

Abstract
Intrauterine growth restriction (IUGR) confers heritable alterations in DNA methylation, rendering risk of adult metabolic syndrome (MetS). Because CpG methylation is coupled to intake of essential nutrients along the one-carbon pathway, we reasoned that essential nutrient supplementation (ENS) may abrogate IUGR-conferred multigenerational MetS. Pregnant Sprague-Dawley rats underwent bilateral uterine artery ligation causing IUGR in F1. Among the F2 generation, IUGR lineage rats were underweight at birth (6.7 vs. 8.0 g, P < 0.0001) and obese by adulthood (p160: 613 vs. 510 g; P < 0.0001). Dual energy X-ray absorptiometry studies revealed increased central fat mass (Δ+40 g), accompanied by dyslipidemic (>30% elevated, P < 0.05) serum triglycerides (139 mg/dl), very-LDLs (27.8 mg/dl), and fatty acids (632 µM). Hyperglycemic-euglycemic clamp studies and glucose tolerance testing revealed insulin resistance. Conversely, IUGR lineage ENS-fed rats did not manifest MetS, with significantly lower body weight (p160: 410 g), >5-fold less central fat mass, normal hepatic glucose efflux, and >70% reduced circulating triglycerides and very-LDLs compared with IUGR control-fed F2 offspring (P < 0.01). Moreover, increased methylation of the IGF-1 P2 transcriptional start site among IUGR lineage F2 offspring was reversed in ENS (P < 0.04). This is an initial demonstration that supplementation along the one-carbon pathway abrogates adult morbidity and associated epigenomic modifications of IGF-1 in a rodent model of multigenerational MetS.—Goodspeed, D., Seferovic, M. D., Holland, W., Mcknight, R. A., Summers, S. A., Branch, D. W., Lane, R. H., Aagaard, K. M. Essential nutrient supplementation prevents heritable metabolic disease in multigenerational intrauterine growth-restricted rats.
Keywords: one-carbon pathway, epigenomics, methyl donors, IGF-1, obesity
The gestational environment is a key arbiter of the relative risk of health and disease in later life. In utero metabolic substrate restriction and uteroplacental insufficiency leading to intrauterine growth restriction (IUGR) is thought to bestow lifelong epigenetic changes that are maladaptive to the postnatal environment. This phenomenon is referred to as the developmental origins of health and disease hypothesis and was originally identified by associating low birth weight neonates with increased mortality from coronary heart disease much later in life (1, 2). IUGR has now been associated with increased incidence of health conditions including vascular dysfunction (3, 4), type II diabetes and dyslipidemia (5, 6), obesity, and hypertension (2, 7, 8). A symptomatic grouping of these IUGR health complications later in life is broadly characterized as metabolic syndrome (MetS), a condition approaching global epidemic prevalence (9). The clinical criteria for MetS is evolving as it struggles to account for large differences in global populations, age, and race (10). Currently, the consensus of clinical criteria includes the following: dyslipidemia, characterized by elevated triglycerides, ApoB lipoproteins, and reduced HDL; elevated blood pressure; and altered glucose homeostasis, with elevated central fat mass/obesity and insulin resistance (10–12).
Maternal influences, including poor nutrition and uteroplacental insufficiency, are associated with IUGR and adult MetS (13–16). Rodent IUGR models that restrict either protein or carbohydrates have demonstrated adult-onset MetS associated with important epigenetic regulatory changes to key metabolic pathways (17–20), and our previous data in primates have demonstrated that a caloric-dense maternal diet can influence epigenetic changes in the fetal liver (21). Although nutrient deprivation is a risk for MetS regardless of IUGR etiology (13, 22–24), maternal deprivation is not a predominant cause of IUGR in all but the most underdeveloped countries. Rather, uteroplacental insufficiency is the predominant cause of fetal nutrient restriction leading to IUGR, which is independent of maternal nutrition. Thus, maternal nutrient restrictive models inadequately replicate the broad deprivation of metabolites and essential nutrients (including methyl donors) in placental insufficiency, as well as other important changes such as hypoxia and acidosis, waste product accumulation, and associated biochemical changes such as inflammation, key regulatory factor alterations, and broad transcriptional changes particularly associated with hypoxia. Ergo, the surgical bilateral uterine artery ligation procedure represents a more appropriate model to study epigenetic changes associated with placental insufficiency as it replicates restriction due to circulatory defects and in this way leads to reduced maternal/fetal exchange. The immediate consequences are persistent in utero hypoinsulinemia, hypoglycemia, acidosis, and hypoxia (25, 26).
Extensive study of animal models has uncovered some specific epigenetic-mediated molecular mechanisms implicated in long-term heath complications (20, 27–35). IGF-1 is the primary driver of late gestational and postnatal growth and has been implicated in altered weight and development (36, 37). Decreased expression is associated with insulin resistance later in life, as well as increased incidence of MetS (38). In a number of model systems, a perturbed in utero environment lending epigenomic modifications (both histone variations and differential methylation) leads to IGF-1 expression changes (33–35, 39). Of interest to our studies herein, an identical bilateral uterine artery ligation procedure results in altered IGF-1 expression changes, which are accompanied by site-specific differential methylation (33). Of the splice variants, the IGF-1B was shown to be particularly variable both in its expression and its methylation (33).
Although DNA methylation is relatively stable, there are developmental time points, such as gamete imprinting, preimplantation development, and post-midgestational development, when methylation changes are active (40). These windows provide intervention points where methylation is particularly plastic and, for imprinting and postimplantation events, amenable to dietary influence. Dietary methylation supplementation is now known to lead to variable gene expression (41, 42). These epigenetic changes associated with methylation supplementation continue to influence biology after weaning (43). For example, glucose tolerance in later life is associated with methyl deficiency during gestation (44), and supplementation of methyl donors alters hepatic promoter methylation and the transcriptional profile in nonalcoholic fatty liver disease (45).
DNA methylation changes as causative agents of disease have long been established. It was discovered that methylation of our DNA is sufficiently sensitive to essential dietary methyl sources and cofactors to cause human disease over a decade ago (46). For example, methyl sources related to seasonal food consumption in Gambia affect DNA methylation patterns (47). This sensitivity is likely caused by methyl group substrates and cofactors that constitute the one-carbon metabolism pathway, many components of which are essential, and therefore can only be derived from dietary sources (48). Methylation of CpG islands is catalyzed by DNA methyltransferases with S-adenosyl methionine as the methyl donor, synthesized from methionine via dietary methyl donors or one-carbon metabolism (49). Notably, the essential amino acid methionine is a methyl donor, itself dependent on B12 as a methionine synthase cofactor, which also uses a downstream product of folic acid as a substrate. In an alternate liver pathway, methionine is synthesized from betaine, which is derived from choline. Further, several of the enzymes are zinc dependent (46, 50). Methylation is further influenced along the renal arginine-dependent nitric oxide pathway (51).
Studies have made use of these essential nutrients to study how epigenetic methylation changes relate to disease. For example, male mice have shown epigenetic reprogramming of sperm that leads to birth defects in offspring when fed a folate-deficient diet (52). In another example, rats using essential nutrient methyl-donor dietary supplementation (ENS) have shown modifications in DNA methylation and decreased hepatic triglyceride accumulation and nonalcoholic fatty liver disease, which are one of the first hepatic alterations of metabolic syndrome (53). In humans, methylation changes to IGF-1 that were associated with nonalcoholic fatty liver disease were partially reversed as a result of nutritional adjustment following bariatric surgery (54).
