 colocalizes with VE-cadherin-positive early endosomes to promote the recycling of VE-cadherin to the IEJ, thus enhancing endothelial barrier function in vitro and in vivo.
LPS increases Src-dependent tyrosine phosphorylation of p18
p18 has been identified as a potential substrate for Src-mediated tyrosine phosphorylation (25). An elevation in activated cellular Src levels has been observed in LPS-induced models of ALI (26), yet Src-induced phosphorylation of VE-cadherin at the AJ is not sufficient to cause Src-mediated barrier disruption (27). Furthermore, the endosomal adaptor protein, β-arrestin, tethers Src to the endosome lipid raft (28). Thus, our final set of experiments studied whether, in experimental settings mimicking ALI, activated Src at the endosome induced tyrosine phosphorylation of p18.
Following exposure to LPS, we observed an increase in Src activation, as measured by levels of phosphorylated Src at tyrosine residue Tyr416; an effect that was abrogated in the presence of the Src inhibitor PP2 (Fig. 8A and Supplemental Fig. S8). In addition, we noted no change in the level of Src tethered to the early endosome following treatment with LPS (Fig. 8B). We further observed a significant increase in p18 tyrosine phosphorylation in LMVECs exposed to LPS, which was attenuated in the presence of PP2 (Fig. 8C and Supplemental Fig. S8). Finally, we noted sequential tyrosine phosphorylation of VE-cadherin and p18 following LPS exposure from 30 to 120 min (Supplemental Fig. S8). Thus, in settings of ALI, activated Src elevates tyrosine phosphorylation of p18, potentially at the early endosome.
Figure 8.
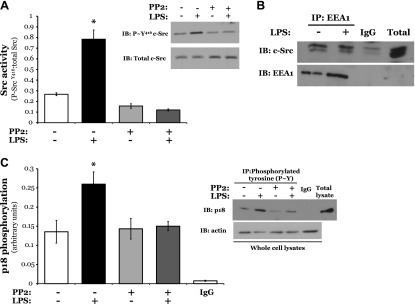
LPS induces Src-mediated tyrosine phosphorylation of p18. LMVECs were exposed to LPS (1 μg/ml, 6 h) in the presence or absence of PP2 pretreatment (12 µM, 30 min). A) LMVECs were lysed and subjected to immunoblot analysis for phosphorylated Src. Blots were stripped and reprobed for total Src levels. A representative blot (inset) and densitometry analysis are shown. B) Early endosomes were isolated from LMVECs via immunoprecipitation with an antibody specific to the EEA1 and subjected to immunoblot analysis with an antibody specific to Src. Blots were stripped and reprobed for EEA1. Representative blots are shown. C) Lysates were immunoprecipitated for tyrosine phosphorylation using the 4G10 clone antibody. Immunocomplexes and whole-cell lysates were subjected to immunoblot analysis with an antibody specific to p18 and actin, respectively. A representative blot (inset) and densitometry analysis are shown. Data are presented as the mean ± SD (n = 4). *P < 0.05 vs. vehicle.
Taken together, our data demonstrate that p18 enhances endothelial barrier function within the pulmonary vasculature through elevated recycling of VE-cadherin back to the IEJ and the formation of AJ structures via early endosome trafficking. We further show that, in settings of ALI, active Src phosphorylates p18, which in turn influences the fate of VE-cadherin-positive endosomes.
DISCUSSION
The AJ protein, VE-cadherin, is vital to the maintenance of endothelial barrier function in the pulmonary vasculature, with increased formation of pulmonary edema following disruption of the homophilic bond at the IEJ (29). Although previous studies have addressed the function of VE-cadherin at the AJ, few have assessed the intracellular regulation of VE-cadherin, specifically, the mechanisms through which the protein is recycled to the cell surface vs. undergoing degradation by the lysosome. In the current study, we demonstrate for the first time that regulation of endocytic trafficking within the endothelium represents an interesting and novel mechanism to enhance pulmonary vasculature function. We show that overexpression of p18 enhances endothelial barrier integrity in vitro and in vivo through increased expression and function of VE-cadherin at the IEJ. Internalization of VE-cadherin and resulting increased monolayer permeability, in response to VEGF or an LPS-induced model of ALI, are attenuated by the overexpression of WT p18. Mutant p18, unable to bind the endosome, exerted no protective effect on in vitro or in vivo endothelial function or VE-cadherin at the IEJ. Enhanced endothelial barrier function and VE-cadherin surface expression, following p18 overexpression, were not affected by inhibition of MAPK or mTOR signaling pathways. In LPS-induced ALI, increased VE-cadherin internalization from the IEJ into p18-positive early endosomes occurs concomitant with elevated Src tethered to the early endosome and increased Src-dependent tyrosine phosphorylation of the endocytic protein p18. Thus, we propose that p18 regulates endocytic trafficking and, thus, the fate of VE-cadherin-positive endosomes. Thus, the effect of p18 on endosome dynamics within the pulmonary vasculature may implicate both the protein and endocytic trafficking as novel targets in the treatment of ALI.
