

Abstract
Helicobacter pylori induces the antiapoptotic protein myeloid cell leukemia 1 (Mcl1) in human gastric epithelial cells (GECs). Apoptosis of oncogenic protein Mcl1-expressing cells is mainly regulated by Noxa-mediated degradation of Mcl1. We wanted to elucidate the status of Noxa in H. pylori–infected GECs. For this, various GECs such as AGS, MKN45, and KATO III were either infected with H. pylori or left uninfected. The effect of infection was examined by immunoblotting, immunoprecipitation, chromatin immunoprecipitation assay, in vitro binding assay, flow cytometry, and confocal microscopy. Infected GECs, surgical samples collected from patients with gastric adenocarcinoma as well as biopsy samples from patients infected with H. pylori showed significant up-regulation of both Mcl1 and Noxa compared with noninfected samples. Coexistence of Mcl1 and Noxa was indicative of an impaired Mcl-Noxa interaction. We proved that Noxa was phosphorylated at Ser13 residue by JNK in infected GECs, which caused cytoplasmic retention of Noxa. JNK inhibition enhanced Mcl1-Noxa interaction in the mitochondrial fraction of infected cells, whereas overexpression of nonphosphorylatable Noxa resulted in enhanced mitochondria-mediated apoptosis in the infected epithelium. Because phosphorylation-dephosphorylation can regulate the apoptotic function of Noxa, this could be a potential target molecule for future treatment approaches for H. pylori–induced gastric cancer.—Rath, S., Das, L., Kokate, S. B., Pratheek, B. M., Chattopadhyay, S., Goswami, C., Chattopadhyay, R., Crowe, S. E., Bhattacharyya, A. Regulation of Noxa-mediated apoptosis in Helicobacter pylori–infected gastric epithelial cells.
Keywords: JNK, cytochrome c, gastric cancer, myeloid cell leukemia 1, mitochondrial fragmentation
Helicobacter pylori infects the human stomach leading to gastritis, gastric and duodenal ulcers, gastric carcinoma, and mucosa-associated lymphoid tissue (MALT) lymphoma (1). The risk for gastric cancer is at least 2-fold greater in infected persons than in uninfected individuals. H. pylori promotes gastric cancer either via its cancer-promoting effects or via creating a carcinogenic environment by inducing host inflammatory responses to chronic infection. Signaling events induced in infected host cells play crucial roles in determining disease pathogenesis. One of the major signaling molecules induced in H. pylori–infected gastric epithelial cells (GECs) is hypoxia-inducible factor 1 (Hif1), a heterodimeric transcription factor (2, 3). Endogenous or H. pylori–induced gastric epithelial reactive oxygen species (ROS) stabilize the oxygen-labile α-subunit of Hif1 in normoxic condition (3, 4), whereas the β-subunit is constitutively expressed (5). Several cellular processes including cell proliferation, apoptosis, and tumor angiogenesis are regulated by Hif1 (5). A Bcl2 family antiapoptotic and tumorigenic protein myeloid cell leukemia 1 (Mcl1) and a proapoptotic Bcl2 homology 3 (BH3)-only tumor suppressor protein Noxa (synonyms are immediate-early response protein APR or phorbol 12-myristate 13-acetate-induced protein 1) both contain hypoxia-response elements (HREs) in their promoters (6, 7). H. pylori infection augments expression of both Mcl1 (2) and Noxa in the infected GECs (8). Noxa is a unique BH3 protein because it binds only with Mcl1 and A1 prosurvival proteins, whereas other BH3 proteins can interact with any Bcl2 family member. However, a recent finding shows that Noxa can bind to other antiapoptotic and proapoptotic proteins as well (9). Mcl1 has potent antiapoptotic and tumorigenic functions, but in effect, it is an extremely short-lived molecule due to its proteasomal degradation. Noxa imparts its apoptotic function mainly by translocating to mitochondria followed by its binding with Mcl1. Noxa-bound Mcl1 is targeted for proteasomal degradation (10). In the presence of glucose, Noxa phosphorylation by Cdk5 sequesters the BH3 protein in the cytoplasm and decreases apoptosis of proliferating leukemia cell lines and primary T cells (11). Understanding the regulation of Mcl1 stability by Noxa is important for developing cancer therapeutics, especially for cancers with up-regulated Mcl1 expression. Because Mcl1 is highly induced in gastric cancer and is associated with a poor prognosis (12), studying the Mcl1-Noxa interaction in H. pylori–infected GECs would help in designing better treatment strategies for H. pylori–induced gastric cancer.
To gain insight into the kinetics of expression of Mcl1 and Noxa in H. pylori–infected GECs, we analyzed expression of Mcl1 and Noxa over a time period. Our results show that although H. pylori simultaneously up-regulates both Mcl1 and Noxa expression in the infected GECs, Mcl1 is not degraded, suggesting phosphorylation of Noxa. We confirm that Noxa is phosphorylated by JNK, a stress-induced MAPK activated in H. pylori–infected GECs. Furthermore, we prove that H. pylori–mediated phosphorylation regulates the apoptotic function of Noxa in the infected GEC.
MATERIALS AND METHODS
Cell culture and bacteria
Gastric epithelial cancer cells AGS, MKN-45, NCI-N87, and KATO III, and H. pylori strain 26695 and 8-1 [a cytotoxin-associated gene (cag) pathogenicity island (PAI) (+) strain (American Type Culture Collection, Manassas, VA, USA) and a cag PAI(−) strain, respectively] were cultured and maintained as previously reported (13).
Plasmids, mutagenesis, and transfections
Human wild-type (WT) Noxa sequence and S13A mutant (mut) sequence were cloned in pcDNA3.1+ vector (Invitrogen, Carlsbad, CA, USA) by using HindIII and XhoI as restriction enzymes. Primers used for WT Noxa cloning were 5′-AAAAGCTTCACCATGCCTGGGAAGAAGGCGCGC-3′ (forward primer) and 5′-GCTCGAGTCAGGTTCCTGAGCAGAAGAGTTTGGATATCAGATTCAG-3′ (reverse primer). Noxa S13A mut was generated by using Noxa WT construct as a template using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions. The primer used for mutagenesis was 5′-AAGAACGCTCAACCGGCC-(AGC)CCCGCGCGGGCTCCA-3′, where the codon for A (underlined and in italics) replaced the Ser13 of WT Noxa (in parentheses, original codon). JNK1 and JNK2 constructs used were described earlier (14, 15). A total of 1 × 106 AGS cells were seeded in 6-well cell culture plates 18–24 h before transfection and were transfected with 2 μg plasmid DNA and 10 μl Lipofectamine 2000 reagent (Invitrogen). Cells were infected after 24 h of transfections.
Stable hif1α knockdown in AGS cells
hif1α was stably knocked down using short hairpin RNA (shRNA) using Lipofectamine 2000 reagent. We also derived stable cells expressing empty negative control shRNA and scrambled negative control shRNA-expressing cells. All constructs (HuSH plasmids) were purchased from OriGene Technologies, Incorporated (Rockville, MD, USA).