Given that DNA methylation is modifiable by ENS of the diet, we hypothesized that supplementation initiated after weaning and continued through gestation and lactation of the F1 generation would abrogate the heritable adult metabolic disease resulting from in utero IUGR among F2 offspring. To study these effects, we rendered uteroplacental insufficiency-induced IUGR by performing late-gestation bilateral uterine artery ligation on grandparental (P1) pregnant Sprague-Dawley rats at e19, with subsequent delivery of the F1 pups at e21. The F1 generation was allocated onto either control or ENS-supplemented diet at weaning (p21) and then allowed to breed spontaneously; the F1 generation was not surgically manipulated during pregnancy. Regardless, the resultant IUGR lineage rats in the F2 generation weighed significantly less at birth than the sham lineage pups. By p160, a sex-specific MetS phenotype was apparent. IUGR lineage males fed a control diet exhibited obesity, increased central fat mass accumulation, glucose intolerance, insulin resistance, and increased triglyceride, very-LDL (VLDL), and fatty acids. ENS-fed IUGR lineage males exhibited no obesogenicity, glucose intolerance, insulin resistance or increases in triglycerides, VLDL, or fatty acids, indicating a diet induced reversal of metabolic disease. Accompanying these notable and significant phenotypic alterations are significant site-specific differences in DNA methylation of the IGF-1 promoter 2 region. Specifically, we demonstrate that there is an observable and significant corresponding increase in methylation among F2 generation IUGR lineage rats relative to sham controls; a finding that reversed with ENS diet supplementation in the F1 and F2 generation. Collectively, we observe a multigenerational transmission of the IUGR phenotype with manifestation of adult MetS and variations in site specific methylation of the P2 promoter of IGF-1, which is abrogated by dietary supplementation of one-carbon intermediates in the F1 generation.
MATERIALS AND METHODS
IUGR rat model development
The experiments were conducted with the review and approval of the University of Utah Animal Care committee. Surgical procedures were performed as previously described (55). Briefly, pregnant Sprague-Dawley rats were obtained on e17 and acclimated to their environment for 2 d prior to surgery. On day e19, pregnant females were anesthetized with an intraperitoneal injection of xylazine (8 mg/kg) and ketamine (40 mg/ml). After confirmation of anesthetization, bilateral uterine artery ligation was performed to produce the uteroplacental insufficiency IUGR model, or sham surgeries were performed for controls. Pups were born via cesarean section after 21 d of gestation, and litters were culled to 8 pups. On p21, suckling F1 offspring were weaned onto an ad libitum lifelong diet of either control rat chow (Harlan Teklad 8640; Harlan Laboratories, Indianapolis, IN, USA) or a custom-formulated isocaloric Harlan Teklad 8640 rat chow supplemented with 15 mg of folic acid, 15 mg of choline, 1.5 mg of B12, 15 g of betaine, 11 g of l-methionine, 50 g of l-arginine, and 150 mg of zinc for every kilogram of Harlan Teklad 8640, constituting the ENS diet. Mating pairs were established by p80, prior to the development of F1 MetS. Mating pairs were shammat × shampat, IUGRmat × shampat, shammat × IUGRpat, and IUGRmat × IUGRpat for both the control diet and ENS diet, giving a total of 8 different mating sets. Four hours after spontaneous delivery of the F2 generation (N = 512), fetal weights were obtained, and litters were again culled to 8 pups (N = 32). On p21, the F2 generation was weaned onto the same diet of their maternal and paternal lineage. Rats were weighed both at birth and at p160.
Dual energy X-ray absorptiometry
Dual energy X-ray absorptiometry (DEXA) Norland XR-36 Bone Densitometer (Norland Products, Inc., Cranbury Township, NJ, USA) was used to assess rat whole body global and regional body composition. DEXA relies on X-ray technology to separate the lean and fat mass based on density and has been previously used extensively for nutritional studies (56, 57). Rats were anesthetized before measurements with an intraperitoneal injection of xylazine (8 mg/kg) and ketamine (40 mg/ml) and then placed in prone position with extremities extended and stabilized with tape to enable full scanning of the radius and ulna. Spatial resolution was 6.0 × 6.0 mm. Scans were performed at a speed of 60 mm/s. The scan field width was 12 cm, and scanning time for each animal was <3 min. Following the scan, rats were immediately euthanized. To determine central fat mass, a region of interest including the torso area was extracted from the whole body scan, and fat mass was determined.
Fasting serum lipid profiling
Adult p160 rats were fasted for 12 h, and then blood was collected by nicking the end of the tail with a scalpel once. Blood flow was maintained throughout by gentle stroking of the tail. Bleeding was stopped after sampling by dipping the tail in alcohol and subsequent application of direct pressure. A coronary risk and lipid profiling panel was run on each serum sample. Triglyceride and VLDL concentrations were obtained by clinical laboratory testing performed by ARUP Laboratories (Salt Lake City, UT, USA) using standard quantitative enzymatic assays. Free fatty acids were measured from serum using the Roche Half-Micro Test Kit (Indianapolis, IN, USA). The kit was scaled down for use in a 96-well plate (one-fifth suggested amount for each reagent; 10 µl serum). All preparation was performed according to the manufacturer’s instructions. Palmitate standard was made by dissolving 74 mg palmitate into a solution of 35 ml ethanol and 65 ml 6% Triton X-100 to make a high standard of 2.878 mM and then serially diluted. Absorbance was assessed on a UV/Vis microplate reader. A standard curve was generated from which total fatty acid contents from serum samples were interpolated using the average of triplicate wells.
Glucose tolerance test
Adult p160 rats were fasted for 6 h and then weighed. Blood samples were collected by tail vein as previously described. A sample of blood was collected before administration of glucose, which was then administered orally to each rat (2.5 g glucose/kg body weight). Blood samples were subsequently collected at 15, 30, 45, 60, 90, and 120 min after glucose challenge from the tail as described. Serum insulin levels were measured using an ELISA kit.
Hyperinsulinemic-euglycemic clamp studies
The use of hyperinsulinemic-euglycemic clamp studies was carried out to determine insulin resistance, as the most accurate and sensitive laboratory method that is specific to hepatic insulin resistance (58). Experiments were performed as described previously (59). Adult p160 rats were fasted for 6 h prior to commencement of the experiment. Following aseptic placement of jugular (for infusion) and carotid (for blood sampling) catheters, animals were allowed to recover to presurgical weight (7–10 d). Clamps were performed in conscious unrestrained animals using swivels and tethers (Instech, Plymouth Meeting, PA, USA) to allow uninterrupted movement of the animal without disruption of infusion lines. Hyperinsulinemia was initiated by intravenous infusion of insulin (4 mU/kg per min). Blood was sampled from arterial lines in 7 min intervals and analyzed with a Beckman Glucose analyzer II (Beckman Coulter, Fullerton, CA, USA). Euglycemia was maintained by variable infusion of 20% dextrose. Steady-state glucose (∼130 mg/dl) was achieved ∼90 min after initiating hyperinsulinemia and maintained for ≥45 min. Glucose infusion rates were calculated as the average glucose infusion rate during the steady-state period. Additional blood samples were taken before initiating hyperinsulinemia and at the end of the clamp for analysis of insulin. [14C]Glucose and [3H]2-deoxyglucose were given as a bolus during the steady state for estimation of hepatic glucose output and 2-deoxyglucose uptake, as previously described (60).
Methylation-specific PCR
To map IGF-1 promoter methylation changes, a standard PCR-based method following bisulfite modification was performed as described previously (33). Briefly, primer sets were designed to probe 3 regions of CpG sites of the IGF-1 P2 transcription start site. These were forward 5′-GGAGGGTTTAATTTATAAAAGATTTTAG, reverse 5′-CCCAAACCACTTCCTTACCTAA; forward 5′-GTTGTTGTTGTTATTGTTYGTGGTA, reverse 5′-CTAAAATCTTTTATAAATTAAACCCTCC; and forward 5′-TTAGGTAAGGAAGTGGTTTGGG, reverse 5′-CACTATCTCAACTACCAAACCAC. PCR conditions were as follows: 95°C for 10 min, and then 94°C for 30 s, annealing at 53°C for 30 s, and 72°C for 30 s for 35 cycles. Primers were designed and figures prepared with the use of University of California, Santa Cruz Genome Browser (61). A subset of 8 p21 animals was used for each condition. PCR products were cloned into the vector pSC-A (Stratagene, Cedar Creek, TX, USA), and 6 colonies from each cloning were inoculated on SeqPrep 96 plates (Edge Biosystems, Gaithersburg, MD, USA). The SeqPrep 96 Plasmid Prep Kit (Edge Biosystems) was used to prepare the plasmid DNA according to manufacturer’s instructions, and the DNA was then sequenced.