Recently, p18 was identified as a membrane-associated type 1 transmembrane matrix metalloprotease-associated protein (18). Further studies observe that p18 binds to the endosome to play a role in development, cholesterol homeostasis, and tumor invasion (20, 22, 30). All the reported functions of p18 stem from its ability to bind the endosome through the N-terminal palmitoylation and myristoylation sites (20, 22). Likewise, we observe that p18wt enhances barrier function in the pulmonary vasculature, whereas the N-terminal p18 mutant unable to bind the endosome exerts no effect on monolayer integrity. Furthermore, p18wt overexpression increases surface expression of VE-cadherin and attenuates LPS- or VEGF-induced internalization of VE-cadherin; however, the p18N39 mutant does not elicit an effect on VE-cadherin recycling. Thus, the ability for p18 to bind to the endosome is vital to its function in regulating the pulmonary vasculature.
At the late endosome, p18 tethers the p14-MP1 complex, a scaffold for MEK1; thus, PC12 cells from p18-deficient mice exhibit abrogated MEK-ERK activity following epidermal growth factor treatment (20). In this cellular location, p18-p14-MP1 also forms the basis for the Ragulator complex, a key activator of mTOR activity (19). mTOR inhibition, via rapamycin, has been observed to mimic the dysregulated late endosomal/lysosomal function observed in p18−/− mouse embryonic fibroblasts (MEFs); however, this role of p18 is independent of MEK/ERK signaling (19). Thus, p18 appears to play different roles at the late endosome depending on the cellular stimulus. In the present study, the role of mTOR activity and MEK1-ERK1/ERK2 signaling on endothelial barrier function was assessed. Although overexpression of p18 increased p38 activation, independently of mTOR activity, p38 inhibition exhibited no effect on enhanced p18-mediated endothelial barrier function and VE-cadherin surface expression. In addition, mTOR or MEK-ERK inhibition both increase barrier permeability and decrease VE-cadherin surface levels; however, p18 overexpression attenuated these effects. Thus, p18 plays a role in the pulmonary endothelium via anchoring at the endosome, which is independent of mTOR or MAPK signaling. We therefore propose that p18 influences barrier function through endosomal trafficking of cargo and not endosomal signaling through the regulation of MAPK complexes.
Previous studies linking monolayer permeability with endocytosis have focused on clathrin-independent, caveola-mediated transcytosis of cargo across the ECs (41). Knockdown of caveolin-1 expression, the key component of caveolae, causes defective albumin endocytosis, increased endothelial monolayer permeability in vitro, and pulmonary edema formation in vivo (42, 43). Caveolin-1 deficiency in pulmonary microvascular cells indicated that approximately 65% of endocytosis is clathrin dependent, indicating that caveolin-mediated endocytosis is not the predominant form of endocytic trafficking within the endothelium (44). Early studies on endocytosis dictate that clathrin-coated endosomes represent a trafficking pathway with distinct cellular compartments, whereas caveola cannot regulate or differentiate between recycling and degradation (45). Although links between endocytosis of clathrin-coated vesicles into the cell and microvascular permeability have been previously established (16), the fate of the endosome has yet to be established. Furthermore, Gavard and Gutkind (16) observed that VE-cadherin-positive early endosomes colocalized with clathrin-, but not caveolin-, positive vesicles. Thus, it is possible that p18 mediates endothelial barrier function through association with clathrin-coated, VE-cadherin-positive endosomes, representing a caveolin-independent novel mechanism to link endocytosis and vascular permeability.
