

Abstract
The retinitis pigmentosa 2 polypeptide (RP2) functions as a GTPase-activating protein (GAP) for ARL3 (Arf-like protein 3), a small GTPase. ARL3 is an effector of phosphodiesterase 6 Δ (PDE6D), a prenyl-binding protein and chaperone of prenylated protein in photoreceptors. Mutations in the human RP2 gene cause X-linked retinitis pigmentosa (XLRP) and cone-rod dystrophy (XL-CORD). To study mechanisms causing XLRP, we generated an RP2 knockout mouse. The Rp2h−/− mice exhibited a slowly progressing rod-cone dystrophy simulating the human disease. Rp2h−/− scotopic a-wave and photopic b-wave amplitudes declined at 1 mo of age and continued to decline over the next 6 mo. Prenylated PDE6 subunits and G-protein coupled receptor kinase 1 (GRK1) were unable to traffic effectively to the Rp2h−/− outer segments. Mechanistically, absence of RP2 GAP activity increases ARL3-GTP levels, forcing PDE6D to assume a predominantly “closed” conformation that impedes binding of lipids. Lack of interaction disrupts trafficking of PDE6 and GRK1 to their destination, the photoreceptor outer segments. We propose that hyperactivity of ARL3-GTP in RP2 knockout mice and human patients with RP2 null alleles leads to XLRP resembling recessive rod-cone dystrophy.—Zhang, H., Hanke-Gogokhia, C., Jiang, L., Li, X., Wang, P., Gerstner, C. D., Frederick, J. M., Yang, Z., Baehr, W. Mistrafficking of prenylated proteins causes retinitis pigmentosa 2.
Keywords: rod-cone dystrophy, ARL3, PDE6D, RP2, XLRP
Retinitis pigmentosa is a group of inherited retinal diseases with night blindness and progressive vision loss affecting approximately 1 in 4000 people worldwide (1, 2). XLRP, a form of retinal degeneration affecting mostly males, presents with symptoms of night blindness and decreased visual function early in life; by midlife, affected males are legally blind (3, 4). Two major XLRP genes have been identified, RP2 and RP3, encoding RP2 (5) and retinitis pigmentosa GTPase regulator (RPGR) polypeptides (6), respectively. RPGR mutations are the major cause of XLRP; fewer than 20% of XLRP cases are caused by mutations in RP2 (7). Clinical symptoms of XLRP caused by RP2 gene mutations range from recessive severe rod-cone dystrophy in males to semidominant XLRP in some affected females. Null mutations in the RP2 gene primarily affect the function of rod and cone photoreceptors. RP2 mutations are also associated with macular atrophy and cone-rod dystrophy (8, 9). Male patients with XLRP also show an increase in abnormal sperm tails with axoneme defects (10).
Multiple missense and nonsense mutations in 4 of the RP2 gene’s 5 exons account for less than one quarter of all XLRPs (11, 12). The human RP2 polypeptide consists of 350 amino acids with limited sequence, but high structural, similarity to tubulin-binding cofactor C (TBCC), which is involved in β-tubulin folding (13, 14). RP2 has GAP activity for tubulin in the presence of TBCD (13). A second domain, of unknown function, in the C-terminal half is homologous to diphosphate nucleoside kinase (DNK) (13). RP2 has 3′-5′ exonuclease activity perhaps conferred through the DNK domain and translocates to the nucleus in response to DNA damage (15). RP2 localizes to the cytoplasmic face of the plasma membrane of cells throughout the retina, including photoreceptors and synaptic terminals as well as inner retina cells (16, 17). The RP2 N terminus is acylated by myristoylation at G2 and palmitoylation at C3, and acylation is required for plasma membrane targeting (16, 18). G2A-RP2 and Δ6S-RP2 mutants lack acyl side chains and are cytosolic (18).
RP2 interacts with the small GTPase ARL3 (13), and cocrystallized with ARL3-GMPPNP (19, 20). Biochemical analysis showed RP2 is a GAP for ARL3-GTP, stimulating ARL3 GTPase more than 90,000-fold under saturating RP2 conditions (20). The crystal structure of RP2 revealed the presence of an N-terminal β-helix that binds ARL3 through β-sheet interactions providing the active center for GTPase acceleration (20). Many RP2 mutations identified in patients, including the catalytically important arginine finger R118, are located in this area (19, 20). Other known RP2-interacting partners are polycystin 2 (21), N-ethylmaleimide–sensitive factor, a protein promoting vesicle-membrane fusion (17), transducin β (Tβ) (22, 23) and UNC119, an acyl-binding protein (24–27).
Upon activation by a guanine nucleotide exchange factor (GEF), ARL3 switches from an inactive GDP- to an active GTP-bound form. ARL3-GTP, acting as a GDI displacement factor (GDF), promotes cargo release from UNC119 (25, 26) and PDE6D (28, 29). UNC119 regulates ciliary targeting of acylated G proteins, such as ODR-3 and GPA-13 in Caenorhabditis elegans olfactory neurons (27, 30), and transducin α (Tα) to the outer segment following light-induced translocation to the inner segment (27). PDE6D is a prenyl-binding protein interacting with prenylated Ras-like small GTPases, phototransduction components cGMP phosphodiesterase (PDE6) and GRK1 (31), and with unprenylated RPGR and ARL2/3 (28, 29, 32). Both UNC119 and PDE6D feature an immunoglobulin-like β-sandwich structure into which lipid side chains insert to form soluble complexes (27, 28, 32). PDE6D mediates postsynthesis trafficking of PDE6 and GRK1 to outer segments, and trafficking is impeded in Pde6d−/− retina (33). An Rp2h null mouse line with an in-frame deletion of Rp2h exon 2 was shown to develop rod-cone dystrophy, accompanied by mistrafficking of M/L cone opsin (34).
We generated an Rp2h null mouse line in which RP2 is truncated after exon 1 of the Rp2h gene. Deletion of RP2 impeded trafficking of prenylated cone PDE6 and GRK1, and to a lesser extent rod PDE6. We propose a mechanism in which in the absence of the ARL3 GAP RP2, hyperactive ARL3-GTP accumulates and in complex with PDE6D impedes binding and trafficking of prenylated proteins. The Rp2h phenotype closely resembles that of the PDE6D knockout mouse (33) and simulates human X-linked rod-cone dystrophy.
MATERIALS AND METHODS
Animals
All procedures were approved by the University of Utah Institutional Animal Care and Use Committee and were conducted under the guidelines of the U.S. National Institutes of Health Guide for Care and Use of Laboratory Animals. Mice were maintained in a 12:12 h light-dark cycle. For dark adaptation, the mice were placed in the dark for ≥12 h. For light adaptation, the pupils of the mice were dilated with 1% tropicamine and the mice were exposed to light of 2000 lux for 40 min. Mutant mice were outbred to C57BL/6 mice from The Jackson Laboratory (Bar Harbor, ME) to remove the rd8 mutation (35).
Generation of Rp2 gene knockout mouse
A mouse embryonic stem cell line harboring a gene trap cassette in the first intron of the Rp2h gene was purchased from the European Conditional Mouse Mutagenesis Program (EUCOMM; Helmholtz Zentrum Muenchen, Munich, Germany). Trap presence and integrity of short and long arms were verified by PCR according to EUCOMM specifications. Chimeric mice were generated by the University of Utah transgenic mouse core facility. The wild-type (WT) allele was genotyped by PCR using primer pair RP2-F1 (5′-CTCCCTTGAATAGTGATTGAC) and RP2-R (5′-CCTAGCTGGCTTCAACTAAG), yielding a 400-bp amplicon. The gene trap allele was genotyped using primer pair RP2-F5 (5′-CTAGACAATCGGACAGACAC) and RP2-R, yielding a 550-bp amplicon. The rd8 mutation was identified by PCR as described elsewhere (35).
Subretinal injection and electroporation
A full-length mouse Rp2h cDNA was amplified using RT-PCR from a mouse retina cDNA library using RP2-F (5′-AAGGATCCACCAATGGGCTGCTGCTTCAC) and RP2-R (5-AACTCGAGTATCCCCATCTGGATCTCAGC) and cloned in-frame into the eGFP-N1 vector (Takara Clontech, Mountain View, CA, USA), with eGFP fused to the C terminus of the Rp2h gene. The RP2-eGFP expression plasmid was injected into the subretinal space of a neonatal C57BL6 mouse using a 32-gauge needle and syringe (Hamilton, Reno, NV, USA). The retinas were electroporated to transfect photoreceptors by standard procedures (36).