Infections and treatments
Cells were infected with various multiplicities of infection (MOIs) of H. pylori for specified periods. We found that an MOI of 200 for 5 h was optimum to induce Hif1α and Noxa in GECs. When required, AGS cells were pretreated with 150 nM Echinomycin (Sigma-Aldrich, St. Louis, MO, USA), MEK1/2 inhibitor PD98059, p38 MAPK inhibitor SB203580, and JNK inhibitor II (all from Calbiochem, San Diego, CA, USA) at 25 µM concentration for 1 h prior to infection.
Human GEC isolation from mucosal biopsy specimens
Gastric biopsy samples from the gastric antral mucosa were collected from individuals undergoing esophagogastroduodenoscopy, as per a University of Virginia (UVA) institutional review board-approved protocol. Epithelial cells were isolated, cultured, and infected with an MOI of 200 of H. pylori 26695 or 8-1 for 5 h as mentioned previously (13).
Mitochondrial and cytosolic lysate preparation
A total of 2 × 106 cells were collected by centrifugation at 1300 × g for 3 min at 4°C. Cells were resuspended in 150 µl ice-cold extraction buffer containing 20 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (pH 7.5), 10 mM KCl, 150 mM sucrose, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT supplemented with protease inhibitor cocktail (Sigma-Aldrich) and kept for 10 min at 4°C. Cells were homogenized by 20 passages through a 26-gauge needle. Homogenates were centrifuged at 10,000 × g at 4°C for 5 min to remove nuclei and unbroken cells. Supernatant was centrifuged at 12,000 × g for 30 min at 4°C to obtain the cytosolic fraction in supernatant. The mitochondria-enriched pellet was resuspended in cold 10 µl lysis buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, and 1 mM DTT along with protease inhibitor cocktail followed by 20 min incubation on ice and finally centrifuged at 15,000 × g for 5 min. Mitochondrial lysate was collected and boiled with an equal volume of 2× Laemmli buffer (HiMedia, Mumbai, Maharastra, India).
Immunoprecipitation, Western blotting, and antibodies
Whole-cell lysates were prepared from GECs after treatments and were separated by SDS-PAGE followed by Western blotting. Blots were probed with antibodies to phosphoserine and phosphotyrosine (Sigma-Aldrich), CagA (Santa Cruz Biotechnology, Santa Cruz, CA, USA), caspase-3 (Sigma-Aldrich), Noxa, Hif1α, Mcl1, and Cdk5 (all from Abcam, Cambridge, MA, USA), p53 (Calbiochem), phospho-JNK, JNK, phospho-ERK, ERK, phospho-p38, p38, cytochrome c, phospho-p53 (Ser15) Cdk5, and Bax (all from Cell Signaling Technology, Danvers, MA, USA). α-Tubulin (Abcam) and histone deacetylase 1 and Cox IV (both from Cell Signaling Technology) antibodies were used for normalization of protein loading. Noxa was immunoprecipitated to analyze interaction between Noxa and JNK. Mcl1 was immunoprecipitated from mitochondrial fraction to assess its interaction with Bax and Noxa. Proteins were detected by using the Thermo Scientific SuperSignal West Femto Chemiluminescent Substrate kit (Thermo Fisher Scientific, Kalamazoo, MI, USA). Western blot images were taken with ChemiDoc XRS (Bio-Rad Laboratories, Hercules, CA, USA) equipped with Quantity One 1-D Analysis software version 4.6.9 (Bio-Rad Laboratories).
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed using the QuikChIP Kit (Imgenex, San Diego, CA, USA) as described before (13). Following treatments, chromatin was sonicated into fragments with an average length of 0.5–3 kb. After centrifugation at 12,000 × g for 10 min, supernatants were diluted in dilution buffer, and immunoprecipitation was performed using Hif1α antibody (Novus Biologicals, Littleton, CO, USA). ChIP DNA was detected by PCR with the primers 5′-GACGGG- GTTTCACCATATTGGCAAG-3′ (forward primer) and 5′-TGAGAGCCGCTTCATGCTAAGGACTT-3′ (reverse primer) targeting a 346 bp region from the noxa promoter containing the HRE region. Specificity of the reaction was assessed by using a pair of negative control primers (5′-GCACGTTTCATCAATTTGAAGAAAGACTGC-3′ and 5′-AACAGCAACAACAACA ATGCACTGAACTGT-3′) from the 5′-upstream region of noxa promoter. After reversing the DNA-protein cross-links in the immunocomplexes, the noxa promoter sequence in the oligonucleotide containing the HRE region (91 bp) was quantitated by real-time PCR using the DNA Engine Opticon continuous fluorescence detector (Bio-Rad Laboratories). Preoptimized FAM-labeled primer-probe sets for noxa and 18S rRNA (Applied Biosystems, Foster City, CA, USA) were used for the study.
In vitro binding assay
Nuclear extracts were prepared from treated cells using the Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Rockford, IL, USA). Streptavidin-coated superparamagnetic beads (Dynabeads M-280 Streptavidin, Dynal; Invitrogen) were used to capture 5′-biotinylated double-stranded noxa HRE oligonucleotide, and binding assays were performed as described previously (13). The noxa HRE WT oligos were 5′-TGGGA TTACAGGCGTGAGCCACCGCGT-3′ and 5′-ACGCGGTGGCTCACGCCTGTAATCCCA-3′, whereas mut primers were 5′-TGGGATTACAGGGCACAGCCACCGCGT-3′ and 5′-ACGCGGTGGCTGTGCCCTGTAATCCCA-3′. These primers were biotinylated at the 5′ end. Bound proteins were dissociated by boiling in 1× Laemmli sample buffer and analyzed by Western blotting.
Immunohistochemistry
Immunohistochemistry (IHC) staining was performed for Hif1α, Mcl1, and Noxa proteins. Paraffin-embedded infected or uninfected biopsy samples were collected following a UVA institutional review board-approved protocol. Surgical sections of gastric adenocarcinomas were obtained from the UVA Biorepository and Tissue Research Facility. Sections were stained as previously reported (13). Dilutions of primary antibodies were as follows: Hif1α (Novus Biologicals), Mcl1 (BD Biosciences, Franklin Lakes, NJ, USA), and Noxa (ProSci, Poway, CA, USA). Digital images were captured using the Aperio ScanScope XT Slide Scanner (Aperio Technologies, Vista, CA, USA).
Flow cytometry
AGS cells were either infected with H. pylori for 10 h or left uninfected. Cells were harvested and washed twice with chilled PBS. Cells were then stained with Annexin V PE/7-AAD dyes (BD Biosciences) as per manufacturer’s instruction. In a separate set of experiments, AGS cells were transfected with either Noxa WT or Noxa S13A plasmid constructs or empty vector followed by infection with H. pylori for 10 h. Cells were stained with Annexin V PE/7-AAD dyes. A total of 10,000 cells were acquired by the FACSCalibur Flow Cytometer (BD Biosciences) and analyzed by CellQuest Pro software (BD Biosciences).