Rab proteins are small GTPases that regulate the spatial and temporal trafficking of endosomes within cells. Rab proteins tightly regulate endosomal fate by binding to the endosome in the Rab-GTP active form and directing its transport via specific interactions with multiple Rab effector proteins. Rabs 1–9, 11, 13–15, 22, and 30 have all been identified in ECs (46). MEFs from p18 knockout mice display disrupted localization of Rab proteins, such as prodegradative Rab7 and prorecycling Rab11; thus, the absence of p18 results in aberrant endocytic dynamics within the cell (20). We therefore hypothesize that p18 modulates trafficking of the VE-cadherin-positive endosome and thus endothelial barrier function, through regulation/maintenance of Rab-endosome associations.
Immunoaffinity profiling of the tyrosine kinase Src identified p18 as a substrate of Src-mediated tyrosine phosphorylation at residue 40 (25). Through interaction with the endosome-regulating protein, β-arrestin, Src is tethered to the endosome (28). In settings of ALI, Src is activated within the ECs (26, 47). In agreement with this, we observe an increase in Src phosphorylation at tyrosine 416 following exposure to LPS, which is attenuated by the Src family kinase inhibitor PP2. Src activation plays a role in VE-cadherin endocytosis by initiating phosphorylation and internalization of VE-cadherin (16). However, Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to disrupt p120- and β-catenin binding with VE-cadherin or increase endothelial monolayer permeability (27). Thus, activated Src is acting through an alternate mechanism to influence endothelial monolayer permeability. In the present study, we observe Src tethered to the early endosome and Src-mediated tyrosine phosphorylation of p18 in settings of ALI. The trafficking of E-cadherin-positive endosomes to the lysosome is regulated by Src-induced activation of the late endosomal, degradative Rab7 GTPase (48). Although we do not address the phosphorylation of p18 following overexpression of the protein, it is possible that, in settings of ALI, Src tethered to the VE-cadherin-positive endosome phosphorylates p18, potentially at tyrosine 40. We speculate that phosphorylation of p18 may then decrease association of the endosome to prorecycling Rab GTPases, or increase association of VE-cadherin-positive endosomes to prodegradation Rab-GTPases, resulting in late endosomal/lysosomal degradation of VE-cadherin. However, further studies are necessary to elucidate the site of tyrosine phosphorylation and the resulting effect on p18 function within the ECs.
The AJ protein, p120-catenin, is an important regulator of AJ stability, with knockdown of p120 expression causing rapid internalization and lysosomal degradation of VE-cadherin (9, 11). Recently, Nanes et al. (10) demonstrated that the p120-binding region of VE-cadherin is comprised of a highly conserved endocytic signal through which p120 binds to and thus inhibits cadherin endocytosis, by physically occupying the signal motif. In the present study, we demonstrate enhanced VE-cadherin levels at the AJ and association with p120- and β-catenin following overexpression of p18wt. Thus, p18-mediated increases in p120-catenin:VE-cadherin protein associations likely stabilize the AJ structurally and prevent VE-cadherin internalization by p120 masking the endocytic signal in the cadherin. Association of AJs with the vimentin cytoskeleton in ECs, via γ-catenin and desmoplakin, strengthens and anchors it at the IEJ (49). Although we do not directly address the VE-cadherin:cytoskeleton association in the present study, association of VE-cadherin with the cytoskeleton is essential for the maintenance of endothelial junction integrity and vascular permeability. Furthermore, p18 has been observed to colocalize with punctate actin structures within lung metastatic tumor cells (30). Interestingly, endocytic trafficking and actin dynamics overlap because the actin cytoskeleton is known to participate in endocytic trafficking (50). Indeed, disrupting filamentous actin results in the redistribution of late endosomes to the cell membrane, altered endosome motility, and inhibition of cargo degradation (50). Thus, we propose that p18 increases catenin-binding VE-cadherin at the IEJ, therefore enhancing the anchoring of the AJ to the EC cytoskeleton and maintaining endothelial barrier integrity.
Studies with anti-VE-cadherin antibodies and edemagenic agents such as TNF, thrombin, and VEGF demonstrate disruption of VE-cadherin homophilic bonds concomitant with lung edema formation (12, 29). Likewise, we observe a breakdown of the p120- and β-catenin association with VE-cadherin following LPS exposure, resulting in VE-cadherin internalization, barrier disruption, and pulmonary edema formation. Genetic manipulation to enhance VE-cadherin-catenin interactions has been previously observed to reduce protein and leukocyte bronchoalveolar lavage leak in LPS-challenged mice (14, 51). Overexpression of p18 mimics these effects by enhancing levels of VE-cadherin at the IEJ, stabilizing VE-cadherin-catenin interactions, and protecting against LPS-induced monolayer permeability in vitro and in vivo. Thus, p18-mediated stabilization of the AJ and enhanced pulmonary vasculature indicates that targeting both endocytic trafficking and p18 will have pathogenic implications for ALI.