Generation of RP2 antibody
An oligopeptide corresponding to the C terminus of RP2 (CKMFVSEKKETASGD) was synthesized and conjugated to keyhole limpet hemocyanin by the University of Utah core facility. Antibody generation in rabbits was outsourced to Covance (Princeton, NJ, USA). Sera were tested by immunoblotting after the first boost, and final sera were collected after the second boost. Polyclonal antibodies directed against RP2 were isolated by affinity purification using immobilized antigen (Sulfo-Link kit; Thermo Fisher Scientific, Rockford, IL, USA).
Immunolabeling of mouse retina
Retina cryosections were labeled as described previously (37) using the following antibodies: VPP (polyclonal anti-rhodopsin, 1:1000) (38); UUTA (anti-rod Tα, 1:1000, from Dr. Ching-Kang Chen, Baylor College of Medicine, Houston, TX, USA); anti-Tβ (1:200, from Dr. Barry M. Willardson, Brigham Young University, Provo, UT, USA); G8 (monoclonal anti-GRK1, 1:1000) and IS4 [monoclonal anti-guanylate cyclase 1 (GC1), 1:1000, from Dr. Kris Palczewski, Case Western Reserve University, Cleveland, OH, USA]; anti-cone PDE6 (1:500, from Dr. Tiansen Li, NEI, Bethesda, MD, USA); anti–ML-opsin (1:500; Merck Millipore, Temecula, CA, USA) and MOE (anti-rod PDE6, 1:800; Cytosignal, Irvine, CA, USA). FITC-conjugated donkey anti-rabbit and donkey anti-mouse, and Cy3-conjugated goat anti-mouse, secondary antibodies were diluted 1:300. Images were acquired on an Olympus Fluoview 1000 or Leica confocal microscope (Leica, Wetzlar, Germany).
Immunoblotting
Proteins of retina lysates were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane processed as described (37). Primary antibodies were diluted 1:1000 for anti-RP2; 1:3000 for anti-PDE6α (Thermo Fisher Scientific); 1:10,000 for each anti-UUTA, VPP, G8, and β-actin (Sigma-Aldrich, St. Louis, MO, USA).
Electroretinography
Electroretinography (ERG) was performed using a UTAS E-3000 (LKC, Gaithersburg, MD, USA) visual diagnostic system. For scotopic ERGs, the mice were dark-adapted overnight and anesthetized by intraperitoneal injection (10 µl/g body weight) of a mixture of ketamine (10 mg/ml) and xylazine (1 mg/ml). The mice were kept warm by using a heating pad. Single-flash responses were recorded at stimulus intensities of −40 db (−0.6 log cd∣s∣m−2 [log candela seconds per square meter]) to 10 db (1.48 log cd∣s∣m−2). For photopic ERGs, the mice were light-adapted under background light of 1.48 log cd∣s∣m−2 for 10 min. Single-flash responses were recorded at stimulus intensities of −4 db (−0.6 log cd∣s∣m−2) to 15 db (1.86 log cd∣s∣m−2).
Fundus/optical coherence tomography
Mice were anesthetized by intraperitoneal injection (10 µl/g body weight) of a mixture of ketamine (10 mg/ml) and xylazine (1 mg/ml) and transferred to a heated water pad (37°C). Pupils were dilated with a 1% tropicamide solution. Corneas were hydrated periodically with saline solution to prevent corneal desiccation. Imaging was accomplished using an HRA-optical coherence tomography (OCT) Spectralis (Heidelberg, Germany) with a 488 nm argon blue laser with a standard 500 nm long-pass filter. Images were acquired from both eyes with a 30° lens. Then animals were transferred to a recovery heat pad maintained at 37°C and monitored until the mice regained full consciousness.
Immunostaining of C. elegans olfactory neurons
C. elegans strain RB1550 with a null mutation in the rpi-2 gene (human Rp2 gene ortholog) was provided by the C. elegans Gene Knockout Project at the Oklahoma Medical Research Foundation, which is part of the international C. elegans Gene Knockout Consortium. The staining of C. elegans olfactory cells using an ODR-3 antibody followed a procedure described elsewhere (27). Anti–ODR-3 antibody was a gift from Dr. G. Jansen, Center for Biomedical Genetics, Utrecht, The Netherlands.
Extraction of PDE6 from photoreceptor outer segments
Three bovine retinas were homogenized in 3 ml H buffer (29% sucrose, 65 mM NaCl, 2 mM MgCl2, 10 mM Tris-HCl, 1 mM DTT, 0.5 mM PMSF, 0.1 mM EDTA) and centrifuged at 1500 × g for 5 min at 4°C (39, 40). The resulting supernatant was diluted with an equal volume of isotonic buffer (100 mM NaCl, 2 mM MgCl2, 10 mM Tris-HCl, 1 mM DTT, 0.5 mM PMSF, 0.1 mM EDTA), and centrifuged at 6000 × g for 10 min. The pellet was washed with 500 μl of isotonic buffer 5 times. After the final wash, the pellet was resuspended in 500 μl of dilution buffer (10 mM Tris HCL, pH 7.4, 1 mM EDTA) and used as crude ROS. To test effect of ARL3 on the extraction of PDE6 from the membrane by PDE6D, 25 μl ROS prep was incubated with 10 μg purified GST-PDE6D and 10 μg purified recombinant ARL3 for 30 min prior to addition of GTP or GMPPNP (a final concentration of 0.2 mM in a volume of 30 μl for both GTP and GMPPNP). The extraction reactions were incubated with GTP for various lengths of time ranging from 1 min to 10 h at 4°C. The reaction was stopped by centrifugation at 20,000 g for 5 min. The supernatants were analyzed by Western blot using a polyclonal anti-PDE6α antibody (Bioaffinity). Purification of GST-PDE6D fusion protein followed a procedure described previously (41).
Recombinant ARL3
Bovine ARL3 was cloned into a pGEX-2T vector (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, United Kingdom). The GST-ARL3 fusion protein was expressed in BL21 bacteria and purified by a GSTrap column (GE Healthcare Life Sciences). ARL3 was separated from the GST tag by in-column cleavage using biotinylated thrombin (Merck Millipore). Biotinylated thrombin was removed by immobilized streptavidin. Protein was quantified using the Bradford method (42).
RESULTS
Generation of the Rp2h knockout mouse
We used a gene trapped embryonic stem cell line provided by EUCOMM (Helmholtz Zentrum Muenchen, Germany) to generate an Rp2h null mouse (Fig. 1A). The gene trap cassette is located in the first intron of the mouse Rp2h gene, upstream of most truncation mutations associated with XLRP in the human gene (Fig. 1B). Genotyping with WT and trap-specific primers confirmed the trap’s presence in intron 1 (Fig. 1C). Because the Rp2h gene is located on the X-chromosome, the hemizygous male mouse (Rp2h−/y) is an RP2 knockout mouse. Immunoblotting using an anti-RP2 antibody verified gene disruption and ablation of RP2 expression. The RP2 polypeptide (40 kDa) was present in WT mouse retina lysates, but absent in the Rp2h−/y retina (Fig. 1D). Rp2hy/− males and Rp2h−/− females developed normally and were fertile. In vivo expression of RP2-eGFP fusion protein by neonatal electroporation localized the protein to the plasma membrane of photoreceptor inner segments and synaptic terminals (Fig. 1E), confirming previous results with polyclonal antibodies in human retina (16). Rhodopsin, prominently present in the outer segments, does not overlap with RP2.
Figure 1.