Cell viability assay using trypan blue
AGS cells were kept uninfected or infected with H. pylori for 10 h. In another set, AGS cells were transfected with either Noxa WT or Noxa S13A plasmid constructs or empty vector followed by infection with H. pylori for 10 h. A total of 20 μl cell suspension was mixed with 180 μl trypan blue (Invitrogen), and cell counting was done using a Neubauer hemocytometer (Camlab Ltd., Cambridge, United Kingdom).
Confocal microscopy
Noxa WT and S13A mutant or empty vector constructs were transfected in pDsRed2 (Clontech, Mountain View, CA, USA)-expressing AGS stable cells. Cells were fixed with 4% paraformaldehyde at 37°C for 15 min followed by DAPI (dilactate; Invitrogen) treatment for 20 min. Mitochondrial integrity was assessed by a laser-scanning confocal microscope (LSM 780; Carl Zeiss, Jena, Germany) with argon laser 488 excitation and a 63×/1.4 NA oil objective (Carl Zeiss). The microscope was equipped with an LSM-TPMT camera system and ZEN 2010 software (both from Carl Zeiss). Images were analyzed using LSM software (Carl Zeiss). All images were acquired at room temperature and digitally processed for presentation using Adobe Photoshop CS4 (Adobe Systems Incorporated, San Jose, CA, USA). Mitochondrial fragmentation (measured by length, circularity, and roundness) was quantified by ImageJ software (NIH, Bethesda, MD, USA). Roundness [4 × (surface area)/(π × major axis2)] and circularity [4π × (surface area/perimeter2)] are indices of sphericity, with values of 1 indicating perfect spheroids.
Statistics
Values are given as the mean SE. Student’s t test was performed for comparisons of 2 groups. Statistical significance was determined at P < 0.05. 2-way ANOVA was performed to compare various transfection groups. Tukey test was applied for post hoc comparisons.
RESULTS
H. pylori infection induces Noxa but does not degrade Mcl1 in the human GECs
The present study aimed to assess the effect of H. pylori infection on Noxa expression because the Mcl1/Noxa axis plays a crucial role in regulating the life span of a cell. For this, AGS cells were infected with a cag PAI(+) H. pylori strain 26695 at an MOI of 200 for various time periods. Representative Western blot results (n = 3) showed that H. pylori time-dependently increased both Mcl1 and Noxa expression (Fig. 1A), reaching a maximal level at 5 h postinfection (p.i.). We observed that strain 26695 at an MOI of 200 and 300 equally induced Hif1α (data not shown), but lower MOIs had no significant effect on Hif1α expression (2). Accordingly, all future experiments were performed with the 26695 strain at an MOI of 200 for 5 h unless stated otherwise. We also performed IHC analyses on human gastric biopsy samples obtained from uninfected (n = 5) and infected (n = 5) subjects. Both Noxa and Mcl1 were significantly increased in H. pylori–infected samples (Fig. 1B), which corroborated our observations in cultured AGS cells. Noxa and Mcl1 are induced in various cancers (16, 17). We also observed induced expression of both Noxa and Mcl1 in surgically resected human gastric adenocarcinoma samples as compared to nonneoplastic gastric tissue sections (Supplemental Fig. S1). Other gastric cancer cell lines, MKN-45, NCI-N87, and Kato III, were assessed along with AGS cells for their ability to express Mcl1 and Noxa. Western blot results revealed that expression of Noxa and Mcl1 and their transcriptional activator Hif1α was induced in all of these GECs (Fig. 1C). Hif1α, Noxa, and Mcl1 were equally induced by cag PAI(+) and cag PAI(−) H. pylori strains (26695 and 8-1, respectively) in native GECs isolated from uninfected human gastric biopsy samples (Fig. 1D).
Figure 1.
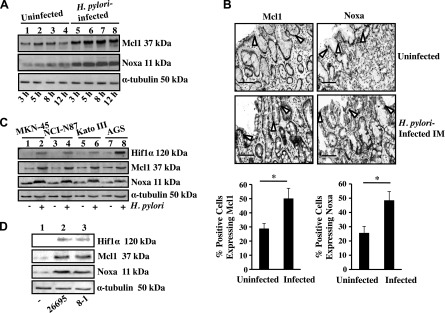
H. pylori–induced Noxa and Mcl1 expression in GECs. A) A representative Western blot (n = 3) of whole-cell lysates prepared from uninfected and infected (3, 5, 8, and 12 h with an MOI of 200) AGS cells showed time-dependent induced expression of Noxa and Mcl1. α-Tubulin was used as a loading control. B) Immunohistochemical staining of biopsy samples from uninfected and H. pylori-infected patients showing expression of Noxa as well as Mcl1 in the epithelium as well as lamina propria (open arrows indicate epithelial cell lining in the gastric mucosa). Quantification is shown of Mcl1+ and Noxa+ cells in the infected and uninfected gastric mucosa (n = 5). Graph shows the mean ± sem. *P < 0.05, Student’s t test. Original magnification, ×50. Scale bars, 100 μm. C) Expression of Hif1α, Mcl1, and Noxa was analyzed by immunoblotting cell lysates prepared from uninfected or H. pylori–infected (an MOI of 200; 5 h) MKN-45, NCI-N87, KATO III, and AGS cells. α-Tubulin was kept as a loading control. D) Western blot analysis of epithelial cells isolated from 3 sets of uninfected human gastric biopsy samples and separately infected with either an MOI of 200 of cag PAI(−) 8-1 strain or (+) 26695 strain for 5 h. IM, intestinal metaplasia.
Hif1α and its coactivator p300 bind to the noxa promoter in H. pylori–infected GECs
Enrichment of the noxa promoter HRE (mean ± sem, n = 3) by ChIP assay with Hif1α revealed Hif1α binding to the noxa promoter (Fig. 2A). In vivo binding of Hif1α to the noxa HRE was analyzed by ChIP assay using Hif1α antibody. The PCR product of DNA in the Hif1α immunocomplex corresponding to the human noxa promoter flanking the HRE sequence demonstrated specific binding of Hif1α that was not present in the PCR product corresponding to the 5′ far upstream region (Fig. 2B). Streptavidin-covered magnetic beads were coated with noxa HRE-specific oligos as well as HRE-mut oligo, and nuclear extracts from uninfected and infected cells were incubated with beads. Hif1α and p300 binding to the noxa HRE (Fig. 2C) was observed only in H. pylori–infected cells. Coincubation of AGS cells with 150 nM Echinomycin (an inhibitor of DNA-binding activity of Hif1 and a peptide antibiotic) at the time of H. pylori infection abrogated Noxa expression induced by H. pylori (Fig. 2D), further indicating the importance of Hif1 in inducing Noxa expression in H. pylori–infected GECs. Because the human Noxa promoter has a p53-binding site, we assessed the expression of p53 and phospho-(Ser15) p53 in H. pylori–infected AGS cells. Infection for 3, 6, and 10 h with an MOI of 200 of H. pylori had no effect on p53 and phospho-p53 expression (data not shown). Comparison of Hif1α shRNA stably expressing cell lines with scrambled shRNA and empty vector-expressed cells after H. pylori infection and Western blot analysis further confirmed the importance of Hif1α in Noxa expression in H. pylori–infected GECs (Fig. 2E).