Statistical analyses
Statistical analyses were performed using Prism v6.0 (GraphPad Software Incorporated, La Jolla, CA, USA). One-way ANOVA with Bonferroni’s post hoc analysis was used to compare mean changes in weight. Student’s t test was used to compare body mass composition and serum lipid profile and mean hepatic glucose output (HGO). Means are reported ± sem. Significance was determined at P < 0.05.
RESULTS
Development of a multigenerational heritable IUGR rat model
P1 Sprague-Dawley female rats fed a control diet underwent either a sham or bilateral uterine artery ligation surgery at day 19 after gestation (Fig. 1A). The rats spontaneously delivered either the F1 sham control or IUGR lineage litters (Fig. 1A) and were thereafter culled (n = 8 pups per litter). On p21, the F1 offspring were weaned onto isocaloric diets of either control or ENS consisting of the control diet with added folic acid, choline, B12, betaine, methionine, arginine, and zinc. To establish the F2 line, F1 rats were mated at approximately day 80 of life (Fig. 1B). IUGR females and males were either fed control or ENS diets and were paired to establish the IUGRmat/IUGRpat lineage groups. Rats with either maternal or paternal IUGR lineage were also mated with sham partners of the same diet. Additionally, sham males and females were paired and fed either control or ENS diets. The F1 dams were fed their lifelong control or ENS diet throughout gestation and suckling. At weaning, the F2 rats were placed on the same diet as their F1 parents. All experiments were performed on the F2 generation.
Figure 1.

Mating scheme for heritable growth restriction in rats. A) Pregnant female Sprague-Dawley rats (P1) underwent either a bilateral uterine artery ligation or sham surgery at gestation day e19. Uterine ligation F1 rats exhibiting IUGR or sham control rats were then assigned to a lifelong diet of either control chow (control) or ENS chow at weaning (p21). B) At P80, males and females were mated in all combinations of both maternal and paternal IUGR lineage with and without ENS. The resultant F2 generation was assigned the parental diet of either ENS or control at p21, generating 8 F2 study groups (n = 512 animals).
Multigenerational heritable growth restriction modifiable by ENS in F1 and F2
After spontaneous delivery, fetal weights were obtained and litters were culled (n = 8 pups per litter). Pups derived from the Shammat/Shampat lineage were significantly larger at birth than the pups derived from the IUGRmat/IUGRpat lineage (Fig. 2A, B). Quantification of birth weights revealed IUGRmat/IUGRpat pups from the maternal control diet to be 16% less at birth than the Shammat/Shampat lineage (P < 0.0001; 6.7 vs. 8.0 g, respectively). Pups from either IUGRmat/Shampat (5.8 g) or Shammat/IUGRpat (6.1 g) lineage also weighed 27% and 24% less at birth, respectively, than the Shammat/Shampat lineage (P < 0.0001). With maternal ENS-fed rats, a 30% decrease in birth weight with the IUGRmat/IUGRpat lineage was observed, which was statistically identical to the nonsupplemented IUGRmat/IUGRpat lineage rats. Similarly, Shammat/IUGRpat ENS-treated rats showed a marked decrease in birth weight (35%, P < 0.0001), whereas IUGRmat/Shampat ENS-treated rats did not show any significant decrease in weight compared with Shammat/Shampat controls (Fig. 2B). These results suggest a multigenerational effect of low birth weight for the F2 pups, resultant from uteroplacental insufficiency of the F1 generation.
Figure 2.
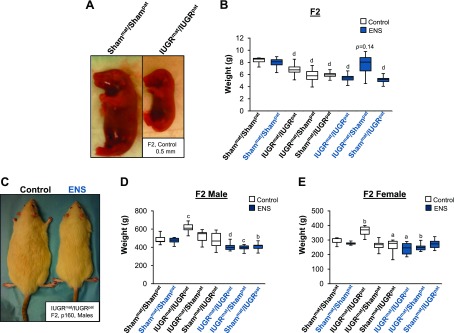
Uterine artery ligation in rats confers heritable growth restriction and adult obesity. A) F2 pups at birth from F1 IUGRpat/IUGRmat lineage and F1 Shampat/Shammat lineage show substantial birth weight differences. B) Differences in mean birth weight of the F2 generation from the F1 lineage fed either a control or ENS diet. Significant differences from Shampat/Shammat control diet are indicated. C) F2 males at p160 from the IUGRpat/IUGRmat lineage fed either a control or ENS diet. Mean p160 weights for F2 male (D) and female (E) rats, bred from the F1 lineage that were fed either a control or ENS diet. Significant differences from Shampat/Shammat control diet are indicated. (aP < 0.05, bP < 0.01, cP < 0.001, dP < 0.0001, 1-way ANOVA with Bonferroni’s post hoc test).
Male and female pups reassessed at p160 demonstrated a significant effect of ENS treatment. Figure 2C shows the IUGRmat/IUGRpat males fed an ENS diet were smaller than the IUGRmat/IUGRpat males fed a control diet, which was true for females as well. Males derived from IUGRmat/IUGRpat fed a control diet weighed ∼17% greater than males from the Shammat/Shampat (Fig. 2D; P < 0.0001). Similarly, the females from the IUGRmat/IUGRpat fed a control diet weighed ∼19% more than the Shammat/Shampat females (Fig. 2E; P < 0.0001). Interestingly however, both male and female IUGRmat/IUGRpat rats reported an average ∼ 20% reduction in weight gain when fed an ENS diet compared with the Shammat/Shampat control diet (Fig. 2D, E; P < 0.0001). For IUGRmat/Shampat rats fed a control diet, there was no statistically significant difference in weights from the Shammat/Shampat and only a small 8% decrease for Shammat/IUGRpat, in female rats only (P < 0.05; Fig. 2D, E). However, when fed an ENS diet, the IUGRmat/Shampat males and females weighed significantly less than the Shammat/Shampat (P < 0.0001 and 0.01, respectively), but Shammat/IUGRpat was significantly decreased for males only (P < 0.0001; Fig. 2D, E). Whereas the Shammat/Shampat showed no significant change in mean weight when fed an ENS diet, the obesity evident in the IUGRmat/IUGRpat control fed was abolished by ENS, with a more pronounced effect apparent in the males.
Multigenerational heritable obesity and dyslipidemia modifiable by ENS in F1 and F2
To determine the basis for the weight changes in IUGR, the relative body fat and lipid profile of the animals was characterized as an indication of metabolic health. IUGRmat/IUGRpat rats fed a control diet were observed to have visibly more fat mass than either IUGRmat/IUGRpat rats fed an ENS diet or Shammat/Shampat rats (Fig. 3A). Lean mass and central fat mass were quantified on adult animals (p160) using DEXA on a clinical caliber Norland scanner, for which representative scans are shown in Fig. 3B. No significant difference in lean mass in either males or females was apparent (Fig. 3C, D). The IUGRmat/IUGRpat males fed a control diet, however, had 3 times more central fat mass than Shammat/Shampat control fed (P = 0.05), which was also significantly greater than either IUGRmat/IUGRpat and Shammat/Shampat males fed the ENS diet (P = 0.01; Fig. 3E). IUGRmat/IUGRpat females fed a control diet also had significantly more central fat mass than the IUGRmat/IUGRpat females fed an ENS diet (Fig. 3F; P < 0.01). The increased central fat mass observed in IUGRmat/IUGRpat males and females suggests that ENS prevents central fat deposition.