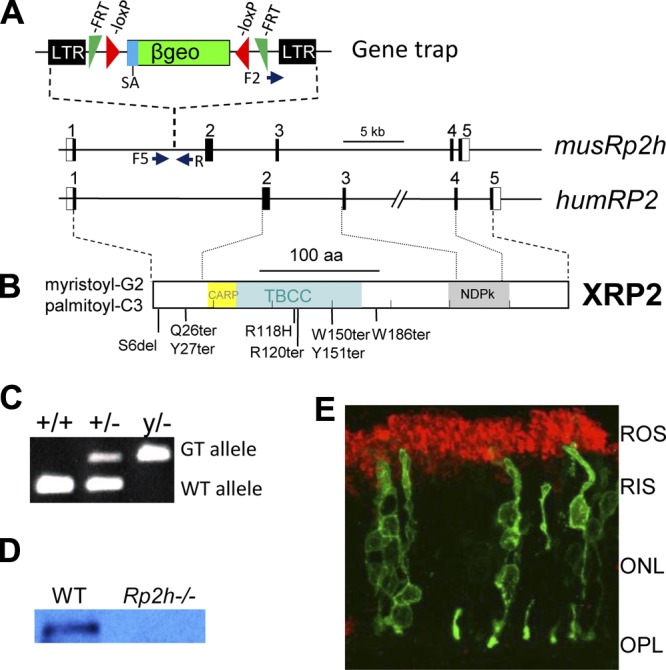
The mouse Rp2h gene knockout. A) The mouse Rp2h gene contains 5 exons. A gene trap located in intron 1 of mouse Rp2h truncates RP2 after lysine 32 (K31). The trap contains long terminal repeats (LTR), a splice acceptor (SA), a β-Geo selective cassette containing β-gal and neo genes. A pair of FRT and loxP recombination signals in antisense orientation flanks the β-Geo cassette. RP2-F1, RP2-F5, and RP2-R are the primers used for genotyping. Underneath, the human RP2 gene; positions of some of the most common RP-associated mutations are shown. B) The human RP2 polypeptide that is myristoylated and palmitoylated at the N-terminal. CARP, cyclase-associated domain; TBBC, tubulin binding cofactor C domain; NDP, nucleoside diphosphate kinase domain. C) Genotyping. The WT allele was genotyped using the primers of RP2-F1 and RP2-R. The mutant Rp2h allele carrying the gene trap was genotyped using the primers of RP2-F5 and RP2-R. D) Immunoblot of WT and knockout retina lysates using an anti-RP2 antibody. E) Localization of RP2-eGFP in WT mouse photoreceptors. Mouse photoreceptors expressing RP2-eGFP (green) were colabeled with rhodopsin (red). ROS, rod outer segment; RIS, rod inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer.
Progressive rod-cone dystrophy in the Rp2h knockout mouse
A comprehensive study with human RP2 patients showed that 9 of 10 patients displayed severe rod-cone dystrophy, suggesting that RP2 is required for normal cone function as well (8). To evaluate the rod and cone function in the Rp2h knockout mouse, we performed single-flash scotopic and photopic ERGs. Scotopic a-wave (Fig. 2A, B) and photopic b-wave amplitudes (Fig. 2D, E) were reduced in the Rp2h knockout animals as early as 1 mo of age, indicating that RP2 depletion affected the function of both rod and cone photoreceptors. This is consistent with observation of human patients and a previous report for Rp2h null mice (34). Both rod and cone photoreceptors of Rp2h null mice at 6 mo of age suffered severe vision loss, compared with 1-mo-old mice, as indicated by the ERG recordings (Fig. 2C, F). WT control mice at 6 mo of age showed little change in photoreceptor function. We followed the progression of retina degeneration up to 2 yr by immunohistochemistry (Fig. 3A–D), histology at 3 and 19 mo (Fig. 3E–G), and optical coherence tomography (Fig. 3H). At 3 mo and even 12 mo of age the outer nuclear layer thickness shows little change. Retina thickness at 2 yr by OCT is 261 μm for WT and 232 μm for mutant retina. These results demonstrate that ablation of the Rp2h gene caused slowly progressing rod-cone dystrophy.
Figure 2.

Scotopic and photopic electroretinography. A) Representative scotopic ERG traces recorded from WT and Rp2h knockout (KO) mice at 1 mo of age. B) Scotopic a-wave amplitudes from WT (n = 3) and Rp2h knockout (n = 3) mice at 1 mo of age. Each data point represents mean ± sd. C) Single-flash scotopic a-wave amplitudes at 0 db for WT and Rp2h knockout mice at the age of 1 mo (gray bars, average of 3 independent values) and 6 mo (white bars, average of 2 independent values). The difference between the WT and Rp2h knockout mice at 6 mo of age is statistically significant (t test, P < 0.05). D) Representative photopic ERG traces recorded from the WT and Rp2h knockout mice at 1 mo of age. E) Photopic b-wave amplitudes from the WT (n = 3) and Rp2h knockout (n = 3) mice at 1 mo of age. Each data point represents mean ± sd. The differences between the WT and Rp2h knockout mice from −4 db to 10 db (included) are statistically significant (t test, P < 0.05). F) Single-flash photopic b-wave amplitudes recorded at 10 db for WT and Rp2h knockout mice at the age of 1 mo (gray bars, average of 3 independent values) and 6 mo (white bars, average of 2 independent values). The differences between the WT and Rp2h knockout mice at one and 6 mo of age are statistically significant (t test, P < 0.05).
Figure 3.
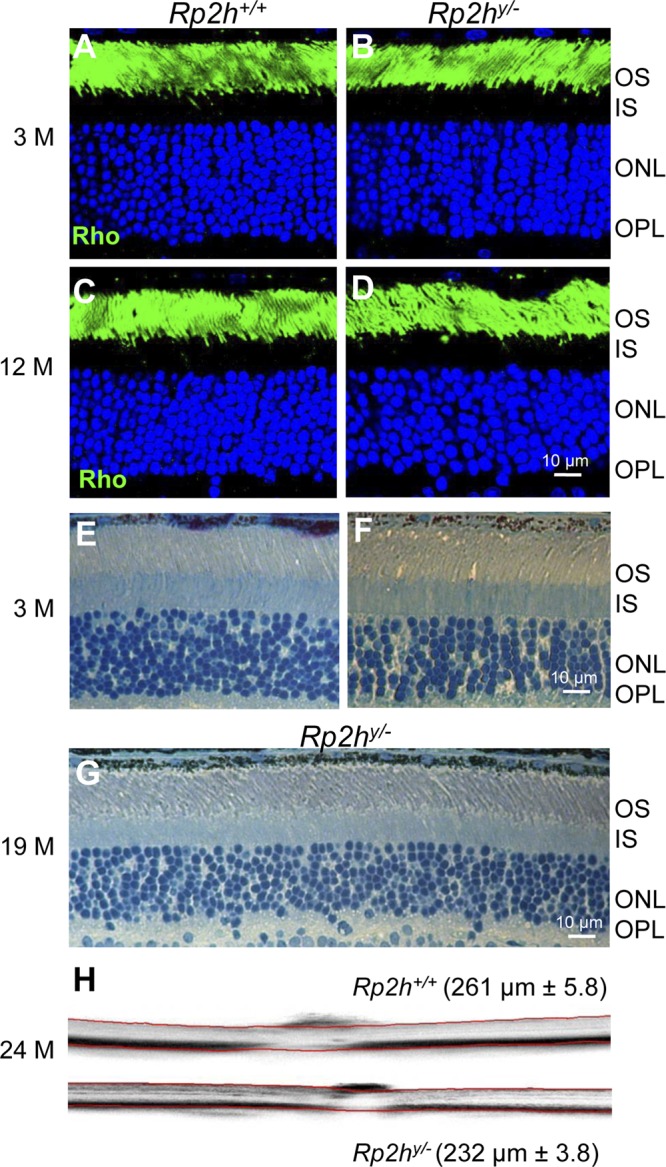
Slow rate of Rp2h−/− retina degeneration. A–D) Frozen sections of WT and mutant retina were probed with anti-rhodopsin antibodies (Rho, green) at 3 mo (3M) (A, B) and 12 mo of age (C, D). E–G) Plastic sections of WT and mutant retina at 3 mo (E, F) and of a 19-mo-old Rp2hy/− breeder (G). H) OCT of 2-yr-old WT (top) and mutant (bottom) retina. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer. Scale bar, 10 μm.
Cone pigments, rhodopsin, and GC1 target normally to Rp2h null outer segments
A previous report presenting an Rp2h knockout mouse suggested that cone opsins mislocalize to synaptic pedicles and cone opsin mislocalization represents an early step in RP2-XLRP (34). However, in our Rp2h knockout mice, ML-opsin trafficked normally to the cone outer segments at 1 mo (Fig. 4A, B) and 14 mo of age (Fig. 4C–F). Localization of S-opsin was also normal in the Rp2h knockout retina (Fig. 4G, H). Distributions of rhodopsin and GC1 in the Rp2h knockout retinas were indistinguishable from that in WT (Fig. 4I–L). Normal localizations of the transmembrane proteins GC1, as well as ML- and S-opsins, in the Rp2h−/− retina excludes RP2 as a factor in trans-Golgi sorting, trafficking and intraflagellar transport. Thus, cone opsin mistrafficking is unlikely the cause for slow photoreceptor degeneration in our Rp2h null mice.