Figure 2.
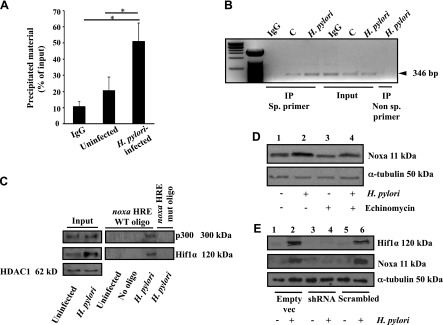
Hif1α and its coactivator p300 interacted with the promoter region of noxa and enhanced Noxa expression after H. pylori infection. A) Enrichment of the noxa promoter HRE by ChIP with Hif1α antibody relative to input DNA measured by real-time PCR (mean ± sem, n = 3). *P < 0.05. B) ChIP analysis of Hif1α immunocomplex for the noxa promoter HRE. Specificity of Hif1α binding to the noxa HRE was confirmed by comparing the 5′ far upstream region (nonspecific [sp.] primer) with the HRE-flanking region of the noxa promoter. C) Western blotting (n = 3) showed binding of Hif1α and its transcriptional coactivator p300 to the noxa promoter HRE (coated on streptavidin beads) after infection with an MOI of 200 of H. pylori for 5 h. D) Hif1α-induced Noxa expression was validated by pretreating AGS cells with 150 nM Echinomycin prior to infection with an MOI of 200 of H. pylori for 5 h. α-Tubulin was the loading control. E) Western blotting (n = 3) showed expression of Noxa and Hif1α in the negative control shRNA, scrambled negative control shRNA, and Hif1α-shRNA stably expressing AGS cells. IP, immunoprecipitation; vec, vector.
Noxa is phosphorylated by JNK at Ser13 in H. pylori–infected GECs
Because the Mcl1 level was maintained in H. pylori–infected GECs despite sustained expression of Noxa (Fig. 1A), we wanted to assess the phosphorylation status of Noxa in infected AGS cells. Western blot (n = 3) analysis clearly indicated that Noxa was maximally phosphorylated at Ser residues at 5 h of H. pylori infection (Fig. 3A). We also found that Noxa phosphorylation by H. pylori was a cag PAI-independent event (Supplemental Fig. S2C). The cag PAI encodes proteins for a type IV secretion system (T4SS) that injects virulence factors into the infected host cell cytoplasm. H. pylori CagA protein is one such factor that is directly injected into the infected GECs, and upon phosphorylation, it enhances cell motility. To assess the cag PAI status of strains 26695 and 8-1, we infected AGS cells with an MOI of 200 of respective strains for 5 h. Western blotting of whole-cell lysates clearly showed CagA expression and its phosphorylation in 26695-infected cells, but not in cells infected with strain 8-1 (Supplemental Fig. S2D). GPS2.1 software (18) was used to predict potential phosphorylation sites of Noxa (Supplemental Fig. S2A) and to predict kinases involved in its phosphorylation. Only Cdk5 and JNK appeared to have significant potential to phosphorylate at Ser13; p38 MAPK and ERKs phosphorylated with a much less predicted effect. However, we did not observe any expression of Cdk5 in GECs, which corroborated findings of other studies showing that the human stomach does not express Cdk5 (19, 20).
Figure 3.
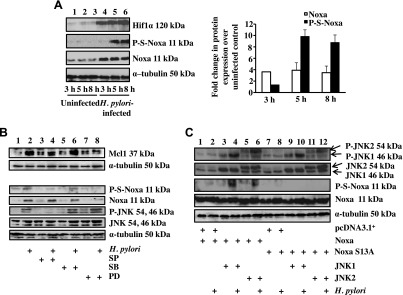
JNK induced Noxa phosphorylation at Ser13 residue in H. pylori–infected GECs. A) Western blot analysis of whole-cell lysates prepared from AGS cells infected with an MOI of 200 of H. pylori for 3, 5, and 8 h showed a time-dependent expression of P-S-Noxa. Densitometric analysis of Noxa and P-Noxa bands shown in (A) is represented as normalized Noxa and P-Noxa (mean ± sem, n = 3). Bar graphs are normalized as fold increase over uninfected controls. B) Immunoblot analysis of whole-cell lysates derived from AGS cells infected with an MOI of 200 of H. pylori for 5 h showing the expression status of P-JNK and total JNK. Pretreatment with 25 µM JNK inhibitor II [SP600125 (SP)], 25 µM p38 MAPK inhibitor [SB203580 (SB)], and MEK 1/2 inhibitor [PD98039 (PD)] was done as indicated. C) Western blot analysis of P-S-Noxa and total Noxa after cotransfecting JNK1 or JNK2 constructs along with empty vector, WT, or S13A mut constructs of Noxa followed by infection with an MOI of 200 of H. pylori for 5 h. α-Tubulin was used as the loading control.
MAPKs have broad-tissue distributions except for JNK 3, which is expressed only in neurons, cardiac muscles, and testis. Phosphorylation of ERK1/ERK2, p38, and JNK occurs as early as 30 min after H. pylori infection and remains up-regulated until 24 h (21). We also found activation of these 3 MAPKs in infected GECs at 5 h of infection with an MOI of 200 (Fig. 3B and Supplemental Fig. S2E). MEK1 inhibitor PD98059 (25 μM, 1 h pretreatment prior to infection) or p38 MAPK inhibitor SB203580 (25 μM, 1 h pretreatment prior to infection) partly suppressed total Noxa and phospho-Ser-Noxa (P-S-Noxa) expression. This result corroborated findings of other studies showing that inhibition of ERK and p38 MAPK causes suppression of Noxa expression (22). In contrast, JNK inhibitor II or SP600125 (25 μM, 1 h pretreatment) inhibited only P-S-Noxa formation in the infected GECs without suppressing total Noxa, indicating that Noxa expression was not regulated by the JNK pathway, whereas Noxa phosphorylation was regulated by the JNK pathway in the H. pylori–infected gastric epithelium. Because JNK is known to induce Mcl1 phosphorylation, stabilization, and antiapoptotic function (23), JNK inhibition should reduce the Mcl1 protein level in H. pylori–infected AGS cells. Because Noxa expression has been reported to cause activation of JNK and p38 (24), we wanted to identify the effect of WT Noxa and a Ser13 nonphosphorylatable mut (S13A) on H. pylori–mediated activation of MAPKs. For this, transfected cells were either infected with H. pylori or left uninfected. A representative Western blot (n = 3) indicated that WT and S13A Noxa constructs highly induced H. pylori–mediated JNK phosphorylation and slightly induced p38 phosphorylation but had no effect on ERK1/ERK2 phosphorylation (Supplemental Fig. S3A).