Figure 3.
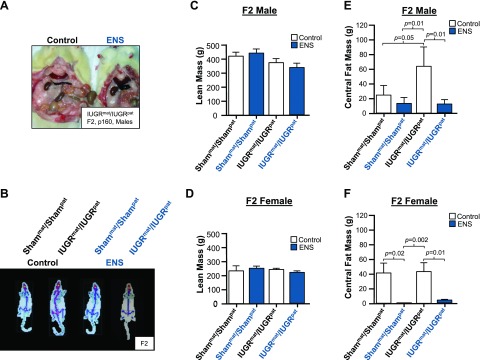
ENS prevents adult obesity. A) Representative image of the central fat found in IUGR males fed either a control or ENS diet. B–F) DEXA scans for body fat analysis based on tissue density. B) Representative DEXA scans of Shampat/Shammat and IUGRpat/IUGRmat males fed either a control or ENS diet. Mean lean mass in F2 males (C) and females (D) fed either a control or ENS diet are indicated compared with Shampat/Shammat control. Differences in mean central fat mass of Shampat/Shammat and IUGRpat/IUGRmat F2 males (E) and females (F) fed either a control or ENS diet compared with IUGRmat/IUGRpat control fed. (aP < 0.05 considered significant, independent Student t test).
Fasting serum lipid profiles were then assessed as markers of dyslipidemia, which accompanies atherosclerotic disease and is an indication of MetS. Triglycerides were elevated in IUGRmat/IUGRpat control fed males and females (P < 0.04), with a significant reduction in both Shammat/Shampat and the IUGRmat/IUGRpat ENS fed for both males and females (P < 0.003; Fig. 4A, B). Similarly, VLDL appeared elevated for both males and females. Although it did not reach statistical significance (P = 0.13 and 0.09, respectively), VLDL was nevertheless lowered to normal levels with ENS supplementation for both sexes (P < 0.005; Fig. 4C, D). Serum free fatty acid levels also exhibited the trend of lineage-induced elevation in IUGRmat/IUGRpat control diet, with males showing ∼50% elevation (P = 0.009), which was reversed in both IUGRmat/IUGRpat and Shammat/Shampat ENS-fed rats (P < 0.01; Fig. 4E, F). The alterations in serum lipid concentrations suggest that ENS supplementation reverses the IUGR-induced increases that characterize metabolic disease.
Figure 4.
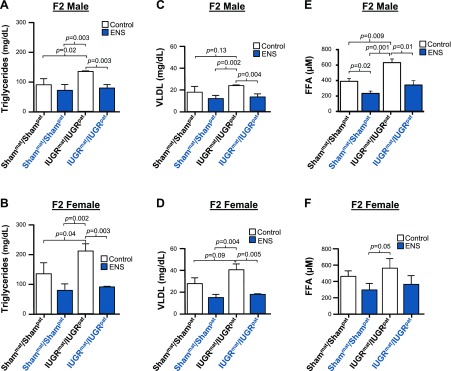
ENS prevents adult dyslipidemia. Mean triglyceride levels in F2 males (A) and females (B) fed either a control or ENS diet compared with IUGRmat/IUGRpat control fed. Mean VLDL levels in F2 males (C) and females (D) fed either a control or ENS diet compared with IUGRmat/IUGRpat control fed. Mean free fatty acid levels in F2 males (E) and females (F) fed either a control or ENS diet compared with IUGRmat/IUGRpat control fed. (P < 0.05 considered significant and all significant values annotated, independent Student t test).
Multigenerational heritable glucose intolerance and dysglycemia modifiable by ENS in F1 and F2
Given the strong relationship between insulin resistance and MetS, hyperinsulinemic-euglycemic clamp studies with carbon 14-labeled glucose and tridium-labeled 2-deoxy glucose tracers were performed on adult animals. These clamp studies were used to discern the hepatic insulin resistance, as opposed to overall or peripheral, as a highly specific indication of metabolic dysregulation. HGO was significantly higher in IUGRmat/IUGRpat males than Shammat/Shampat males fed a control diet (Fig. 5A; P < 0.001), indicative of impaired hepatic insulin sensitivity. When fed an ENS diet, IUGRmat/IUGRpat males exhibited enhanced insulin-mediated suppression of HGO, reversing them to Shammat/Shampat control levels (Fig. 5A; P < 0.02). Similarly significantly distinct HGO profiles were observed among female offspring (Fig. 5B).
Figure 5.
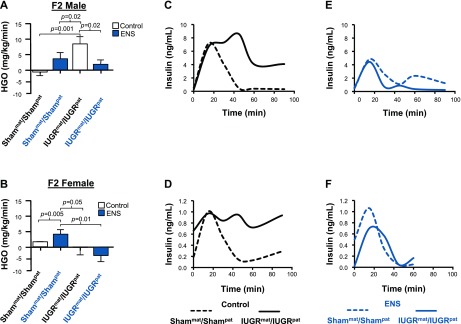
ENS prevents adult insulin resistance. Mean HGO of Shampat/Shammat and IUGRpat/IUGRmat F2 males (A) and females (B) fed either a control or ENS diet compared with IUGRpat/IUGRmat control. Representative insulin response of Shampat/Shammat and IUGRpat/IUGRmat F2 male and female without (C, D) and with (E, F) ENS supplementation, respectively, after glucose challenge. Insulin was measured by ELISA (P < 0.05 considered significant and all significant values annotated, independent Student t test).
To test for insulin responsiveness, glucose tolerance tests were also performed. Serum insulin levels were increased in the IUGRmat/IUGRpat rats fed a control diet compared with Shammat/Shampat following a glucose challenge (Fig. 5C, D). At 50 min, the insulin levels of both male and female IUGRmat/IUGRpat rats failed to return to control baseline, and the levels were still severalfold elevated at 90 min compared with sham (Fig. 5C, D). For both male and female rats, their ENS-fed insulin response was very different. For males, insulin was not discernibly elevated in IUGRmat/IUGRpat rats fed the ENS diet compared with Shammat/Shampat (Fig. 5E). For females, there appeared to be a significant amelioration of the insulin response for ENS-fed IUGRmat/IUGRpat rats compared with control diet; however, insulin remained elevated above baseline at 50 min (Fig. 5F). Overall, the changes associated with IUGR and also the abrogation of the phenotype are considerably more pronounced in the male rats compared with the females.
IGF-1 promoter methylation in IUGR abrogated by ENS
To test whether ENS was acting to affect methylation of critical metabolic regulatory genes, we mapped the methylation status of the second promoter (P2) of the IGF-1 from liver tissue of the variously treated groups. The promoter map is depicted in Fig. 6A, where vertical lines are indicative of individual CpG dinucleotides, and amplicons from each of the areas I–III of the P2 promoter are shown. Sequencing of bisulfite-modified hepatic genomic DNA revealed site (CpG dinucleotide) specific differential methylation. Specifically, among IUGRmat/IUGRpat control-fed F2 offspring destined to manifest adult MetS, we observed significantly increased P2 methylation compared with Shammat/Shampat control-fed F2 offspring (P = 0.03). Consistent with our phenotypic analysis, there was no apparent effect of ENS alone on the non-IUGR lineage animals (Shammat/Shampat ENS fed; Fig. 6B, C). Promoter area II amplicons retained CpG sites where significant methylation changes occurred, whereas areas I and III showed minimal changes (Fig. 6B). When fed a diet supplemented with essential nutrients inclusive of the one-carbon intermediates, methylation of the P2 promoter CpG dinucleotides were significantly decreased in the IUGRmat/IUGRpat rats compared with control-fed offspring (P = 0.04; Fig. 6C). The altered methylation patterning in a site (CpG dinucleotide and region-specific manner) suggests that IUGR-mediated methylation changes in a critical metabolic organ are neither global nor indiscriminate. Further, the changes in promoter methylation appear in agreement with IGF-mediated restricted growth in F1 as previously reported (33).