Figure 4.

Localization of transmembrane proteins in photoreceptors. A–F) WT and Rp2h knockout mouse retina frozen sections (age of 1 mo, A, B; and age 14 mo, C–F were stained with anti-M/L-opsin antibody [green]). The 14-mo-old retinas (C–F) were counterstained with DAPI (blue) to contrast the outer nuclear layer. G, H) Distribution of S-opsin in the retina of the Rp2h−/− mouse. Mouse retina frozen sections at the age of 1 mo were stained with anti–S-opsin (red). I–L) One-month-old retina sections stained with anti-rhodopsin (I, J), and anti-GC1 (K, L). Scale bar, 10 μm.
RP2 is not required for ciliary targeting of transducin in photoreceptors
RP2 is present in cilia of cultured cells and may mediate the targeting of acylated proteins to cilia by regulating UNC119/ARL3 activity. Other in vitro experiments suggested that RP2 directly interacts with Tβ, but not Tβγ, facilitating trafficking of Tβ independently from Tγ to the destination membrane where assembly of Tβγ occurs (22, 23). Deletion of RP2 should therefore severely impede transducin trafficking. We examined the distribution of transducin in the Rp2h null retina and found that Tα (Fig. 5A) and Tβ (Fig. 5E) subunits (and by association Tβγ) localized prominently to the photoreceptor outer segments in the dark-adapted Rp2h null retina, indistinguishable from their localization in the WT retina. Furthermore, absence of RP2 had no effect on translocation of Tα to the inner segment in light (Fig. 5B) or its return from inner to outer segments during dark adaptation (Fig. 5C). These results suggest that RP2 and its ARL3-GAP activity are not involved in trafficking of acylated Tα or prenylated Tβγ to photoreceptor outer segments (see Discussion).
Figure 5.
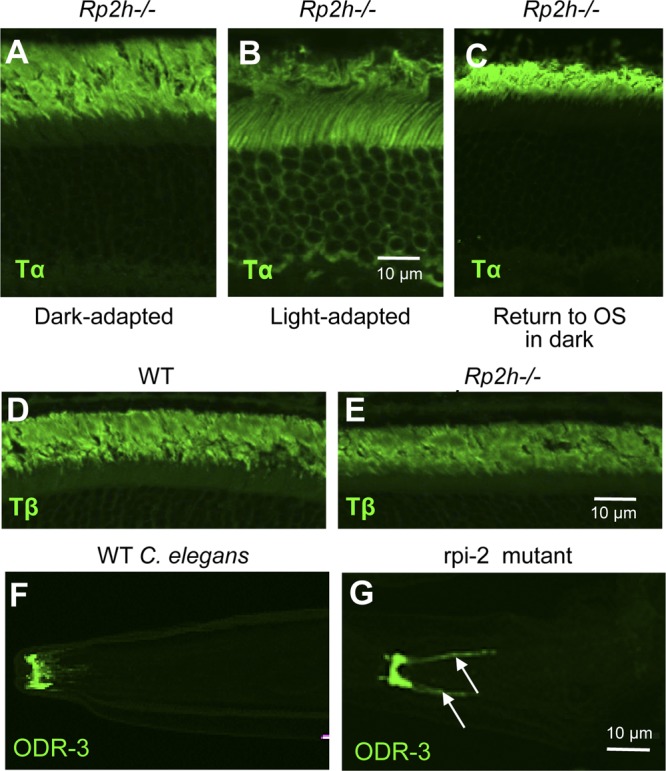
Trafficking of transducin in Rp2h−/− photoreceptors and of ODR-3 in mutant C. elegans. A–C) Normal trafficking to rod outer segments of Tα in dark-adapted retina (A), translocation to the inner segments in light (B), and return of Tα to photoreceptor outer segments during dark adaptation (C). Rp2h−/− mice were illuminated with light of 2000 lux for 40 min and returned to the dark as described previously (27). After 10 h in the dark, mouse eyeballs were harvested. Frozen sections were stained with anti-Tα antibody. D, E) WT and Rp2h−/− retina frozen sections (age of 1 mo) were stained with anti-Tβ antibody. F, G) Defect in transport of ODR-3 to olfactory cilia in rpi-2 mutant C. elegans. The WT and rpi-2 mutant C. elegans were stained with anti-ODR-3. Arrows point at mislocalized ODR-3 in the cell membrane.
Targeting the Gα homolog, ODR-3, to C. elegans olfactory neuron cilia requires rpi-2
ODR-3 is an N-myristoylated G-protein α-subunit involved in the olfaction cascade of C. elegans olfactory neurons (27). The C. elegans ortholog of the human RP2 gene is rpi-2. In WT C. elegans, GFP-tagged RPI-2 associates with sensory neuron membranes, enriched as a ringlike pattern at or near the basal body (43). We asked whether rpi-2 worm mutants would show trafficking defects of ODR-3 to cilia in olfactory neurons. C. elegans rpi-2 null olfactory neurons labeled with anti-ODR-3 antibody showed that ODR-3 mislocalized to the cell membrane (Fig. 5G, arrows). In contrast, ODR-3 locates correctly to WT olfactory cilia (Fig. 5F). Therefore, differing from mouse photoreceptors, RP2 is a regulator for ciliary transport of acylated G-protein in C. elegans.
RP2 is required for trafficking of PDE6 and GRK1 to cone outer segments
RP2 GAP activity is intrinsically linked to ARL3-GTP and its effector, PDE6D. Additionally, ARL3-GTP is known to act as GDF to release prenylated cargo from PDE6D (28). For this reason, we expected trafficking defects of prenylated proteins in Rp2h−/− rods and cones. As shown in Fig. 6A, rod PDE6 localizes exclusively to WT outer segments, but mislocalized in part to the inner segments and the synaptic region in the Rp2h−/− mouse. However, the bulk of rod PDE6 is located correctly in the mutant ROS. Weak mislocalization of rod PDE6 was also observed in Pde6d−/− rods (33). In contrast to rod PDE6, defects in OS transport of cone PDE6 (Fig. 6C, D) and GRK1 (Fig. 6E, F) were much more evident. Cone PDE6 and GRK1 polypeptides were nearly absent in cone outer segments of the Rp2h−/− retina (Fig. 6D, F), respectively. Immunoblots with WT and Rp2h−/− retina lysates documented normal levels of GRK1, PDE6α, suggesting that these proteins are not targeted for degradation (Fig. 6G).
Figure 6.
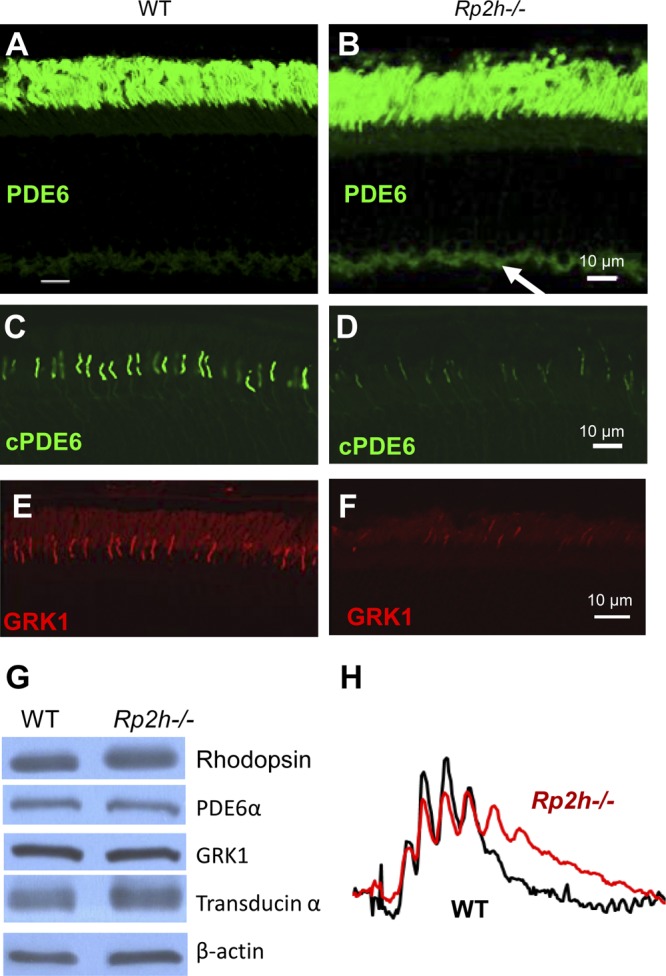
Trafficking defects of peripheral membrane proteins in RP2h−/− photoreceptors. A–F) Frozen sections were labeled with anti-PDE6 (A, B), anti-cone PDE6α (C, D), and anti-GRK1 (E, F). Scale bar, 10 μm. Arrow in (B) points at mislocalized PDE6 in the synaptic region. G) Western blot of WT and Rp2h−/− retina lysates. Lysates were probed with anti-rhodopsin, anti-PDE6A, anti-GRK1, anti-transducin, and anti–β-actin antibodies. PDE6 and GRK1 are present at normal levels, but fail to target to the OS. H) Comparison of normalized representative photopic ERG traces recorded at 10 db from the WT and Rp2h gene knockout mice at 1 mo of age. Recovery is delayed because GRK1 levels are reduced in outer segments.