To determine whether Noxa binds to phospho-JNK (P-JNK) in H. pylori–infected GECs, we immunoprecipitated whole-cell extracts with Noxa antibody and probed for P-JNK and total Noxa. A representative result (Supplemental Fig. S3B) showed that interaction of P-JNK with Noxa was induced by H. pylori. The interaction of P-JNK with Noxa was equally enhanced by ectopic expression of WT or S13A Noxa construct, indicating that Noxa mutation did not impair its interaction with P-JNK. To further establish the role of JNK in Noxa phosphorylation, we cotransfected AGS cells with either empty vector along with WT or S13A Noxa constructs, or WT Noxa along with JNK1 or JNK2 constructs or S13A Noxa with JNK1 or JNK2 constructs and infected with H. pylori for 5 h or left uninfected. JNK1/JNK2 overexpression resulted in enhanced phosphorylation of WT Noxa but not of S13A mut in the infected GECs (Fig. 3C, compare lanes 2, 4, and 6 with lanes 8, 10, and 12, respectively). Together, Fig. 3C and Supplemental Fig. S3B showed that Noxa mutation did not change its interaction with P-JNK, but JNK-mediated Noxa phosphorylation could not take place in Noxa S13A-expressing cells. These results established the importance of Ser13 residue in JNK-mediated phosphorylation of Noxa. We also found that cag PAI(+) and cag PAI(−) strains equally induced JNK phosphorylation (Supplemental Fig. S2B).
Blocking Noxa phosphorylation induces Mcl1 degradation and mitochondrial apoptosis events
To examine the effect of Noxa phosphorylation on the Mcl1 level, AGS cells were transiently transfected with control vector, WT Noxa, and S13A Noxa constructs followed by infection with H. pylori and Western blotting of whole-cell lysates (n = 4). H. pylori–mediated induction of Mcl1 expression was significantly blocked in S13A mut-transfected cells (Fig. 4A). These data indicated that although WT Noxa enhanced H. pylori infection-mediated Mcl1 expression, the nonphosphorylatable S13A mut Noxa was unable to do so.
Figure 4.
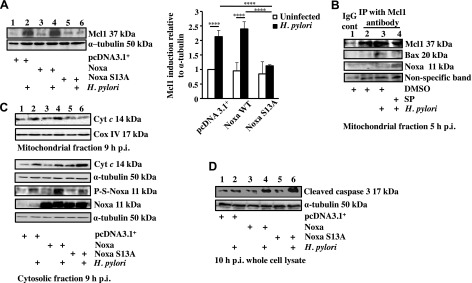
Mcl1 degradation, cytochrome c release, and cleaved caspase-3 formation were enhanced in S13A Noxa-expressing H. pylori–infected GECs. A) AGS cells were transfected with either empty vector, Noxa WT or Noxa S13A construct followed by infection for 5 h with an MOI of 200 of H. pylori, and Mcl1 status was analyzed by immunoblotting of whole-cell lysates. Data were analyzed by 2-way ANOVA with Tukey’s post hoc test (n = 4). Error bars, sem. ****P < 0.0001. B) Immunoprecipitation experiment assessing Mcl1-Noxa interaction and Mcl1-Bax interaction in the mitochondrial fraction of infected AGS cells. Mitochondrial extracts were immunoprecipitated with Mcl1 antibody after various treatments as indicated. A nonspecific band was used to estimate protein loading. C) Western blot showing phosphorylation status of Noxa and cytochrome (Cyt) c in the cytosol in 9 h H. pylori–infected AGS cells expressing either empty vector or Noxa WT or Noxa S13A. Corresponding mitochondrial fraction assessed cytochrome c retention status 9 h p.i. Cox IV was used as a loading control for mitochondrial fraction. D) Immunoblot of whole-cell lysates showing the status of cleaved caspase-3 in Noxa WT and S13A Noxa-transfected AGS cells infected with an MOI of 200 of H. pylori for 10 h. α-Tubulin was used as a loading control.
Reduced Mcl1 level is associated with mitochondrial apoptotic events (25). We assessed the effect of JNK inhibition on Mcl1-Noxa interaction. Mitochondrial lysates were prepared from vehicle (DMSO)-treated, 5 h H. pylori–infected, or JNK inhibitor II pretreated and infected AGS cells followed by immunoprecipitation with Mcl1. We observed Mcl1-Noxa interaction exclusively in JNK inhibitor-treated and -infected mitochondria, but not in infected cells without the inhibitor (Fig. 4B). Furthermore, this experiment showed that although Bax was bound with Mcl1 in H. pylori–infected AGS cells, JNK inhibitor II pretreatment completely abrogated Mcl1-Bax interaction.
Cytochrome c release is the hallmark of the intrinsic apoptotic pathway, and Noxa displaces apoptotic effectors such as Bak and Bax from Mcl1 so that these effectors can undergo oligomerization resulting in cytochrome c release (26). To identify the effect of Noxa phosphorylation on cytochrome c release in H. pylori–infected GECs, AGS cells were transfected with WT Noxa, S13A Noxa, or the empty vector. Cytosolic and mitochondrial fractions were Western blotted. H. pylori–induced cytochrome c release in all transfected groups, but S13A-expressing H. pylori–infected cells showed substantially higher cytochrome c release (n = 3) (Fig. 4C). This result suggests that the apoptotic potential of Noxa is reduced by its phosphorylation. Because the release of cytochrome c into the cytosol leads to the activation of caspase-3 in apoptotic cells, we sought to understand the effect of Noxa and P-S-Noxa on caspase-3 activation in H. pylori–infected GECs. For this, AGS cells were transfected with the aforementioned constructs or transfected with empty vector followed by infection with H. pylori for 10 h or left uninfected. Western blot of cytosolic lysates (n = 3) showed that H. pylori maximally cleaved caspase-3 in S13A mut-expressing cells compared to the infected counterparts from empty vector and WT Noxa-transfected groups (Fig. 4D).
AGS cells are very small, and their mitochondrial network is easily destroyed with mitochondrial stress (27). Confocal microscopy was performed to assess whether the cell death caused by H. pylori was linked with mitochondrial morphology changes. pDsRed2 stably expressing AGS cells were transiently transfected with empty vector or Noxa WT or S13A mut followed by infection with H. pylori for 8 h. Mitochondrial phenotype was then assessed by confocal microscopy. The reticulotubular network of healthy mitochondria found in empty vector-transfected cells was partially disintegrated and appeared smaller in H. pylori–infected cells (Fig. 5A). Mitochondria lose their membrane potential and undergo fragmentation (form round and small spheroidal or punctate structures) before releasing cytochrome c. We found significantly greater roundness and circularity in the mitochondria from the infected S13A-expressing cells as compared to the mitochondria from pcDNA3.1+ and WT Noxa-expressing cells (Fig. 5B).
Figure 5.