Figure 6.

IGF-1 promoter methylation mediated by IUGR and ENS. A) The 3 analyzed regions of promoter 2 of IGF-1 are depicted together with the amplified areas sequenced. CpG dinucleotides are represented as vertical lines. In total, among the 3 indicated regions, the methylation status of 11 CpG dinucleotides was determined in bisulfite-treated hepatic tissue. B) Methylation patterns of CpG islands on the P2 transcription start site. Open circles are unmethylated CpGs, whereas closed circles indicate methylated. Promoter site in indicated on the horizontal axis. C) Quantitation of overall methylation changes at the 11 CpG sites for IUGR and Sham linage mice with and without supplementation. Differences from IUGRmat/IUGRpat are indicated (independent Student t test, sem).
DISCUSSION
In this study, we used our well-established bilateral uterine artery ligation model of uteroplacental insufficiency to demonstrate that IUGR induces MetS in the F1 generation (33, 34) and in the F2 generation. Our novel findings are consistent with other findings of multigenerational effects in the human population and a single prior maternal lineage rodent study (15, 62). Remarkably, the adult F2 generation exhibited excessive weight with increased central adiposity and a highly transformed metabolism. Additionally, the adult F2 phenotype included dyslipidemia, significant insulin resistance, and altered glucose metabolism, which altogether are characteristics of MetS. The observed changes were significant in the males, who exhibited much greater heritability compared with females. Most interestingly, the phenotypes were nearly completely reversed with ENS along the one-carbon metabolic pathway regardless of sex or amplitude. IGF-1 methylation was also shown to be reversed by ENS. These results suggest that adaptive epigenomic changes are modifiable and sufficiently plastic to reverse or prevent an inherited diseased epigenome.
The phenotypic consequences of bilateral intrauterine artery ligation to the F1 generation rats are well established and include the MetS phenotype of insulin resistance, diabetes, hypertriglyceridemia, dyslipidemia, and hypertension (13, 55, 62, 63). Additionally, a myriad of physiologic and biochemical changes underlying the phenotype have been found. These include metabolic changes related to altered islet development and dysglycemia (20), increased hepatic glucocorticoid activity (27), altered GLUT1 and GLUT2 expression (28), hepatic glucose production, hepatic one-carbon metabolism, altered hepatic fatty acid metabolism, muscle mitochondrial lipid metabolism, and cerebral chromatin structure (29–31, 64). There is evidence of compromised kidney development and function and an altered intestinal morphology (32, 65, 66). Further, important molecules related to development and to a MetS phenotype, such as IGF-1, PGC-1, CPTI, and PDX1, have been shown to be epigenetically altered (33, 34, 67, 68).
In these extensive and multigenerational experiments, we were able to enrich our deep phenotyping data across both the F1 and F2 generations with initial interrogations into key regulatory genes, such as IGF-1 (33, 34, 67, 68). We were guided in our decision of which genes and promoters to interrogate by the established biology, as well as the known role of methylation in regulating alternate splice sites for transcription of the IGF-1 P2 promoter (33). We show that CpG site-specific methylation of the IGF-1 P2 promoter was altered specifically and significantly in the liver with inclusion of one-carbon metabolism intermediates as dietary supplements. Of note, these modifications occurred only among animals arising from the IUGR lineage and thus accompany reversal of the adult MetS phenotype. Although at first glance it might be expected that ENS would lead to diffuse methylation effects in multiple organ and tissue types, it is particularly significant that IGF-1 (a central anabolic and developmental regulator with systemic endocrine function) is altered by ENS; this is consistent with prior findings in a single generation model of uterine artery ligation in the rat (33).
Hepatic expression of IGF-1 has diverse and systemic roles in development, growth, and glucose homeostasis, and it is known to be adaptively regulated in IUGR (69–71). Increased promoter methylation in the F1 IUGR rat liver dampens IGF-1 expression in what is also thought to be an additional epigenetically mediated adaptive mechanism (33). It is well established that the IGF-1 gene sequence has variable epigenetic regulation, and accompanying transcriptional changes have been shown repeatedly and by multiple investigators (33, 35, 39, 54). Despite this, the mechanisms for multigenerational inheritance observed here are thus far unclear. Aside from its role in growth and development, low levels of IGF-1, like those seen accompanying increased methylation following placental insufficiency (33), is critical to later glucose homeostasis and insulin resistance (37, 72, 73). Our findings herein significantly contribute to the role of epigenomic modifications in both regulating IUGR-mediated adult MetS and an accompanying variation in IGF-1 expression and demonstrate a favorable impact of one-carbon intermediate supplementation.
Although the metabolic effects to those born of IUGR pregnancies are well documented, fewer studies have reported on the multigenerational metabolic effects of IUGR. Recently, a study focusing on the F2 generation uncovered that rats with low maternal birth weight due to late-gestation (e19) uteroplacental insufficiency exhibited a sex-specific reduced insulin response and altered pancreatic morphology (55). In another study, female F2 rats exhibited increased hepatic weight with aberrant glucose and insulin metabolism (74). Natural experiments and comparisons to human populations have borne out the validity of the rodent IUGR models, finding similarities in epigenetic metabolic regulation (23, 33, 55, 65). Of particular interest, the Dutch famine studies have identified transgenerational epigenetic modifications and a propensity for increased adiposity and poor health in F2 as a result of F1 in utero metabolic restriction (23, 24). Therefore, although some aspects of metabolic dysfunction have been demonstrated, it was previously unclear that MetS can be passed down from F1.
The heritable MetS found in our IUGR model of uteroplacental insufficiency indicates there are sex-specific phenotypes for heritability. For instance, males are more severely affected than females in our model, which is highlighted by IUGRmat/IUGRpat lineage males developing the entire spectrum of MetS by adulthood. However, we are able to ameliorate the metabolic syndrome apparent in the IUGRmat/IUGRpat lineage male rats with feeding of an ENS diet. Conversely, although IUGRmat/IUGRpat lineage females show a trend toward MetS, the distinction between IUGRmat/IUGRpat compared with Shammat/Shampat was most evident with respect to adult weight. Similarly, the metabolic end points of insulin dyslipidemia and insulin resistance were considerably more robust for males. Nevertheless, there was both evidence of significant multigenerational inheritance of the adult MetS phenotype in IUGR lineage offspring, as well as a statistical amelioration when fed the ENS diet, regardless of sex. These results are consistent with reports from others that show sex-specific differences in IUGR (27, 62, 75–77). A suggested mechanism to account for the protected phenotype in females is thought to be through estrogen signaling, where estrogen regulates levels of choline, a critical downstream substrate of methionine synthesis. Higher levels of choline have been shown to be protective of metabolic deficits, potentially decreasing the effects of heritable epigenetic aberrations in females (78, 79).
We find the effectiveness of ENS supplementation remarkable in nearly reversing IUGR-associated adult metabolic morbidities. We intentionally initiated the ENS diet intervention in the F1 at the time of weaning and prior to the onset of adult MetS, in an effort to both introduce supplementation prior to adolescence, as well as prior to evidence of metabolic dysfunction. We demonstrated the development of the multigenerational phenotype and the adult MetS phenotypic reversal (but not multigenerational IUGR per se) is abrogated with an early in life ENS intervention.