GRK1 phosphorylates opsins and plays a key role in the shutdown of the rod and cone cascades (44). Insufficient GRK1 levels in cone outer segments delay shutdown of the photoresponse in Grk1−/− mice. As expected, photopic b-wave amplitudes of Rp2h−/− mice showed a delay in return to the baseline consistent with depletion of GRK1 (Fig. 6H). The delay is subtle because GRK1 levels are only reduced, not absent.
ARL3-GTP regulates association of prenylated PDE6 with PDE6D
Previously, we postulated that PDE6D escorts a subset of prenylated proteins as diffusible complexes to the target membrane, either a Trans-Golgi Network vesicle containing other outer segment proteins, or directly the proximal ciliary membrane where cargo for intraflagellar transport is assembled (33, 45). Discharge at the destination membrane requires interaction of ARL3-GTP with PDE6D forming a transient ternary complex (29). ARL3-GTP forces PDE6D to adopt a “closed pocket” conformation, by which PDE6D is unable to associate with a prenyl group (28). Closure of the binding pocket allows discharge of cargo to the destination membrane. To show that PDE6D in the presence of ARL3-GTP is compromised for interaction with prenylated proteins, we designed a biochemical protocol in which we tested the ability of PDE6D to extract PDE6 from ROS membranes in the presence of ARL3/GTP and ARL3/GMPPNP. In the presence of ARL3 and GMPPNP, a nonhydrolyzable analog of GTP, the amount of PDE6 extracted from ROS membranes was significantly reduced (Fig. 7, lanes 11–13), compared with that in the absence of ARL3 (Fig. 7, lane 8). However, in the presence of hydrolysable GTP, ARL3 did not interfere with the function of PDE6D (Fig. 7, lanes 3–7) even after 12 h of incubation, because ARL3-GTP is unstable and converts to inactive ARL3-GDP. ARL3-GMPNP forms a complex with PDE6D resulting in a closed conformation of the β-sandwich structure, which is unable to accommodate PDE6D. The experiment verifies biochemically that upon interaction with ARL3-GTP, PDE6D assumes a closed pocket conformation, which compromises interaction with PDE6 and other prenylated proteins.
Figure 7.
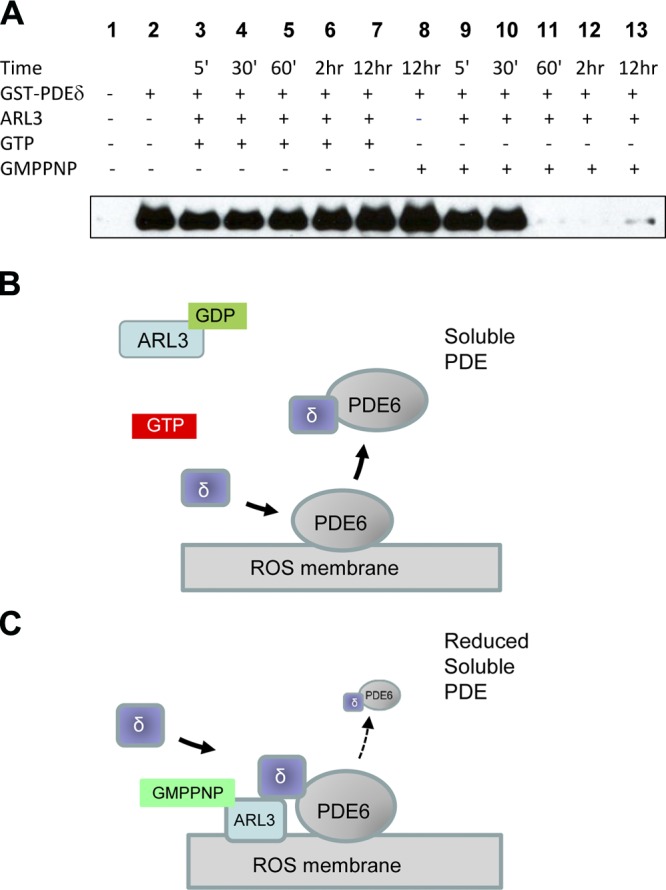
Inhibition of PDE6 extraction from rod outer segments by GMPPNP. A) Extraction protocol. The extraction was performed in the absence (lane 2) or presence of ARL3 (lanes 3–7 and 9–13). Addition of GTP to the reaction for a time from 5 min to 12 h had little effect on the extraction (lanes 3–7) compared with lane 2. Addition of GMPPNP (lanes 9–13) inhibited the extraction starting at 60 min (lane 11) of a 12 h incubation. GMPPMP alone did not affect the extraction (lane 8). B) Cartoon of PDE6 extraction. In the presence of ARL3-GDP and GTP, PDE6D is in the open conformation and extracts PDE6. C) In the presence of GMPPNP, ARL3, and PDE6D form a closed pocket conformation that is less potent in extracting PDE6.
DISCUSSION
Mutations in the human RP2 gene were originally associated with XLRP, a dystrophy that affects primarily the function of rod photoreceptors. A careful recent examination of RP2 patients revealed that RP2-XLRP also affected cones (8); 9 of 10 patients exhibited severe rod-cone degeneration, and 1 patient was diagnosed with cone-rod dystrophy. In 2 Rp2h gene knockout mouse models (Rp2h−/−, this work, and Rp2hΔx2/Δx2) (34), the disease symptoms clearly fall in the category of slowly progressing rod-cone dystrophy. In the Rp2hΔx2/Δx2 mouse, mislocalization of cone opsin was reported as the cause for disease, suggesting that RP2 was regulating opsin trafficking in cone photoreceptors (34). Opsin mislocalization was also reported in the zebra fish with Rp2 morpholino knockdown (46, 47). In our Rp2h−/− mouse, however, a gene trap truncates RP2 after exon 1 (K31), and mislocalization of cone opsin was not observed even at 14 mo of age (Fig. 4D, F). The reason for this discrepancy is unclear, but differences in phenotypes may reflect the complexity associated with the disease heterogeneity observed in patients. The RP2 knockout in Li et al. (34) was achieved by an in-frame deletion of exon 2 containing the TBCC domain, leaving intact the N- and C-terminal ends of RP2, including membrane association by acylation at G2 and the nucleoside diphosphate kinase domain. A dominant-negative effect of a membrane-bound protein consisting of exons 1, 3–5 containing a functional nucleoside diphosphate kinase domain on the canonical secretory pathway of cone pigments is not obvious but cannot be ruled out.
The present work indicates RP2 and its target, ARL3-GTP, play a key role in transport of prenylated proteins (PDE6 and GRK1), but apparently not acylated Tα or prenylated Tβγ from photoreceptor inner segments to outer segments. This is surprising, as RP2 was shown to form a complex with ARL3-GTP and UNC119 (24), an acyl-binding protein involved in trafficking of Tα by diffusion (27) but not with PDE6D. UNC119b, an UNC119 isoform, is a chaperone of N-acylated nephrocystine-3 (NPHP3), and in an Unc119b knockdown of IMCD3 cells, NPHP3 failed to reach cilia (26). Tα transport to photoreceptor outer segments in 2 independent Rp2h−/−mouse lines (Fig. 5A) (34) was not impeded, most likely because TαTβγ cotraffic with rhodopsin in vesicular transport carriers following the secretory pathway. The function of UNC119 as a Tα chaperone appears to be limited to return of Tα to the outer segments after massive translocation to the inner segment in constant light (27). Translocation of Tα in light and return to Rp2h−/− outer segments in a diffusible Tα-UNC119 complex proceeded normally (Fig. 5B, C). Normal return suggests that discharging Tα does not require RP2 GAP activity. Alternatively, the intrinsic GTPase activity of ARL3 is sufficient for hydrolysis of GTP bound to ARL3, at least on the time scale of transducin’s return (hours), or other nonspecific GAPs are available in rod cilia that can interact with ARL3. In contrast to Tα in rods, the acylated olfactory Gα subunit ODR-3 mistrafficked in rpi-2 C. elegans mutants (Fig. 5F, G), as has been observed for ODR-3 and GPA-13 in unc-119 C. elegans mutants (27).