Non–P-Noxa increases H. pylori–induced mitochondrial fragmentation. A) Mitochondrial morphology was examined by confocal microscopy after transfecting empty vector (pcDNA3.1+), Noxa WT, and Noxa S13A constructs in pDsRed2 stably expressing AGS cells followed by infection with H. pylori for 8 h, with an MOI of 200. Scale bars, 5 µm. B) Mitochondrial length, roundness, and circularity information was collected from 4 cells (from 4 independent experiments), and 5 mitochondria were measured from each cell for statistical analysis. Circular mitochondrial appearance indicative of mitochondrial stress was more apparent in infected Noxa S13A-transfected cells when compared with the pcDNA3.1+or Noxa WT construct-expressing infected cells. Data were analyzed by 2-way ANOVA with Tukey’s post hoc test. Error bars, sem. *P < 0.05; **P < 0.01; ****P < 0.0001.
Noxa dephosphorylation enhances apoptosis in H. pylori–infected GECs
To determine the extent of cell death by H. pylori, AGS cells were infected with an MOI of 200 for 10 h. We stained cells with Annexin V PE to detect apoptotic cells, and 7-AAD was used to detect necrotic population. Flow cytometric analysis revealed that H. pylori significantly induced apoptotic cell death (Fig. 6A and Supplemental Fig. S4A). To assess the role of Noxa WT and S13A mut transfections on apoptosis, AGS cells were transiently transfected with empty vector, WT Noxa, or S13A Noxa followed by infection with an MOI of 200 of H. pylori for 10 h. Flow cytometric analysis showed that S13A-transfected and H. pylori–infected cells significantly induced apoptosis compared with their counterparts from empty vector-transfected and Noxa WT-transfected groups (Fig. 6B and Supplemental Fig. S4B). Cell mortality was also calculated with trypan blue assay (data not shown).
Figure 6.

Non–P-Noxa enhances H. pylori–induced apoptosis in AGS cells. A) AGS cells were either infected with H. pylori or left uninfected. Cells were harvested and stained with Annexin V PE/7-AAD dyes. Representative flow cytometry data show that H. pylori induced apoptosis in infected cells (n = 6). Apoptotic cells are in the lower-right quadrant. B) AGS cells were transfected with either Noxa WT or Noxa S13A plasmid constructs or empty vector followed by infection with H. pylori for 10 h. A representative dot plot indicates a marked increase in H. pylori–induced apoptosis in S13A Noxa construct-transfected cells as compared to WT and empty vector-transfected cells (n = 6).
Overall, our data confirmed that although H. pylori–mediated induction of Noxa-induced apoptosis, impairment of Noxa phosphorylation enhanced the apoptotic potential of Noxa in H. pylori–infected GECs. The effect of Noxa phosphorylation status on H. pylori–mediated GEC apoptosis is graphically summarized in Fig. 7.
Figure 7.

Regulation of apoptotic potential of Noxa in H. pylori–infected GECs. Hif1α expressed in H. pylori infected GECs enhances Noxa expression. H. pylori also induces Bax translocation to mitochondria, which can bind with Mcl1 in the mitochondria. If Noxa manages to stay as non–P-Noxa, it translocates to the mitochondria, displaces Bax from Mcl1, and binds with Mcl1. As a result, Mcl1 is degraded, and free Bax is oligomerized to help in the cytochrome c release, resulting in apoptosis. When Noxa is phosphorylated at Ser13 by JNK activated in H. pylori–infected GECs, P-Noxa is unable to translocate to mitochondria, resulting in impairment of apoptosis. Curved arrow indicates translocation.
DISCUSSION
The chronic imbalance between host epithelial cell death and proliferation is a major mechanism that contributes to H. pylori–mediated gastric cancer because it allows accumulation of mutations in host cells (28). The BH3-only apoptotic protein Noxa binds mainly with the antiapoptotic protein Mcl1, a complex that is targeted for proteasomal degradation. Thus, Noxa functions as a critical regulator of apoptosis in Mcl1-expressing cells (29). Here, we report that although Noxa is up-regulated in H. pylori–infected GECs, its apoptotic function is regulated by phosphorylation. The stress-induced kinase JNK, which mediates apoptosis in the H. pylori–infected gastric epithelium (30, 31), is responsible for Noxa phosphorylation. This also indicates that H. pylori can mold host apoptotic effectors toward antiapoptosis.
H. pylori infection induces gastric epithelial ROS generation and apoptosis (32). The indirect activation model of apoptosis suggests that the interaction of BH3-only proteins with their antiapoptotic partners induces conformational changes in the multi-BH domain-containing proteins Bax and Bak. This leads to auto-oligomerization of these “apoptosis effectors” in the outer membrane of the mitochondria, resulting in the release of cytochrome c (33). BH3-only proteins show selectivity in binding with antiapoptotic members of the Bcl2 family. As a “sensitizer,” BH3-only protein Noxa was earlier thought to have a weak proapoptotic function. However, Lowman et al. (11) showed that S13A Noxa-expressing leukemia cells die from glucose stress. These investigators were unable to generate stable S13A-expressing Jurkat cells. We also failed in our attempt to generate GECs with Noxa S13A mutation, which strengthened our belief that Noxa phosphorylation by H. pylori has some essential protective functions against apoptosis. Previously, induction of Noxa was reported to be essential but not sufficient for apoptosis (34). Thus, we surmise that Noxa phosphorylation could be one of the explanations of these findings.
H. pylori induces MAPKs and regulates various cellular processes, including cell cycle, proliferation, and apoptosis (21). JNK plays a role in regulating the intrinsic apoptotic pathway because activation of JNK increases cytochrome c release (35). Our results reveal a new phenomenon that Noxa is phosphorylated by JNK in H. pylori–infected GECs and regulates the apoptotic potential of Noxa. Because JNK can induce invasiveness and motility of H. pylori–infected cells (36), we speculate that Noxa phosphorylation by JNK also helps in the survival of invasive gastric cancer cells. Further studies are needed to explore this hypothesis.