Supplementary Material
Acknowledgments
This material is the result of work supported with resources and the use of facilities at the Providence Veterans Affairs Medical Center. The work was supported by the U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute Grant R01 HL-67795, American Heart Association Grant 10GRNT4160055, COBRE NIH National Institute of General Medical Sciences Award 5P20 GM103652, and University Medicine Foundation of Rhode Island Hospital (to E.O.H.). H.C. was supported by American Heart Association Grant 13POST16860031. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs.
Glossary
- AJ
adherens junction
- ALI
acute lung injury
- BSA
bovine serum albumin
- CFU
colony-forming unit
- EC
endothelial cell
- EEA1
early endosome antigen 1
- GFP
green fluorescent protein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IEJ
interendothelial junction
- kf
filtration coefficient
- LAMP1
lysosomal-associated membrane protein 1
- LMVEC
lung microvascular endothelial cell
- MEF
mouse embryonic fibroblast
- MP1
MEK partner 1
- mTOR
mammalian target of rapamycin
- MTT
methyl thiazolyl tetrazolium
- ns
nonsilencing
- PA103
Pseudomonas aeruginosa strain 103
- r.c.u.
relative chemiluminescence unit
- siRNA
small interfering RNA
- TJ
tight junction
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Rubenfeld G. D., Caldwell E., Peabody E., Weaver J., Martin D. P., Neff M., Stern E. J., Hudson L. D. (2005) Incidence and outcomes of acute lung injury. N. Engl. J. Med. 353, 1685–1693 [DOI] [PubMed] [Google Scholar]
- 2.Gao X., Kouklis P., Xu N., Minshall R. D., Sandoval R., Vogel S. M., Malik A. B. (2000) Reversibility of increased microvessel permeability in response to VE-cadherin disassembly. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L1218–L1225 [DOI] [PubMed] [Google Scholar]
- 3.Xiao K., Allison D. F., Kottke M. D., Summers S., Sorescu G. P., Faundez V., Kowalczyk A. P. (2003) Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J. Biol. Chem. 278, 19199–19208 [DOI] [PubMed] [Google Scholar]
- 4.Breviario F., Caveda L., Corada M., Martin-Padura I., Navarro P., Golay J., Introna M., Gulino D., Lampugnani M. G., Dejana E. (1995) Functional properties of human vascular endothelial cadherin (7B4/cadherin-5), an endothelium-specific cadherin. Arterioscler. Thromb. Vasc. Biol. 15, 1229–1239 [DOI] [PubMed] [Google Scholar]
- 5.Lampugnani M. G., Corada M., Caveda L., Breviario F., Ayalon O., Geiger B., Dejana E. (1995) The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin). J. Cell Biol. 129, 203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taddei A., Giampietro C., Conti A., Orsenigo F., Breviario F., Pirazzoli V., Potente M., Daly C., Dimmeler S., Dejana E. (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 7.Nelson W. J. (2008) Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem. Soc. Trans. 36, 149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiasson C. M., Wittich K. B., Vincent P. A., Faundez V., Kowalczyk A. P. (2009) p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Mol. Biol. Cell 20, 1970–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis M. A., Ireton R. C., Reynolds A. B. (2003) A core function for p120-catenin in cadherin turnover. J. Cell Biol. 163, 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nanes B. A., Chiasson-MacKenzie C., Lowery A. M., Ishiyama N., Faundez V., Ikura M., Vincent P. A., Kowalczyk A. P. (2012) p120-catenin binding masks an endocytic signal conserved in classical cadherins. J. Cell Biol. 199, 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao K., Garner J., Buckley K. M., Vincent P. A., Chiasson C. M., Dejana E., Faundez V., Kowalczyk A. P. (2005) p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol. Biol. Cell 16, 5141–5151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corada M., Mariotti M., Thurston G., Smith K., Kunkel R., Brockhaus M., Lampugnani M. G., Martin-Padura I., Stoppacciaro A., Ruco L., McDonald D. M., Ward P. A., Dejana E. (1999) Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA 96, 9815–9820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gotsch U., Borges E., Bosse R., Böggemeyer E., Simon M., Mossmann H., Vestweber D. (1997) VE-cadherin antibody accelerates neutrophil recruitment in vivo. J. Cell Sci. 110, 583–588 [DOI] [PubMed] [Google Scholar]