A key role for ARL3-GTP, its GAP (RP2), and GEF (still unknown) in trafficking of prenylated proteins in photoreceptors was suspected early (48). The field accelerated dramatically in the last few years with crystallization of RP2-ARL3 (19, 20), RHEB-PDE6D (25, 28), UNC119-ARL3 (25), UNC119-Tα-peptide (27), and the RPGR-PDE6D complex (29). In the PDE6D knockout (33), absence of PDE6D severely impairs transport of GRK1 to rod and cone outer segments and cone PDE to cone outer segments. Autosomal and X-linked recessive rod-cone dystrophies caused by null mutations of the Pde6d and Rp2h genes in mouse, respectively, have similar phenotypes because the downstream targets (PDE6 and GRK1) are identical. In mouse, the RP2-XLRP is milder because trafficking of prenylated proteins to RP2h−/− cones is impeded, but not halted. Interestingly, a PDE6D null allele in human was associated with a severe syndromic ciliopathy (Joubert syndrome) (49).
To summarize transfer of prenylated GRK1 from the ER to its destination membrane, we formulated a model shown in Fig. 7. PDE6D extracts prenylated proteins from the ER forming a soluble complex in which the prenyl side chain inserts into the hydrophobic pocket of PDE6D (28, 32). The soluble GRK1-PDE6D complex forms a transient ternary complex with ARL3-GTP and docks to the destination membrane (Fig. 8A). The docking station has not been determined, but we propose that PDE6D docks to RPGR, which is known to interact with its N-terminal region (RCC1 domain) (29, 50). ARL3-GTP forces PDE6D to assume a closed pocket conformation to release its GRK1 cargo (Fig. 8A). By forcing cargo release, ARL3-GTP functions as a GDF (25). ARL3-GDP is a soluble protein present in inner segments, but a GEF-activated replacement of GDP by GTP in ARL3 shifts distribution of ARL3 from the cytosol to the membrane (26). Membrane association of ARL3-GTP/PDE6D-cargo is essential for RP2 to execute its GAP activity. It is likely that the ARL3-GEF is membrane-associated and located near RP2 and RPGR. RP2 catalyzes hydrolysis of GTP bound to ARL3 to GDP by accelerating ARL3’s intrinsic GTPase activity, up to 90,000-fold (20). Failing this, ARL3-GTP becomes abundant and PDE6D assumes a closed pocket conformation incompetent to extract prenylated proteins from membrane (Fig. 8B). We demonstrated biochemically that the closed pocket of PDE6D in complex with ARL3-GTP, indeed, “loses” its ability to extract PDE6 from ROS membranes (Fig. 7).
Figure 8.
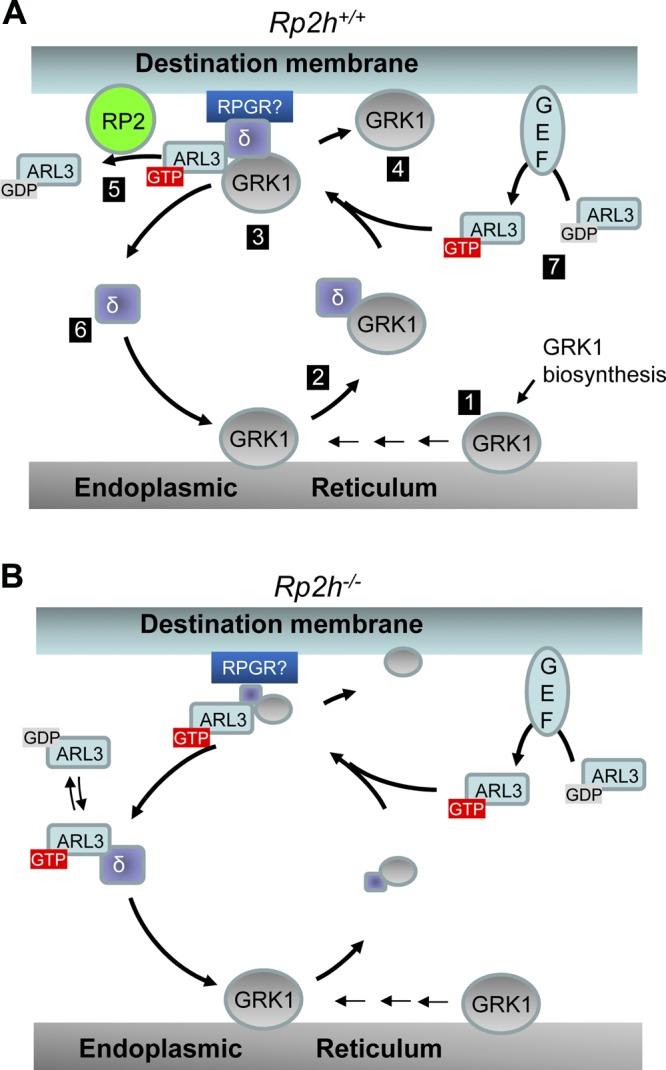
GRK1 trafficking from the ER to the destination membrane. A) Model of ARL3/RP2-dependent trafficking in WT photoreceptors. After biosynthesis and prenylation in the cytosol, GRK1 and other prenylated proteins dock to the ER membrane (position 1). Following CAAX processing (AAX cleavage, Cys-carboxymethylation), PDE6D extracts GRK1 forming a soluble complex (position 2). PDE6D-GRK1 then combines with ARL3-GTP to form a ternary complex, which docks to the membrane. The docking station is likely RPGR as PDE6D is known to interact with RPGR’s N-terminal region (29, 50, 54) (position 3). ARL3-GTP alters the open conformation of PDE6D to a closed conformation, evicting cargo (GRK1) to bind to membranes (position 4). ARL3-GTP dissociates from PDE6D and binds to RP2 accelerating GTP hydrolysis (position 5). PDE6D is released to begin another round of GRK1 transport (position 6). GDP/GTP exchange catalyzed by a GEF regenerates ARL3-GTP (position 7). B) Trafficking in Rp2h knockout photoreceptors. Absence of RP2 shuts down hydrolysis of GTP bound to ARL3 in step 5. Step 1 is therefore inefficient, as the ARL3-GTP/PDED complex inefficiently extracts GRK1, and steps 2–4 are carried out with low efficiency.
GEFs stimulate G-protein signaling by catalyzing GDP/GTP exchange, and GAPs terminate signaling by accelerating GTPase activity. One of the best-characterized GEFs is light-activated rhodopsin, which catalyzes GDP/GTP exchange on transducin as a first step in the mammalian phototransduction cascade. The GAP of transducin, a membrane-bound complex of RGS9/Gβ5L and R9AP, accelerates the intrinsic GTPase activity of ∼1/min (51) several thousand-fold to ∼6000/min (52). Absence of photoreceptor GAP activity causes bradyopsia (slow vision) in patients with null mutations in RGS9 or R9AP genes (53). Prominent features of these patients are photophobia and difficulty in seeing fast-moving objects (e.g., a baseball thrown toward a bradyopsia patient). The intrinsic GTPase activity of ARL3 is much lower than that of the Tα-GAP, with a kcat of 0.007/min (1.2 × 10−5 s−1). This low activity accelerates over 6000-fold with catalytic RP2 and 90,000-fold at saturating conditions of RP2 (20).