One of the main steps in the intrinsic apoptotic pathway is Bax translocation to mitochondria. H. pylori enhances Bax translocation to mitochondria followed by mitochondrial fragmentation (27). Although the Mcl1-Bak interaction is well known, a direct Mcl1-Bax interaction is not commonly observed. However, it has been shown that following Bax translocation to mitochondria, Mcl1 can inhibit Bax-induced cytochrome c release (37). Although the same group reports that direct interaction with Mcl1 is not required for Bax inhibition in that study, there is evidence that a Mcl1-Bax interaction exists (38). We report here that H. pylori induces direct interaction of Mcl1 with Bax. Our results also confirm that abrogation of JNK-mediated phosphorylation of Noxa in infected GECs results in direct Mcl1-Noxa interaction in the H. pylori–infected gastric epithelium. We presume that in the presence of nonphosphorylated Noxa (P-Noxa), which has a very high affinity and priority to bind with Mcl1 (39, 40), Bax is displaced from Mcl1, oligomerizes, and helps in cytochrome c release. p53 is crucial in regulating cell survival, apoptosis, as well as cell cycle and is a very highly mutated gene in human cancers (41). Although it is one of the main transcription factors that induce Noxa, p53-independent mechanisms of Noxa induction also exist and are targeted to treat cancers (42, 43). Almost 40% of cases of gastric cancer have p53 mutations, and H. pylori can cause p53 mutations as well (28, 44). A recent study suggests that p53 can even be inhibited by H. pylori in a mutation-independent mechanism (45). Tumor suppressor function and transcriptional activity of p53 remain inhibited in B cells of patients with MALT lymphoma (46). In our study, Hif1α was highly induced and shown to be a major inducer of Noxa in H. pylori–infected GECs. Because Hif1α (47) and H. pylori (48) individually can contribute to gastric cancer chemoresistance, it is likely that P-Noxa induction by H. pylori further increases the risk for treatment resistance in gastric cancer. We believe that the accumulation of P-Noxa resulting in antiapoptosis of the H. pylori–infected GECs would contribute to accumulation of mutations, leading to the increased probability of developing gastric cancer. Because Noxa and P-Noxa both are induced by H. pylori, it is difficult to assess a direct cause-effect relationship as the P-Noxa:Noxa ratio in gastric cancer tissue samples has not been studied yet. Further research is required to identify the prevalence of Noxa Ser13 mutation and P-JNK expression pattern in H. pylori–infected gastric biopsy samples.
Given the frequency of gastric cancer metastasis and treatment resistance, developing new targeted treatment strategies is very important. Because phosphorylation-dephosphorylation can regulate apoptotic functions of Noxa, this is a potential target molecule for future treatment approaches in H. pylori–induced gastric cancer. Noxa phosphorylation may also be implicated in carcinogenic events in other types of cancer.
Supplementary Material
Acknowledgments
This work was supported in part by a Rapid Grant for Young Investigators (RGYI), Department of Biotechnology, India, and a Fast Track Grant, Science and Engineering Research Board, India (to A.B.); the U.S. National Institutes of Health Grant R01-DK61769 and an American Gastroenterological Association (AGA) Funderburg Research Scholar Award (to S.E.C.); an Indian Council of Medical Research (ICMR) fellowship (to S.R.); and a fellowship from Department of Atomic Energy (DAE), India (to L.D. and S.B.K.).
Glossary
- cag
cytotoxin-associated gene
- ChIP
chromatin immunoprecipitation
- GEC
gastric epithelial cell
- Hif1
hypoxia-inducible factor 1
- HRE
hypoxia-response element
- IHC
immunohistochemistry
- MALT
mucosa-associated lymphoid tissue
- Mcl1
myeloid cell leukemia 1
- MOI
multiplicity of infection
- mut
mutant
- PAI
pathogenicity island
- p.i.
postinfection
- P-JNK
phospho-JNK
- P-Noxa
phosphorylated Noxa
- P-S-Noxa
phospho-Ser-Noxa
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- UVA
University of Virginia
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Peek R. M. Jr, Blaser M. J. (2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2, 28–37 [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharyya A., Chattopadhyay R., Hall E. H., Mebrahtu S. T., Ernst P. B., Crowe S. E. (2010) Mechanism of hypoxia-inducible factor 1 alpha-mediated Mcl1 regulation in Helicobacter pylori-infected human gastric epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G1177–G1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J. H., Kim T. Y., Jong H. S., Kim T. Y., Chun Y. S., Park J. W., Lee C. T., Jung H. C., Kim N. K., Bang Y. J. (2003) Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clin. Cancer Res. 9, 433–440 [PubMed] [Google Scholar]
- 4.Wenger R. H., Stiehl D. P., Camenisch G. (2005) Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, re12. [DOI] [PubMed] [Google Scholar]
- 5.Wang G. L., Jiang B. H., Rue E. A., Semenza G. L. (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 92, 5510–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piret J. P., Minet E., Cosse J. P., Ninane N., Debacq C., Raes M., Michiels C. (2005) Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J. Biol. Chem. 280, 9336–9344 [DOI] [PubMed] [Google Scholar]
- 7.Kim J. Y., Ahn H. J., Ryu J. H., Suk K., Park J. H. (2004) BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J. Exp. Med. 199, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei J., O’Brien D., Vilgelm A., Piazuelo M. B., Correa P., Washington M. K., El-Rifai W., Peek R. M., Zaika A. (2008) Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology 134, 1412–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vela L., Gonzalo O., Naval J., Marzo I. (2013) Direct interaction of Bax and Bak proteins with Bcl-2 homology domain 3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J. Biol. Chem. 288, 4935–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez-Bougie P., Ménoret E., Juin P., Dousset C., Pellat-Deceunynck C., Amiot M. (2011) Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochem. Biophys. Res. Commun. 413, 460–464 [DOI] [PubMed] [Google Scholar]
- 11.Lowman X. H., McDonnell M. A., Kosloske A., Odumade O. A., Jenness C., Karim C. B., Jemmerson R., Kelekar A. (2010) The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol. Cell 40, 823–833 [DOI] [PubMed] [Google Scholar]
- 12.Likui W., Qun L., Wanqing Z., Haifeng S., Fangqiu L., Xiaojun L. (2009) Prognostic role of myeloid cell leukemia-1 protein (Mcl-1) expression in human gastric cancer. J. Surg. Oncol. 100, 396–400 [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharyya A., Chattopadhyay R., Burnette B. R., Cross J. V., Mitra S., Ernst P. B., Bhakat K. K., Crowe S. E. (2009) Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology 136, 2258–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dérijard B., Hibi M., Wu I. H., Barrett T., Su B., Deng T., Karin M., Davis R. J. (1994) JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76, 1025–1037 [DOI] [PubMed] [Google Scholar]
- 15.Kallunki T., Su B., Tsigelny I., Sluss H. K., Dérijard B., Moore G., Davis R., Karin M. (1994) JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 8, 2996–3007 [DOI] [PubMed] [Google Scholar]