Although the findings here present an interesting model of the principle of nongenomic coded inheritance and its implications for human health, there are nevertheless limitations. In humans, as in mice, a variety of factors, including a nonstandard environment, variable parental care, and unique nonepigenetically determined biology of the mother, can influence the development of the fetus and confer health altering changes that are not genetically heritable (80). Likewise, it must also be considered that the F2 germ line was exposed to relative restriction and rescuing ENS supplementation, whereas their F1 mothers were undergoing growth restriction in utero. There are examples of germ-line inherited changes that disappear in F3 (81). It is conceivable that the F2 effects and reversal thereof came about through epigenetic modifications in meiosis. Nevertheless, this does not diminish the significance that epigenetically programmed changes regardless of when they occurred are subject to intervention and mitigation with appropriate diet. It has been suggested that supplementation along the one-carbon pathway alters the biochemical balance of reactions for carbohydrate metabolism. As with most experiments of this kind, it is not possible to fully isolate the desired readouts resultant from genetic regulatory changes from the direct effects of treatment on metabolism. Although it is suggested here that maternal supplementation of ENS diet may stave off later metabolic disease, this conclusion is tempered by the use of an animal model. Further, our model of uteroplacental insufficiency is a particularly severe one.
To our knowledge, we are the first group to show a near-complete reversal of heritable MetS through dietary supplementation, as well as a multigenerational transmission down both maternal and paternal lineages. Using robust metrics of central fat mass, glucose tolerance, hepatic glucose efflux, serum lipid, and free fatty acids, we show that supplementation with essential nutrients along the one-carbon pathway prevents adult obesity, dyslipidemia, and insulin resistance, indicative of metabolic disease. The novel demonstration of a paternal inheritance of adult MetS has substantial translational implications, as it underlines the role of paternal, as well as maternal, factors as important to noncoded heritability of MetS. Identifications of the mechanisms and genetic targets of heritable epigenetic-mediated metabolic changes and the molecular mechanisms of phenotypic reversal through ENS supplementation will reveal insights into the causes of the developmental origins of health and disease as they relate to IUGR. Supplementation of IUGR offspring, as first identified at birth, may be a viable intervention to mitigate the risks conferred by multigenerational inheritance of metabolic-related diseases.
Supplementary Material
Acknowledgments
The authors thank Xing Yu for the technical contributions made during this project. Funding for this project was provided by U.S. National Institutes of Health (NIH) Grant DP21DP2OD001500-01 (“Mapping the Fetal Primate Epigenome and Metabolome”), and the U.S. NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development Reproductive Scientist Development Program Grant 5K12HD00849 (“Transgenerational Growth Restriction through Altered Placental Epigenetics”) (to K.A.).
Glossary
- DEXA
dual energy X-ray absorptiometry
- ENS
essential nutrient supplementation
- HGO
hepatic glucose output
- IUGR
intrauterine growth restriction
- MetS
metabolic syndrome
- VLDL
very-LDL
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Barker D. J., Winter P. D., Osmond C., Margetts B., Simmonds S. J. (1989) Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580 [DOI] [PubMed] [Google Scholar]
- 2.Barker D. J., Osmond C., Golding J., Kuh D., Wadsworth M. E. (1989) Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ298, 564–567 [DOI] [PMC free article] [PubMed]
- 3.Skilton M. R., Evans N., Griffiths K. A., Harmer J. A., Celermajer D. S. (2005) Aortic wall thickness in newborns with intrauterine growth restriction. Lancet 365, 1484–1486 [DOI] [PubMed] [Google Scholar]
- 4.Koklu E., Ozturk M. A., Gunes T., Akcakus M., Kurtoglu S. (2007) Is increased intima-media thickness associated with preatherosclerotic changes in intrauterine growth restricted newborns? Acta Paediatr. 96, 1858. [DOI] [PubMed] [Google Scholar]
- 5.Barker D. J. P., Hales C. N., Fall C. H., Osmond C., Phipps K., Clark P. M. (1993) Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36, 62–67 [DOI] [PubMed] [Google Scholar]
- 6.Barker D. J., Martyn C. N., Osmond C., Hales C. N., Fall C. H. (1993) Growth in utero and serum cholesterol concentrations in adult life. BMJ 307, 1524–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curhan G. C., Chertow G. M., Willett W. C., Spiegelman D., Colditz G. A., Manson J. E., Speizer F. E., Stampfer M. J. (1996) Birth weight and adult hypertension and obesity in women. Circulation 94, 1310–1315 [DOI] [PubMed] [Google Scholar]
- 8.Curhan G. C., Willett W. C., Rimm E. B., Spiegelman D., Ascherio A. L., Stampfer M. J. (1996) Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation 94, 3246–3250 [DOI] [PubMed] [Google Scholar]
- 9.Ervin R. B. (2009) Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003-2006. National Health Statistics Report13, 1–7 [PubMed]
- 10.Kassi E., Pervanidou P., Kaltsas G., Chrousos G. (2011) Metabolic syndrome: definitions and controversies. BMC Med. 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornier M.-A., Dabelea D., Hernandez T. L., Lindstrom R. C., Steig A. J., Stob N. R., Van Pelt R. E., Wang H., Eckel R. H. (2008) The metabolic syndrome. Endocr. Rev. 29, 777–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckel R. H., Alberti K. G., Grundy S. M., Zimmet P. Z. (2010) The metabolic syndrome. Lancet 375, 181–183 [DOI] [PubMed] [Google Scholar]
- 13.Simmons R. A., Templeton L. J., Gertz S. J. (2001) Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50, 2279–2286 [DOI] [PubMed] [Google Scholar]
- 14.Benyshek D. C., Johnston C. S., Martin J. F. (2006) Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 49, 1117–1119 [DOI] [PubMed] [Google Scholar]
- 15.Jimenez-Chillaron J. C., Isganaitis E., Charalambous M., Gesta S., Pentinat-Pelegrin T., Faucette R. R., Otis J. P., Chow A., Diaz R., Ferguson-Smith A., Patti M. E. (2009) Intergenerational transmission of glucose intolerance and obesity by in utero undernutrition in mice. Diabetes 58, 460–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallo L. A., Tran M., Moritz K. M., Mazzuca M. Q., Parry L. J., Westcott K. T., Jefferies A. J., Cullen-McEwen L. A., Wlodek M. E. (2012) Cardio-renal and metabolic adaptations during pregnancy in female rats born small: implications for maternal health and second generation fetal growth. J. Physiol. 590, 617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohi G., Marchand K., Revesz A., Arany E., Hardy D. B. (2011) Maternal protein restriction elevates cholesterol in adult rat offspring due to repressive changes in histone modifications at the cholesterol 7alpha-hydroxylase promoter. Mol. Endocrinol. 25, 785–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gong L., Pan Y.-X., Chen H. (2010) Gestational low protein diet in the rat mediates Igf2 gene expression in male offspring via altered hepatic DNA methylation. Epigenetics 5, 619–626 [DOI] [PubMed] [Google Scholar]
- 19.Zheng S., Rollet M., Pan Y.-X. (2011) Maternal protein restriction during pregnancy induces CCAAT/enhancer-binding protein (C/EBPβ) expression through the regulation of histone modification at its promoter region in female offspring rat skeletal muscle. Epigenetics 6, 161–170 [DOI] [PubMed] [Google Scholar]