In summary, mutant photoreceptors lacking RP2 protein are unable to accelerate hydrolysis of GTP bound to ARL3. This defect results in persistently high concentration of hyperactive ARL3-GTP locking PDE6D in a closed conformation, which impedes its interaction with prenylated cargo, specifically GRK1 and PDE6. In human RP2-XLRP, the resulting photoreceptor degeneration is severe, whereas in mouse the RP2-XLRP is relatively mild and slowly progressing, affecting both rods and cones. The reason for this distinction is unknown, however a possible explanation may be provided by the ubiquitous presence of nonspecific GAP activities. In particular, RP2 is known to be a GAP for tubulin-GTP in the presence of TBCD (13). In mouse photoreceptors, additional ARL3 GAP activities maybe present reducing the level of ARL3-GTP and the severity of disease.
Acknowledgments
This work was supported by the U.S. National Institutes of Health Grants EY020969 (to H.Z.), EY08123, EY019298 (to W.B.), and EY014800-039003 (NEI core grant); National Science Foundation Grant NSFC81371030 (to H.Z.); and unrestricted grants to the University of Utah Department of Ophthalmology from Research to Prevent Blindness (to R.P.B.). W.B. is the recipient of an RPB Senior Investigator and an RPB Nelson Trust Award.
Glossary
- ARL3
ADP-ribosylation factor (Arf)-like 3 protein
- eGFP
enhanced green fluorescent protein
- GAP
GTPase activating protein
- GRK1
G-protein coupled receptor kinase 1
- log cd∣s∣m−2
log candela seconds per square meter
- PDE
cGMP phosphodiesterase
- PDE6D
phosphodiesterase 6 Δ
- RP2
retinitis pigmentosa 2 polypeptide
- RPGR
retinitis pigmentosa GTPase regulator
- T
transducin
REFERENCES
- 1.Hamel C. (2006) Retinitis pigmentosa. Orphanet J. Rare Dis. 1, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartong D. T., Berson E. L., Dryja T. P. (2006) Retinitis pigmentosa. Lancet 368, 1795–1809 [DOI] [PubMed] [Google Scholar]
- 3.Andréasson S., Ponjavic V., Abrahamson M., Ehinger B., Wu W., Fujita R., Buraczynska M., Swaroop A. (1997) Phenotypes in three Swedish families with X-linked retinitis pigmentosa caused by different mutations in the RPGR gene. Am. J. Ophthalmol. 124, 95–102 [DOI] [PubMed] [Google Scholar]
- 4.Fishman G. A., Farber M. D., Derlacki D. J. (1988) X-linked retinitis pigmentosa. Profile of clinical findings. Arch. Ophthalmol. 106, 369–375 [DOI] [PubMed] [Google Scholar]
- 5.Schwahn U., Lenzner S., Dong J., Feil S., Hinzmann B., van Duijnhoven G., Kirschner R., Hemberger M., Bergen A. A., Rosenberg T., Pinckers A. J., Fundele R., Rosenthal A., Cremers F. P., Ropers H. H., Berger W. (1998) Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat. Genet. 19, 327–332 [DOI] [PubMed] [Google Scholar]
- 6.Meindl A., Dry K., Herrmann K., Manson F., Ciccodicola A., Edgar A., Carvalho M. R., Achatz H., Hellebrand H., Lennon A., Migliaccio C., Porter K., Zrenner E., Bird A., Jay M., Lorenz B., Wittwer B., D'Urso M., Meitinger T., Wright A. (1996) A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet. 13, 35–42 [DOI] [PubMed] [Google Scholar]
- 7.Branham K., Othman M., Brumm M., Karoukis A. J., Atmaca-Sonmez P., Yashar B. M., Schwartz S. B., Stover N. B., Trzupek K., Wheaton D., Jennings B., Ciccarelli M. L., Jayasundera K. T., Lewis R. A., Birch D., Bennett J., Sieving P. A., Andreasson S., Duncan J. L., Fishman G. A., Iannaccone A., Weleber R. G., Jacobson S. G., Heckenlively J. R., Swaroop A. (2012) Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest. Ophthalmol. Vis. Sci. 53, 8232–8237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayasundera T., Branham K. E., Othman M., Rhoades W. R., Karoukis A. J., Khanna H., Swaroop A., Heckenlively J. R. (2010) RP2 phenotype and pathogenetic correlations in X-linked retinitis pigmentosa. Arch. Ophthalmol. 128, 915–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dandekar S. S., Ebenezer N. D., Grayson C., Chapple J. P., Egan C. A., Holder G. E., Jenkins S. A., Fitzke F. W., Cheetham M. E., Webster A. R., Hardcastle A. J. (2004) An atypical phenotype of macular and peripapillary retinal atrophy caused by a mutation in the RP2 gene. Br. J. Ophthalmol. 88, 528–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunter D. G., Fishman G. A., Kretzer F. L. (1988) Abnormal axonemes in X-linked retinitis pigmentosa. Arch. Ophthalmol. 106, 362–368 [DOI] [PubMed] [Google Scholar]
- 11.Sharon D., Bruns G. A., McGee T. L., Sandberg M. A., Berson E. L., Dryja T. P. (2000) X-linked retinitis pigmentosa: mutation spectrum of the RPGR and RP2 genes and correlation with visual function. Invest. Ophthalmol. Vis. Sci. 41, 2712–2721 [PubMed] [Google Scholar]
- 12.Breuer D. K., Yashar B. M., Filippova E., Hiriyanna S., Lyons R. H., Mears A. J., Asaye B., Acar C., Vervoort R., Wright A. F., Musarella M. A., Wheeler P., MacDonald I., Iannaccone A., Birch D., Hoffman D. R., Fishman G. A., Heckenlively J. R., Jacobson S. G., Sieving P. A., Swaroop A. (2002) A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 70, 1545–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartolini F., Bhamidipati A., Thomas S., Schwahn U., Lewis S. A., Cowan N. J. (2002) Functional overlap between retinitis pigmentosa 2 protein and the tubulin-specific chaperone cofactor C. J. Biol. Chem. 277, 14629–14634 [DOI] [PubMed] [Google Scholar]
- 14.Chapple J. P., Hardcastle A. J., Grayson C., Willison K. R., Cheetham M. E. (2002) Delineation of the plasma membrane targeting domain of the X-linked retinitis pigmentosa protein RP2. Invest. Ophthalmol. Vis. Sci. 43, 2015–2020 [PubMed] [Google Scholar]
- 15.Yoon J. H., Qiu J., Cai S., Chen Y., Cheetham M. E., Shen B., Pfeifer G. P. (2006) The retinitis pigmentosa-mutated RP2 protein exhibits exonuclease activity and translocates to the nucleus in response to DNA damage. Exp. Cell Res. 312, 1323–1334 [DOI] [PubMed] [Google Scholar]
- 16.Grayson C., Bartolini F., Chapple J. P., Willison K. R., Bhamidipati A., Lewis S. A., Luthert P. J., Hardcastle A. J., Cowan N. J., Cheetham M. E. (2002) Localization in the human retina of the X-linked retinitis pigmentosa protein RP2, its homologue cofactor C and the RP2 interacting protein Arl3. Hum. Mol. Genet. 11, 3065–3074 [DOI] [PubMed] [Google Scholar]
- 17.Holopainen J. M., Cheng C. L., Molday L. L., Johal G., Coleman J., Dyka F., Hii T., Ahn J., Molday R. S. (2010) Interaction and localization of the retinitis pigmentosa protein RP2 and NSF in retinal photoreceptor cells. Biochemistry 49, 7439–7447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapple J. P., Hardcastle A. J., Grayson C., Spackman L. A., Willison K. R., Cheetham M. E. (2000) Mutations in the N-terminus of the X-linked retinitis pigmentosa protein RP2 interfere with the normal targeting of the protein to the plasma membrane. Hum. Mol. Genet. 9, 1919–1926 [DOI] [PubMed] [Google Scholar]
- 19.Kühnel K., Veltel S., Schlichting I., Wittinghofer A. (2006) Crystal structure of the human retinitis pigmentosa 2 protein and its interaction with Arl3. Structure 14, 367–378 [DOI] [PubMed] [Google Scholar]
- 20.Veltel S., Gasper R., Eisenacher E., Wittinghofer A. (2008) The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat. Struct. Mol. Biol. 15, 373–380 [DOI] [PubMed] [Google Scholar]
- 21.Hurd T., Zhou W., Jenkins P., Liu C. J., Swaroop A., Khanna H., Martens J., Hildebrandt F., Margolis B. (2010) The retinitis pigmentosa protein RP2 interacts with polycystin 2 and regulates cilia-mediated vertebrate development. Hum. Mol. Genet. 19, 4330–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz N., Novoselova T. V., Wait R., Hardcastle A. J., Cheetham M. E. (2012) The X-linked retinitis pigmentosa protein RP2 facilitates G protein traffic. Hum. Mol. Genet. 21, 863–873 [DOI] [PubMed] [Google Scholar]