- 14.Broermann A., Winderlich M., Block H., Frye M., Rossaint J., Zarbock A., Cagna G., Linnepe R., Schulte D., Nottebaum A. F., Vestweber D. (2011) Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J. Exp. Med. 208, 2393–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatanaka K., Simons M., Murakami M. (2011) Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am. J. Physiol. Heart Circ. Physiol. 300, H162–H172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavard J., Gutkind J. S. (2006) VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 8, 1223–1234 [DOI] [PubMed] [Google Scholar]
- 17.Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 18.Hoshino D., Tomari T., Nagano M., Koshikawa N., Seiki M. (2009) A novel protein associated with membrane-type 1 matrix metalloproteinase binds p27(kip1) and regulates RhoA activation, actin remodeling, and matrigel invasion. J. Biol. Chem. 284, 27315–27326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi Y., Nada S., Mori S., Soma-Nagae T., Oneyama C., Okada M. (2012) The late endosome/lysosome-anchored p18-mTORC1 pathway controls terminal maturation of lysosomes. Biochem. Biophys. Res. Commun. 417, 1151–1157 [DOI] [PubMed] [Google Scholar]
- 20.Nada S., Hondo A., Kasai A., Koike M., Saito K., Uchiyama Y., Okada M. (2009) The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 28, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soma-Nagae T., Nada S., Kitagawa M., Takahashi Y., Mori S., Oneyama C., Okada M. (2013) The lysosomal signaling anchor p18/LAMTOR1 controls epidermal development by regulating lysosome-mediated catabolic processes. J. Cell Sci. 126, 3575–3584 [DOI] [PubMed] [Google Scholar]
- 22.Guillaumot P., Luquain C., Malek M., Huber A. L., Brugière S., Garin J., Grunwald D., Régnier D., Pétrilli V., Lefai E., Manié S. N. (2010) Pdro, a protein associated with late endosomes and lysosomes and implicated in cellular cholesterol homeostasis. PLoS One 5, e10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knezevic N., Tauseef M., Thennes T., Mehta D. (2009) The G protein betagamma subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J. Exp. Med. 206, 2761–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou M. Y., Lo S. K., Bergenfeldt M., Tiruppathi C., Jaffe A., Xu N., Malik A. B. (1998) In vivo expression of neutrophil inhibitory factor via gene transfer prevents lipopolysaccharide-induced lung neutrophil infiltration and injury by a beta2 integrin-dependent mechanism. J. Clin. Invest. 101, 2427–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X. M., Polakiewicz R. D., Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 26.Severgnini M., Takahashi S., Rozo L. M., Homer R. J., Kuhn C., Jhung J. W., Perides G., Steer M., Hassoun P. M., Fanburg B. L., Cochran B. H., Simon A. R. (2004) Activation of the STAT pathway in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 286, L1282–L1292 [DOI] [PubMed] [Google Scholar]
- 27.Adam A. P., Sharenko A. L., Pumiglia K., Vincent P. A. (2010) Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J. Biol. Chem. 285, 7045–7055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luttrell L. M., Miller W. E. (2013) Arrestins as regulators of kinases and phosphatases. Prog. Mol. Biol. Transl. Sci. 118, 115–147 [DOI] [PubMed] [Google Scholar]
- 29.Vestweber D., Winderlich M., Cagna G., Nottebaum A. F. (2009) Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 19, 8–15 [DOI] [PubMed] [Google Scholar]
- 30.Hoshino D., Koshikawa N., Seiki M. (2011) A p27(kip1)-binding protein, p27RF-Rho, promotes cancer metastasis via activation of RhoA and RhoC. J. Biol. Chem. 286, 3139–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chichger H., Grinnell K. L., Casserly B., Chung C. S., Braza J., Lomas-Neira J., Ayala A., Rounds S., Klinger J. R., Harrington E. O. (2012) Genetic disruption of protein kinase Cδ reduces endotoxin-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 303, L880–L888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buttenschoen K., Kornmann M., Berger D., Leder G., Beger H. G., Vasilescu C. (2008) Endotoxemia and endotoxin tolerance in patients with ARDS. Langenbecks Arch. Surg. 393, 473–478 [DOI] [PubMed] [Google Scholar]