- 20.Schwitzgebel V. M., Somm E., Klee P. (2009) Modeling intrauterine growth retardation in rodents: Impact on pancreas development and glucose homeostasis. Mol. Cell. Endocrinol. 304, 78–83 [DOI] [PubMed] [Google Scholar]
- 21.Suter M., Bocock P., Showalter L., Hu M., Shope C., McKnight R., Grove K., Lane R., Aagaard-Tillery K. (2011) Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB J. 25, 714–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hales C. N., Barker D. J., Clark P. M., Cox L. J., Fall C., Osmond C., Winter P. D. (1991) Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303, 1019–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heijmans B. T., Tobi E. W., Stein A. D., Putter H., Blauw G. J., Susser E. S., Slagboom P. E., Lumey L. H. (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 105, 17046–17049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Painter R. C., Osmond C., Gluckman P., Hanson M., Phillips D. I., Roseboom T. J. (2008) Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG 115, 1243–1249 [DOI] [PubMed] [Google Scholar]
- 25.Ogata E. S., Bussey M. E., Finley S. (1986) Altered gas exchange, limited glucose and branched chain amino acids, and hypoinsulinism retard fetal growth in the rat. Metabolism 35, 970–977 [DOI] [PubMed] [Google Scholar]
- 26.Ogata E. S., Bussey M. E., LaBarbera A., Finley S. (1985) Altered growth, hypoglycemia, hypoalaninemia, and ketonemia in the young rat: postnatal consequences of intrauterine growth retardation. Pediatr. Res. 19, 32–37 [DOI] [PubMed] [Google Scholar]
- 27.Baserga M., Hale M. A., McKnight R. A., Yu X., Callaway C. W., Lane R. H. (2005) Uteroplacental insufficiency alters hepatic expression, phosphorylation, and activity of the glucocorticoid receptor in fetal IUGR rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1348–R1353 [DOI] [PubMed] [Google Scholar]
- 28.Lane R. H., Crawford S. E., Flozak A. S., Simmons R. A. (1999) Localization and quantification of glucose transporters in liver of growth-retarded fetal and neonatal rats. Am. J. Physiol. 276, E135–E142 [DOI] [PubMed] [Google Scholar]
- 29.Lane R. H., Kelley D. E., Ritov V. H., Tsirka A. E., Gruetzmacher E. M. (2001) Altered expression and function of mitochondrial beta-oxidation enzymes in juvenile intrauterine-growth-retarded rat skeletal muscle. Pediatr. Res. 50, 83–90 [DOI] [PubMed] [Google Scholar]
- 30.Lane R. H., MacLennan N. K., Hsu J. L., Janke S. M., Pham T. D. (2002) Increased hepatic peroxisome proliferator-activated receptor-gamma coactivator-1 gene expression in a rat model of intrauterine growth retardation and subsequent insulin resistance. Endocrinology 143, 2486–2490 [DOI] [PubMed] [Google Scholar]
- 31.Ke X., Lei Q., James S. J., Kelleher S. L., Melnyk S., Jernigan S., Yu X., Wang L., Callaway C. W., Gill G., Chan G. M., Albertine K. H., McKnight R. A., Lane R. H. (2006) Uteroplacental insufficiency affects epigenetic determinants of chromatin structure in brains of neonatal and juvenile IUGR rats. Physiol. Genomics 25, 16–28 [DOI] [PubMed] [Google Scholar]
- 32.Baserga M., Hale M. A., Ke X., Wang Z. M., Yu X., Callaway C. W., McKnight R. A., Lane R. H. (2006) Uteroplacental insufficiency increases p53 phosphorylation without triggering the p53-MDM2 functional circuit response in the IUGR rat kidney. Am. J. Physiol. requl. Integr. Comp. Physiol.291, R412–R418 [DOI] [PubMed]
- 33.Fu Q., Yu X., Callaway C. W., Lane R. H., McKnight R. A. (2009) Epigenetics: intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. FASEB J. 23, 2438–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park J. H., Stoffers D. A., Nicholls R. D., Simmons R. A. (2008) Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Invest. 118, 2316–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tosh D. N., Fu Q., Callaway C. W., McKnight R. A., McMillen I. C., Ross M. G., Lane R. H., Desai M. (2010) Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G1023–G1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woods K. A., Camacho-Hübner C., Savage M. O., Clark A. J. (1996) Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 335, 1363–1367 [DOI] [PubMed] [Google Scholar]
- 37.Netchine I., Azzi S., Houang M., Seurin D., Perin L., Ricort J. M., Daubas C., Legay C., Mester J., Herich R., Godeau F., Le Bouc Y. (2009) Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J. Clin. Endocrinol. Metab. 94, 3913–3921 [DOI] [PubMed] [Google Scholar]
- 38.Woods K. A., Camacho-Hübner C., Bergman R. N., Barter D., Clark A. J., Savage M. O. (2000) Effects of insulin-like growth factor I (IGF-I) therapy on body composition and insulin resistance in IGF-I gene deletion. J. Clin. Endocrinol. Metab. 85, 1407–1411 [DOI] [PubMed] [Google Scholar]
- 39.Tan Y., Liu J., Deng Y., Cao H., Xu D., Cu F., Lei Y., Magdalou J., Wu M., Chen L., Wang H. (2012) Caffeine-induced fetal rat over-exposure to maternal glucocorticoid and histone methylation of liver IGF-1 might cause skeletal growth retardation. Toxicol. Lett. 214, 279–287 [DOI] [PubMed] [Google Scholar]
- 40.Morgan H. D., Santos F., Green K., Dean W., Reik W. (2005) Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14, R47–R58 [DOI] [PubMed] [Google Scholar]
- 41.Waterland R. A., Jirtle R. L. (2003) Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 23, 5293–5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waterland R. A., Jirtle R. L. (2004) Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition 20, 63–68 [DOI] [PubMed] [Google Scholar]
- 43.Waterland R. A., Lin J. R., Smith C. A., Jirtle R. L. (2006) Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum. Mol. Genet. 15, 705–716 [DOI] [PubMed] [Google Scholar]
- 44.Jahangir E., Vita J. A., Handy D., Holbrook M., Palmisano J., Beal R., Loscalzo J., Eberhardt R. T. (2009) The effect of L-arginine and creatine on vascular function and homocysteine metabolism. Vasc. Med. 14, 239–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seki Y., Williams L., Vuguin P. M., Charron M. J. (2012) Minireview: Epigenetic programming of diabetes and obesity: animal models. Endocrinology 153, 1031–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van den Veyver I. B. (2002) Genetic effects of methylation diets. Annu. Rev. Nutr. 22, 255–282 [DOI] [PubMed] [Google Scholar]
- 47.Dominguez-Salas P., Moore S. E., Cole D., da Costa K. A., Cox S. E., Dyer R. A., Fulford A. J., Innis S. M., Waterland R. A., Zeisel S. H., Prentice A. M., Hennig B. J. (2013) DNA methylation potential: dietary intake and blood concentrations of one-carbon metabolites and cofactors in rural African women. Am. J. Clin. Nutr. 97, 1217–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cordero P., Campion J., Milagro F. I., Martinez J. A. (2013) Transcriptomic and epigenetic changes in early liver steatosis associated to obesity: effect of dietary methyl donor supplementation. Mol. Genet. Metab. 110, 388–395 [DOI] [PubMed] [Google Scholar]
- 49.Niculescu M. D., Zeisel S. H. (2002) Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J. Nutr. 132(8, Suppl)2333S–2335S [DOI] [PubMed] [Google Scholar]