- 23.Schwarz N., Hardcastle A. J., Cheetham M. E. (2012) Arl3 and RP2 mediated assembly and traffic of membrane associated cilia proteins. Vision Res. 75, 2–4 [DOI] [PubMed] [Google Scholar]
- 24.Veltel S., Kravchenko A., Ismail S., Wittinghofer A. (2008) Specificity of Arl2/Arl3 signaling is mediated by a ternary Arl3-effector-GAP complex. FEBS Lett. 582, 2501–2507 [DOI] [PubMed] [Google Scholar]
- 25.Ismail S. A., Chen Y. X., Miertzschke M., Vetter I. R., Koerner C., Wittinghofer A. (2012) Structural basis for Arl3-specific release of myristoylated ciliary cargo from UNC119. EMBO J. 31, 4085–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright K. J., Baye L. M., Olivier-Mason A., Mukhopadhyay S., Sang L., Kwong M., Wang W., Pretorius P. R., Sheffield V. C., Sengupta P., Slusarski D. C., Jackson P. K. (2011) An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 25, 2347–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H., Constantine R., Vorobiev S., Chen Y., Seetharaman J., Huang Y. J., Xiao R., Montelione G. T., Gerstner C. D., Davis M. W., Inana G., Whitby F. G., Jorgensen E. M., Hill C. P., Tong L., Baehr W. (2011) UNC119 is required for G protein trafficking in sensory neurons. Nat. Neurosci. 14, 874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ismail S. A., Chen Y. X., Rusinova A., Chandra A., Bierbaum M., Gremer L., Triola G., Waldmann H., Bastiaens P. I., Wittinghofer A. (2011) Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat. Chem. Biol. 7, 942–949 [DOI] [PubMed] [Google Scholar]
- 29.Wätzlich D., Vetter I., Gotthardt K., Miertzschke M., Chen Y. X., Wittinghofer A., Ismail S. (2013) The interplay between RPGR, PDEδ and Arl2/3 regulate the ciliary targeting of farnesylated cargo. EMBO Rep. 14, 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Constantine R., Zhang H., Gerstner C. D., Frederick J. M., Baehr W. (2012) Uncoordinated (UNC)119: coordinating the trafficking of myristoylated proteins. Vision Res. 75, 26–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H., Constantine R., Frederick J. M., Baehr W. (2012) The prenyl-binding protein PrBP/δ: a chaperone participating in intracellular trafficking. Vision Res. 75, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanzal-Bayer M., Renault L., Roversi P., Wittinghofer A., Hillig R. C. (2002) The complex of Arl2-GTP and PDE delta: from structure to function. EMBO J. 21, 2095–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H., Li S., Doan T., Rieke F., Detwiler P. B., Frederick J. M., Baehr W. (2007) Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl. Acad. Sci. USA 104, 8857–8862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L., Khan N., Hurd T., Ghosh A. K., Cheng C., Molday R., Heckenlively J. R., Swaroop A., Khanna H. (2013) Ablation of the X-linked retinitis pigmentosa 2 (Rp2) gene in mice results in opsin mislocalization and photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci. 54, 4503–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mattapallil M. J., Wawrousek E. F., Chan C. C., Zhao H., Roychoudhury J., Ferguson T. A., Caspi R. R. (2012) The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci. 53, 2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda T., Cepko C. L. (2007) Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA 104, 1027–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H., Fan J., Li S., Karan S., Rohrer B., Palczewski K., Frederick J. M., Crouch R. K., Baehr W. (2008) Trafficking of membrane-associated proteins to cone photoreceptor outer segments requires the chromophore 11-cis-retinal. J. Neurosci. 28, 4008–4014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avasthi P., Watt C. B., Williams D. S., Le Y. Z., Li S., Chen C. K., Marc R. E., Frederick J. M., Baehr W. (2009) Trafficking of membrane proteins to cone but not rod outer segments is dependent on heterotrimeric kinesin-II. J. Neurosci. 29, 14287–14298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papermaster D. S., Dreyer W. J. (1974) Rhodopsin content in the outer segment membranes of bovine and frog retinal rods. Biochemistry 13, 2438–2444 [DOI] [PubMed] [Google Scholar]
- 40.Baehr W., Devlin M. J., Applebury M. L. (1979) Isolation and characterization of cGMP phosphodiesterase from bovine rod outer segments. J. Biol. Chem. 254, 11669–11677 [PubMed] [Google Scholar]
- 41.Zhang H., Liu X. H., Zhang K., Chen C. K., Frederick J. M., Prestwich G. D., Baehr W. (2004) Photoreceptor cGMP phosphodiesterase delta subunit (PDEdelta) functions as a prenyl-binding protein. J. Biol. Chem. 279, 407–413 [DOI] [PubMed] [Google Scholar]
- 42.Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 43.Blacque O. E., Perens E. A., Boroevich K. A., Inglis P. N., Li C., Warner A., Khattra J., Holt R. A., Ou G., Mah A. K., McKay S. J., Huang P., Swoboda P., Jones S. J., Marra M. A., Baillie D. L., Moerman D. G., Shaham S., Leroux M. R. (2005) Functional genomics of the cilium, a sensory organelle. Curr. Biol. 15, 935–941 [DOI] [PubMed] [Google Scholar]
- 44.Lyubarsky A. L., Chen C., Simon M. I., Pugh E. N. Jr (2000) Mice lacking G-protein receptor kinase 1 have profoundly slowed recovery of cone-driven retinal responses. J. Neurosci. 20, 2209–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karan S., Zhang H., Li S., Frederick J. M., Baehr W. (2008) A model for transport of membrane-associated phototransduction polypeptides in rod and cone photoreceptor inner segments. Vision Res. 48, 442–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shu X., Zeng Z., Gautier P., Lennon A., Gakovic M., Cheetham M. E., Patton E. E., Wright A. F. (2011) Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Invest. Ophthalmol. Vis. Sci. 52, 2960–2966 [DOI] [PubMed] [Google Scholar]
- 47.Patil S. B., Hurd T. W., Ghosh A. K., Murga-Zamalloa C. A., Khanna H. (2011) Functional analysis of retinitis pigmentosa 2 (RP2) protein reveals variable pathogenic potential of disease-associated missense variants. PLoS ONE 6, e21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H., Frederick J. M., Baehr W. (2006) Functional study of photoreceptor PDEdelta. Adv. Exp. Med. Biol. 572, 485–490 [DOI] [PubMed] [Google Scholar]
- 49.Thomas S., Wright K. J., Le Corre S., Micalizzi A., Romani M., Abhyankar A., Saada J., Perrault I., Amiel J., Litzler J., Filhol E., Elkhartoufi N., Kwong M., Casanova J. L., Boddaert N., Baehr W., Lyonnet S., Munnich A., Burglen L., Chassaing N., Encha-Ravazi F., Vekemans M., Gleeson J. G., Valente E. M., Jackson P. K., Drummond I. A., Saunier S., Attie-Bitach T. (2014) A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum. Mutat. 35, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linari M., Ueffing M., Manson F., Wright A., Meitinger T., Becker J. (1999) The retinitis pigmentosa GTPase regulator, RPGR, interacts with the delta subunit of rod cyclic GMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 96, 1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baehr W., Morita E. A., Swanson R. J., Applebury M. L. (1982) Characterization of bovine rod outer segment G-protein. J. Biol. Chem. 257, 6452–6460 [PubMed] [Google Scholar]
- 52.Arshavsky V. Y., Wensel T. G. (2013) Timing is everything: GTPase regulation in phototransduction. Invest. Ophthalmol. Vis. Sci. 54, 7725–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishiguchi K. M., Sandberg M. A., Kooijman A. C., Martemyanov K. A., Pott J. W., Hagstrom S. A., Arshavsky V. Y., Berson E. L., Dryja T. P. (2004) Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature 427, 75–78 [DOI] [PubMed] [Google Scholar]
- 54.Linari M., Ueffing M., Manson F., Wright A., Meitinger T., Becker J. (1999) The retinitis pigmentosa GTPase regulator, RPGR, interacts with the delta subunit of rod cyclic GMP phosphodiesterase. Proc. Natl. Acad. Sci. U. S. A. 96, 1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]