- 16.Ploner C., Kofler R., Villunger A. (2008) Noxa: at the tip of the balance between life and death. Oncogene 27(Suppl 1), S84–S92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warr M. R., Shore G. C. (2008) Unique biology of Mcl-1: therapeutic opportunities in cancer. Curr. Mol. Med. 8, 138–147 [DOI] [PubMed] [Google Scholar]
- 18.Xue Y., Ren J., Gao X., Jin C., Wen L., Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iseki H., Ko T. C., Xue X. Y., Seapan A., Hellmich M. R., Townsend C. M. Jr (1997) Cyclin-dependent kinase inhibitors block proliferation of human gastric cancer cells. Surgery 122, 187–194 [DOI] [PubMed] [Google Scholar]
- 20.Tsai L. H., Takahashi T., Caviness V. S. Jr, Harlow E. (1993) Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 119, 1029–1040 [DOI] [PubMed] [Google Scholar]
- 21.Ding S. Z., Smith M. F. Jr, Goldberg J. B. (2008) Helicobacter pylori and mitogen-activated protein kinases regulate the cell cycle, proliferation and apoptosis in gastric epithelial cells. J. Gastroenterol. Hepatol. 23, e67–e78 [DOI] [PubMed] [Google Scholar]
- 22.Tromp J. M., Geest C. R., Breij E. C., Elias J. A., van Laar J., Luijks D. M., Kater A. P., Beaumont T., van Oers M. H., Eldering E. (2012) Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin. Cancer Res. 18, 487–498 [DOI] [PubMed] [Google Scholar]
- 23.Kodama Y., Taura K., Miura K., Schnabl B., Osawa Y., Brenner D. A. (2009) Antiapoptotic effect of c-Jun N-terminal Kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology 136, 1423–1434 [DOI] [PubMed] [Google Scholar]
- 24.Hassan M., Alaoui A., Feyen O., Mirmohammadsadegh A., Essmann F., Tannapfel A., Gulbins E., Schulze-Osthoff K., Hengge U. R. (2008) The BH3-only member Noxa causes apoptosis in melanoma cells by multiple pathways. Oncogene 27, 4557–4568 [DOI] [PubMed] [Google Scholar]
- 25.Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., Wang X. (2003) Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willis S. N., Adams J. M. (2005) Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17, 617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashktorab H., Frank S., Khaled A. R., Durum S. K., Kifle B., Smoot D. T. (2004) Bax translocation and mitochondrial fragmentation induced by Helicobacter pylori. Gut 53, 805–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumoto Y., Marusawa H., Kinoshita K., Endo Y., Kou T., Morisawa T., Azuma T., Okazaki I. M., Honjo T., Chiba T. (2007) Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat. Med. 13, 470–476 [DOI] [PubMed] [Google Scholar]
- 29.Alves N. L., Derks I. A., Berk E., Spijker R., van Lier R. A., Eldering E. (2006) The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity 24, 703–716 [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto A., Isomoto H., Nakayama M., Hisatsune J., Nishi Y., Nakashima Y., Matsushima K., Kurazono H., Nakao K., Hirayama T., Kohno S. (2011) Helicobacter pylori VacA reduces the cellular expression of STAT3 and pro-survival Bcl-2 family proteins, Bcl-2 and Bcl-XL, leading to apoptosis in gastric epithelial cells. Dig. Dis. Sci. 56, 999–1006 [DOI] [PubMed] [Google Scholar]
- 31.Domek M. J., Netzer P., Prins B., Nguyen T., Liang D., Wyle F. A., Warner A. (2001) Helicobacter pylori induces apoptosis in human epithelial gastric cells by stress activated protein kinase pathway. Helicobacter 6, 110–115 [DOI] [PubMed] [Google Scholar]
- 32.Ding S. Z., Minohara Y., Fan X. J., Wang J., Reyes V. E., Patel J., Dirden-Kramer B., Boldogh I., Ernst P. B., Crowe S. E. (2007) Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect. Immun. 75, 4030–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinou J. C., Youle R. J. (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 21, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu W., Swetzig W. M., Medisetty R., Das G. M. (2011) Estrogen-mediated upregulation of Noxa is associated with cell cycle progression in estrogen receptor-positive breast cancer cells. PLoS One 6, e29466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tournier C., Hess P., Yang D. D., Xu J., Turner T. K., Nimnual A., Bar-Sagi D., Jones S. N., Flavell R. A., Davis R. J. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 [DOI] [PubMed] [Google Scholar]
- 36.Snider J. L., Allison C., Bellaire B. H., Ferrero R. L., Cardelli J. A. (2008) The beta1 integrin activates JNK independent of CagA, and JNK activation is required for Helicobacter pylori CagA+-induced motility of gastric cancer cells. J. Biol. Chem. 283, 13952–13963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Germain M., Milburn J., Duronio V. (2008) MCL-1 inhibits BAX in the absence of MCL-1/BAX Interaction. J. Biol. Chem. 283, 6384–6392 [DOI] [PubMed] [Google Scholar]
- 38.Sedlak T. W., Oltvai Z. N., Yang E., Wang K., Boise L. H., Thompson C. B., Korsmeyer S. J. (1995) Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc. Natl. Acad. Sci. USA 92, 7834–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giam M., Huang D. C., Bouillet P. (2008) BH3-only proteins and their roles in programmed cell death. Oncogene 27(Suppl 1), S128–S136 [DOI] [PubMed] [Google Scholar]
- 40.Zhang L., Lopez H., George N. M., Liu X., Pang X., Luo X. (2011) Selective involvement of BH3-only proteins and differential targets of Noxa in diverse apoptotic pathways. Cell Death Differ. 18, 864–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris S. L., Levine A. J. (2005) The p53 pathway: positive and negative feedback loops. Oncogene 24, 2899–2908 [DOI] [PubMed] [Google Scholar]
- 42.Nikiforov M. A., Riblett M., Tang W. H., Gratchouck V., Zhuang D., Fernandez Y., Verhaegen M., Varambally S., Chinnaiyan A. M., Jakubowiak A. J., Soengas M. S. (2007) Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc. Natl. Acad. Sci. USA 104, 19488–19493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pérez-Galán P., Roué G., Villamor N., Montserrat E., Campo E., Colomer D. (2006) The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 107, 257–264 [DOI] [PubMed] [Google Scholar]
- 44.Wei J., Noto J., Zaika E., Romero-Gallo J., Correa P., El-Rifai W., Peek R. M., Zaika A. (2012) Pathogenic bacterium Helicobacter pylori alters the expression profile of p53 protein isoforms and p53 response to cellular stresses. Proc. Natl. Acad. Sci. USA 109, E2543–E2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei J., Nagy T. A., Vilgelm A., Zaika E., Ogden S. R., Romero-Gallo J., Piazuelo M. B., Correa P., Washington M. K., El-Rifai W., Peek R. M., Zaika A. (2010) Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 139, 1333–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Umehara S., Higashi H., Ohnishi N., Asaka M., Hatakeyama M. (2003) Effects of Helicobacter pylori CagA protein on the growth and survival of B lymphocytes, the origin of MALT lymphoma. Oncogene 22, 8337–8342 [DOI] [PubMed] [Google Scholar]
- 47.Nakamura J., Kitajima Y., Kai K., Mitsuno M., Ide T., Hashiguchi K., Hiraki M., Miyazaki K. (2009) Hypoxia-inducible factor-1alpha expression predicts the response to 5-fluorouracil-based adjuvant chemotherapy in advanced gastric cancer. Oncol. Rep. 22, 693–699 [DOI] [PubMed] [Google Scholar]
- 48.Nardone G., Rocco A., Vaira D., Staibano S., Budillon A., Tatangelo F., Sciulli M. G., Perna F., Salvatore G., Di Benedetto M., De Rosa G., Patrignani P. (2004) Expression of COX-2, mPGE-synthase1, MDR-1 (P-gp), and Bcl-xL: a molecular pathway of H pylori-related gastric carcinogenesis. J. Pathol. 202, 305–312 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.