- 50.Anderson O. S., Sant K. E., Dolinoy D. C. (2012) Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 23, 853–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dominguez-Salas P., Cox S. E., Prentice A. M., Hennig B. J., Moore S. E. (2012) Maternal nutritional status, C(1) metabolism and offspring DNA methylation: a review of current evidence in human subjects. Proc. Nutr. Soc. 71, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lambrot R., Xu C., Saint-Phar S., Chountalos G., Cohen T., Paquet M., Suderman M., Hallett M., Kimmins S. (2013) Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes. Nat. Commun. 4, 2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cordero P., Gomez-Uriz A. M., Campion J., Milagro F. I., Martinez J. A. (2013) Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 8, 105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahrens M., Ammerpohl O., von Schönfels W., Kolarova J., Bens S., Itzel T., Teufel A., Herrmann A., Brosch M., Hinrichsen H., Erhart W., Eqberts J., Sipos B., Schreiber S., Häsler R., Stickel F., Becker T., Krawczak M., Röcken C., Siebert R., Schafmayer C., Hampe J. (2013) DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 18, 296–302 [DOI] [PubMed] [Google Scholar]
- 55.Lane R. H., Kelley D. E., Gruetzmacher E. M., Devaskar S. U. (2001) Uteroplacental insufficiency alters hepatic fatty acid-metabolizing enzymes in juvenile and adult rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R183–R190 [DOI] [PubMed] [Google Scholar]
- 56.Lukaski H. C. (1993) Soft tissue composition and bone mineral status: evaluation by dual-energy X-ray absorptiometry. J. Nutr. 123(2, Suppl), 438–443 [PubMed] [Google Scholar]
- 57.Howie G. J., Sloboda D. M., Kamal T., Vickers M. H. (2009) Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J. Physiol. 587, 905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim J. K. (2009) Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods Mol. Biol. 560, 221–238 [DOI] [PubMed] [Google Scholar]
- 59.Holland W. L., Brozinick J. T., Wang L. P., Hawkins E. D., Sargent K. M., Liu Y., Narra K., Hoehn K. L., Knotts T. A., Siesky A., Nelson D. H., Karathansis S. K., Fonternot G. K., Birnhaum M. J. Summers S. A. (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 5, 167–179 [DOI] [PubMed] [Google Scholar]
- 60.Ye J. M., Doyle P. J., Iglesias M. A., Watson D. G., Cooney G. J., Kraegen E. W. (2001) Peroxisome proliferator-activated receptor (PPAR)-alpha activation lowers muscle lipids and improves insulin sensitivity in high fat-fed rats: comparison with PPAR-gamma activation. Diabetes 50, 411–417 [DOI] [PubMed] [Google Scholar]
- 61.Karolchik D., Hinrichs A. S., Furey T. S., Roskin K. M., Sugnet C. W., Haussler D., Kent W. J. (2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tran M., Gallo L. A., Jefferies A. J., Moritz K. M., Wlodek M. E. (2013) Transgenerational metabolic outcomes associated with uteroplacental insufficiency. J. Endocrinol. 217, 105–118 [DOI] [PubMed] [Google Scholar]
- 63.Baserga M., Bares A. L., Hale M. A., Callaway C. W., McKnight R. A., Lane P. H., Lane R. H. (2009) Uteroplacental insufficiency affects kidney VEGF expression in a model of IUGR with compensatory glomerular hypertrophy and hypertension. Early Hum. Dev 85, 361–367 [DOI] [PMC free article] [PubMed]
- 64.MacLennan N. K., James S. J., Melnyk S., Piroozi A., Jernigan S., Hsu J. L., Janke S. M., Pham T. D., Lane R. H. (2004) Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol. Genomics 18, 43–50 [DOI] [PubMed] [Google Scholar]
- 65.Fu Q., McKnight R. A., Yu X., Wang L., Callaway C. W., Lane R. H. (2004) Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiol. Genomics 20, 108–116 [DOI] [PubMed] [Google Scholar]
- 66.Baserga M., Bertolotto C., Maclennan N. K., Hsu J. L., Pham T., Laksana G. S., Lane R. H. (2004) Uteroplacental insufficiency decreases small intestine growth and alters apoptotic homeostasis in term intrauterine growth retarded rats. Early Hum. Dev. 79, 93–105 [DOI] [PubMed] [Google Scholar]
- 67.Thompson R. F., Fazzari M. J., Niu H., Barzilai N., Simmons R. A., Greally J. M. (2010) Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J. Biol. Chem. 285, 15111–15118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ganu R. S., Harris R. A., Collins K., Aagaard K. M. (2012) Early origins of adult disease: approaches for investigating the programmable epigenome in humans, nonhuman primates, and rodents. ILAR J. 53, 306–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seferovic M. D., Ali R., Kamei H., Liu S., Khosravi J. M., Nazarian S., Han V. K., Duan C., Gupta M. B. (2009) Hypoxia and leucine deprivation induce human insulin-like growth factor binding protein-1 hyperphosphorylation and increase its biological activity. Endocrinology 150, 220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abu Shehab M., Inoue S., Han V. K. M., Gupta M. B. (2009) Site specific phosphorylation of insulin-like growth factor binding protein-1 (IGFBP-1) for evaluating clinical relevancy in fetal growth restriction. J. Proteome Res. 8, 5325–5335 [DOI] [PubMed] [Google Scholar]
- 71.Watson C. S., Bialek P., Anzo M., Khosravi J., Yee S. P., Han V. K. (2006) Elevated circulating insulin-like growth factor binding protein-1 is sufficient to cause fetal growth restriction. Endocrinology 147, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 72.Sjögren K., Wallenius K., Liu J. L., Bohlooly-Y M., Pacini G., Svensson L., Törnell J., Isaksson O. G., Ahrén B., Jansson J. O., Ohlsson C. (2001) Liver-derived IGF-I is of importance for normal carbohydrate and lipid metabolism. Diabetes 50, 1539–1545 [DOI] [PubMed] [Google Scholar]
- 73.Haluzik M., Yakar S., Gavrilova O., Setser J., Boisclair Y., LeRoith D. (2003) Insulin resistance in the liver-specific IGF-1 gene-deleted mouse is abrogated by deletion of the acid-labile subunit of the IGF-binding protein-3 complex: relative roles of growth hormone and IGF-1 in insulin resistance. Diabetes 52, 2483–2489 [DOI] [PubMed] [Google Scholar]
- 74.Thamotharan M., Garg M., Oak S., Rogers L. M., Pan G., Sangiorgi F., Lee P. W., Devaskar S. U. (2007) Transgenerational inheritance of the insulin-resistant phenotype in embryo-transferred intrauterine growth-restricted adult female rat offspring. Am. J. Physiol. Endocrinol. Metab. 292, E1270–E1279 [DOI] [PubMed] [Google Scholar]
- 75.Maloney C. A., Hay S. M., Young L. E., Sinclair K. D., Rees W. D. (2011) A methyl-deficient diet fed to rat dams during the peri-conception period programs glucose homeostasis in adult male but not female offspring. J. Nutr. 141, 95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu Q., McKnight R. A., Yu X., Callaway C. W., Lane R. H. (2006) Growth retardation alters the epigenetic characteristics of hepatic dual specificity phosphatase 5. FASEB J. 20, 2127–2129 [DOI] [PubMed] [Google Scholar]
- 77.Lane R. H., Maclennan N. K., Daood M. J., Hsu J. L., Janke S. M., Pham T. D., Puri A. R., Watchko J. F. (2003) IUGR alters postnatal rat skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1 gene expression in a fiber specific manner. Pediatr. Res. 53, 994–1000 [DOI] [PubMed] [Google Scholar]
- 78.Fischer L. M., Costa K., Kwock L., Galanko J., Zeisel S. H. (2010) Dietary choline requirements of women: effects of estrogen and genetic variation. Am. J. Clin. Nutr 92, 1113–1119 [DOI] [PMC free article] [PubMed]
- 79.Resseguie M., Song J., Niculescu M. D., da Costa K. A., Randall T. A., Zeisel S. H. (2007) Phosphatidylethanolamine N-methyltransferase (PEMT) gene expression is induced by estrogen in human and mouse primary hepatocytes. FASEB J. 21, 2622–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grossniklaus U., Kelly W. G., Ferguson-Smith A. C., Pembrey M., Lindquist S. (2013) Transgenerational epigenetic inheritance: how important is it? Nat. Rev. Genet. 14, 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Waterland R. A., Travisano M., Tahiliani K. G. (2007) Diet-induced hypermethylation at agouti viable yellow is not inherited transgenerationally through the female. FASEB J. 21, 3380–3385 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.