- 33.Stevens T. C., Ochoa C. D., Morrow K. A., Robson M. J., Prasain N., Zhou C., Alvarez D. F., Frank D. W., Balczon R., Stevens T. (2014) The Pseudomonas aeruginosa exoenzyme Y impairs endothelial cell proliferation and vascular repair following lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L915–L924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grinnell K. L., Casserly B., Harrington E. O. (2010) Role of protein tyrosine phosphatase SHP2 in barrier function of pulmonary endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L361–L370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grinnell K. L., Chichger H., Braza J., Duong H., Harrington E. O. (2012) Protection against LPS-induced pulmonary edema through the attenuation of protein tyrosine phosphatase-1B oxidation. Am. J. Respir. Cell Mol. Biol. 46, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uhlig S., Wollin L. (1994) An improved setup for the isolated perfused rat lung. J. Pharmacol. Toxicol. Methods 31, 85–94 [DOI] [PubMed] [Google Scholar]
- 37.Denizot F., Lang R. (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 89, 271–277 [DOI] [PubMed] [Google Scholar]
- 38.Wang Y., Huang L., Yang Y., Xu L., Yang J., Wu Y. (2013) Effects of autocrine vascular endothelial growth factor (VEGF) in non-small cell lung cancer cell line A549. Mol. Biol. Rep. 40, 3093–3099 [DOI] [PubMed] [Google Scholar]
- 39.Stasyk T., Holzmann J., Stumberger S., Ebner H. L., Hess M. W., Bonn G. K., Mechtler K., Huber L. A. (2010) Proteomic analysis of endosomes from genetically modified p14/MP1 mouse embryonic fibroblasts. Proteomics 10, 4117–4127 [DOI] [PubMed] [Google Scholar]
- 40.Oas R. G., Nanes B. A., Esimai C. C., Vincent P. A., García A. J., Kowalczyk A. P. (2013) p120-catenin and β-catenin differentially regulate cadherin adhesive function. Mol. Biol. Cell 24, 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y., Hu G., Zhang X., Minshall R. D. (2009) Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ. Res. 105, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maniatis N. A., Kardara M., Hecimovich D., Letsiou E., Castellon M., Roussos C., Shinin V., Votta-Vellis E. G., Schwartz D. E., Minshall R. D. (2012) Role of caveolin-1 expression in the pathogenesis of pulmonary edema in ventilator-induced lung injury. Pulm. Circ. 2, 452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schubert W., Frank P. G., Woodman S. E., Hyogo H., Cohen D. E., Chow C. W., Lisanti M. P. (2002) Microvascular hyperpermeability in caveolin-1 (−/−) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J. Biol. Chem. 277, 40091–40098 [DOI] [PubMed] [Google Scholar]
- 44.Li H. H., Li J., Wasserloos K. J., Wallace C., Sullivan M. G., Bauer P. M., Stolz D. B., Lee J. S., Watkins S. C., St Croix C. M., Pitt B. R., Zhang L. M. (2013) Caveolae-dependent and -independent uptake of albumin in cultured rodent pulmonary endothelial cells. PLoS One 8, e81903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nichols B. J. (2002) A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol. 4, 374–378 [DOI] [PubMed] [Google Scholar]
- 46.De Leeuw H. P., Koster P. M., Calafat J., Janssen H., van Zonneveld A. J., van Mourik J. A., Voorberg J. (1998) Small GTP-binding proteins in human endothelial cells. Br. J. Haematol. 103, 15–19 [DOI] [PubMed] [Google Scholar]
- 47.Oyaizu T., Fung S. Y., Shiozaki A., Guan Z., Zhang Q., dos Santos C. C., Han B., Mura M., Keshavjee S., Liu M. (2012) Src tyrosine kinase inhibition prevents pulmonary ischemia-reperfusion-induced acute lung injury. Intensive Care Med. 38, 894–905 [DOI] [PubMed] [Google Scholar]
- 48.Palacios F., Tushir J. S., Fujita Y., D’Souza-Schorey C. (2005) Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol. Cell. Biol. 25, 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dejana E., Orsenigo F., Lampugnani M. G. (2008) The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121, 2115–2122 [DOI] [PubMed] [Google Scholar]
- 50.Durrbach A., Louvard D., Coudrier E. (1996) Actin filaments facilitate two steps of endocytosis. J. Cell Sci. 109, 457–465 [DOI] [PubMed] [Google Scholar]
- 51.Schulte D., Küppers V., Dartsch N., Broermann A., Li H., Zarbock A., Kamenyeva O., Kiefer F., Khandoga A., Massberg S., Vestweber D. (2011) Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 30, 4157–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.